

Summary
The scarcity of methods to visualize the activity of individual cell surface proteases in situ has hampered basic research and drug development efforts. In this chapter, we describe a simple, sensitive, and noninvasive assay that uses nontoxic reengineered bacterial cytotoxins with altered protease cleavage specificity to visualize specific cell surface proteolytic activity in single living cells. The assay takes advantage of the absolute requirement for site-specific endoproteolytic cleavage of cell surface-bound anthrax toxin protective antigen for its capacity to translocate an anthrax toxin lethal factor-β-lactamase fusion protein to the cytoplasm. A fluorogenic β-lactamase substrate is then used to visualize the cytoplasmically translocated anthrax toxin lethal factor-β-lactamase fusion protein. By using anthrax toxin protective antigen variants that are reengineered to be cleaved by furin, urokinase plasminogen activator, or metalloproteinases, the cell surface activities of each of these proteases can be specifically and quantitatively determined with single cell resolution. The imaging assay is excellently suited for fluorescence microscope, fluorescence plate reader, and flow cytometry formats, and it can be used for a variety of purposes.
Keywords: Anthrax toxin, β-lactamase, CCF2/AM, Cell surface proteolysis, Flow cytometry, Fluorescence microscopy, Fluorescence plate reader, Furin, Metalloproteinases, Urokinase plasminogen activator
1. Introduction
Proteolysis in the extracellular environment is essential to all aspects of life, including development, homeostasis, tissue repair, tissue remodeling, and reproduction. Dysregulated extracellular proteolysis is also causally linked to the genesis or progression of a large number of human diseases. Important examples include myocardial infarction, stroke, rheumatoid and osteoarthritis, periodontal disease, bacterial infection, and cancer. Physiological and pathological proteolysis within the extracellular environment takes place predominantly on the cell surface, where inactive protease zymogens, protease receptors, protease inhibitors, and other regulatory proteins assemble into complex multiprotein structures that mediate the sequential and spatially restricted activation of protease zymogens and their subsequent cleavage of target proteins (1–7).
Despite its critical physiological and pathological importance, several fundamental aspects of extracellular proteolysis are still quite poorly understood. These include the molecular pathways that initiate the activation and inhibition of many zymogen cascades and the biologically relevant substrates for many, if not the majority, of pericellular proteases. In particular, the lack of imaging agents that can specifically visualize the endogenous activity of individual proteases and monitor the efficacy of their pharmacological inhibition in situ is widely recognized as a serious impediment to both basic research efforts and to the use of proteases as therapeutic targets (8, 9). Considerable efforts therefore have been undertaken to develop assays for the imaging of specific protease activity in biological systems, and a wide variety of approaches have been taken (10, 11).
In this chapter, we describe a simple noninvasive method for imaging specific cell surface proteolytic activity in single living cells that is based on the use of modified bacterial cytotoxins (12). The assay takes advantage of: (a) the absolute requirement for site-specific endoproteolytic cleavage of anthrax toxin protective antigen (PrAg) for toxin activation (13), (b) the ability to reengineer the protease cleavage site in PrAg to be specifically cleaved by various different cell surface-associated proteases (14–18), (c) the ability of anthrax toxin lethal factor (LF) to shuttle heterologous proteins to the cytoplasm in a PrAg cleavage-dependent manner (19, 20), and (d) the development of highly sensitive optical assays for the detection of cytoplasmic β-lactamase activity (21). The imaging assay combines high protease specificity – obtained by the incorporation of the target peptide cleavage sequence in a macromolecular context and the confinement of the cleavage reaction to the cell surface (16, 22) – with the exquisite sensitivity that is associated with enzymatic signal amplification via β-lactamase (21). We provide protocols for the application of the assay for fluorescence microscopy, fluorescent plate reader, and flow cytometry formats.
The devised assay can be tailored to detect the activity or inhibition of any cell surface protease for which a specific peptide substrate can be derived (16, 22–25). Examples of applications for which the imaging assay is particularly suited include: (1) automated high-throughput screening of large chemical libraries for protease inhibitory or stimulating compounds (26); (2) expression cloning-based strategies for the identification of new activators and inhibitors of specific cell surface proteolytic pathways; (3) siRNA and shRNA-based high-throughput screens for the identification of pathways regulating specific cell surface protease activity.
2. Materials
2.1. Modified Anthrax Toxins
The generation and purification of recombinant wild-type PrAg, PrAg-33 (164RKKR167 of PrAg changed to RAAR) (14), PrAg-U2 (164RKKR167 of PrAg changed to GSGRSA) (17, 22), PrAg-L1 (164RKKR167 of PrAg changed to GPLGMLSQ), and PrAg-U7 (164RKKR167 changed to GG) (16) is described in Chapter ?, ?TITLE?, page ?-?. Store at −80°C in aliquots of 50–500 μL at concentrations >500 μg/mL.
The LF/β -lactamase fusion protein (LF/β-Lac) contains DNA sequences encoding the PrAg-binding region of LF (amino acids 1–254) fused 5′ of DNA sequences encoding amino acids 19–286 of the TEM-1 β-lactamase gene from pBluescript (Stratagene, La Jolla, CA). To generate LF/β-Lac, these DNA sequences were fused downstream of a DNA sequence encoding a glutathione-S-transferase (GST) protein with a thrombin cleavage site linking GST and LF/β-Lac (27, 12). BL21 competent bacteria (Invitrogen, Carlsbad, CA) transformed with the expression plasmid encoding the GST- LF/β-Lac fusion protein are grown in Super Broth Medium (Biosource, Camarillo, CA) supplemented with 50 μg/mL ampicillin. Cultures are grown at 37°C until an absorbance of 0.8 at 600 nm. The cells are then induced with isopropyl β-D-thiogalactopyranoside (Sigma, St. Louis, MO) to a final concentration of 0.5 mM for 5 h at 37°C. The cells are pelleted by centrifugation at 3,000 × g for 15 min in a 4°C high speed centrifuge and resuspended in ice-cold bacterial lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, and a protease inhibitor cocktail (Amersham Biosciences, Piscataway, NJ). Bacterial pellets are lysed by five rounds of freeze/thawing in liquid nitrogen, followed by sonication for 30 s using a probe sonicator. The lysate is clarified by centrifugation 3,000 × g for 10 min in a 4°C high-speed centrifuge and incubated with 1 mL glutathione-Sepharose resin slurry (Amersham Biosciences) for 2 h at 4°C. The resin is washed twice with ice-cold lysis buffer without protease inhibitors, and the purified protein is released from the resin by incubation with 2 mL thrombin cleavage buffer (20 mM Tris-HCl, pH 8.4, 150 mM NaCl, 2.5 mM CaCl2 containing 100 Units thrombin/mL). Aliquots of 0.5 mg/mL LF/β-Lac are stored at −80°C.
2.2. Tissue Culture and Imaging Assay
Phenol red-free DMEM (Gibco-BRL, Gaithersburg, MD) is stored at 4°C.
Gentamicin reagent solution is purchased as a liquid 50× solution from Gibco-BRL. Store at 4°C.
L-Glutamine is purchased as a 100× solution from Gibco-BRL. Store at −20°C.
0.05% trypsin 1 mM EDTA solution is purchased as a 1× solution from Gibco-BRL and stored at 4°C.
Poly-D-lysine is purchased from Sigma. Make a sterile 0.1 mg/mL solution in PBS. Store at 4°C.
Ten cm tissue culture plates, eight-well chamber slides, and 96-well black walled tissue culture plates are purchased from Corning, Lowell, MA.
Hanks' balanced salt solution is purchased as a sterile 1× solution from Sigma. Store at 4°C.
Probenicid is purchased from Sigma.
Human pro-uPA (single-chain uPA, no. 107) and human Glu-plasminogen is purchased from American Diagnostica, Inc. (Greenwich, CT).
2.3. β-Lactamase Substrate and Substrate Loading Solutions
“Solution A” [Coumarin cephalosporin fluorescein acetoxymethyl ester (CCF2/AM) in dimethyl sulfoxide (DMSO)]. CCF2/AM is purchased as a dried powder from Invitrogen (Carlsbad, CA). Store at −80°C and protect from light. Dissolve 1 mg CCF2/AM in 924 μL DMSO. Store protected from light in 100 μL aliquots at −80°C. Bring the vial of CCF2/AM to room temperature before removing the desired amount of reagent to reduce intrusion of moisture into the stock solution.
“Solution B” (100 mg/mL Pluronic®-F127 surfactant in DMSO with 0.1% acetic acid) is purchased from Invitrogen. Store at room temperature. Protect from direct light.
“Solution C” (24% w/w PEG 400, 18% TR-40 w/w in water) is purchased from Invitrogen. Store at room temperature. Protect from direct light.
Preparation of 6× “Standard substrate loading solution” (must be prepared immediately before use): In an opaque test tube, add 1 volume “Solution A” to 5 volumes “Solution B” and vortex. Add the combined “Solution A” and Imaging Specific Cell Surface Protease Activity in Living Cells “Solution B” to 77 volumes “Solution C” and vortex. Reagents should be exposed to as little direct light as possible.
Preparation of 6× “Alternative substrate loading solution”: Dissolve 25 g probenecid in 219 mL of 400 mM NaOH in a flask using a stir bar. Add 219 mL of sodium phosphate buffer (pH 8.0). Adjust the pH to 8.0 using 1 M HCl and/or 1 M NaOH. If a precipitate forms, bring into solution by continuous stirring. Freeze the probenecid solution in 1 mL aliquots at −20°C. Immediately before use in an opaque test tube, add 1 volume “Solution A” to 5 volumes “Solution B” and vortex. Add the combined “Solution A” and “Solution B” to 77 volumes “Solution C” and vortex. Add 4 volumes probenecid solution to combined “Solution A,” “Solution B,” and “Solution C.” Reagents should be exposed to as little direct light as possible.
3. Methods
The principle of the imaging assay is shown schematically in Fig. 1. Live cell imaging of furin (Fig 2), uPA (Fig. 3), and metalloproteinase activity (Fig 4) can be performed using a wide variety of cultured primary cells and cell lines, by treating the cells with PrAg-33, PrAg-U2, or PrAg-L1, respectively, in combination with LF/β–Lac. Cleavage of PrAg-33, PrAg-U2, or PrAg-L1 by, respectively, furin, uPA, or metalloproteinases allows the translocation of LF/β-Lac to the cytoplasm. Cytoplasmic LF/β–Lac is imaged by the incubation of cells with the fluorogenic β-lactamase substrate, CCF2/AM, and detection of hydrolyzed CCF2/AM by a fluorescence microscope, a fluorescence plate reader, or a flow cytometer. CCF2/AM fluoresces green before its hydrolysis by β-lactamase and fluoresces blue after hydrolysis by β-lactamase. The noncleavable PrAg variant, PrAg-U7 combined with LF/β–Lac serves as a negative control, and wild-type PrAg, which is cleaved by furin as well as many furin-like proprotein convertases, combined with LF/β–Lac serves as a positive control. The basic protocols for imaging cell surface protease activity by fluorescence microscopy, fluorescence plate reader, and flow cytometry are given below. The cells can be subjected to a large variety of manipulations and treatments prior to imaging and during the imaging process depending on the specific purpose of the experiment (see Subheading 1).
Fig. 1.
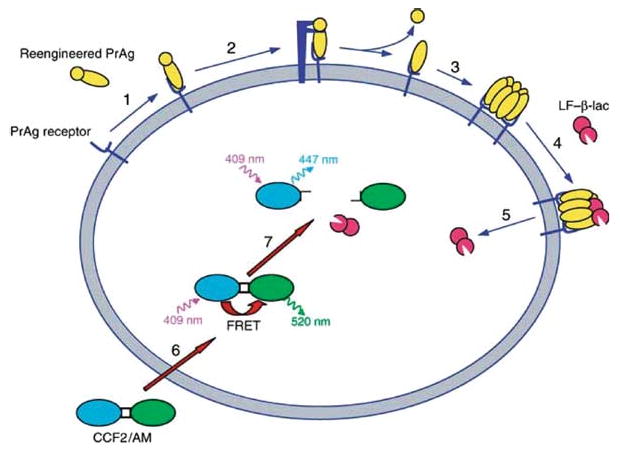
Principle of cell surface protease activity imaging assay. (1) PrAg mutants with reengineered protease cleavage specificity bind to ubiquitous high affinity cell surface receptors. (2) The PrAg mutants are cleaved by the cell surface proteolytic activity to be imaged. (3) The PrAg fragment that remains on the cell surface heptamerizes. (4) High-affinity binding sites for LF/β-Lac are generated by heptamerization of the PrAg. (5) LF/β-Lac is translocated to the cytoplasm after endocytosis of the PrAg-LF/β-Lac complex (not shown). (6) CCF2/AM is added to cells and diffuses into the cytoplasm where it is trapped by hydrolysis of its hydroxethyl ester groups by nonspecific cytoplasmic esterases. (7) LF/β-Lac hydrolyses the cephalosporin ring of CCF2/AM, causing a shift in fluorescence emission from 520 nm (green light) to 447 nm (blue light) after excitation of cells at 409 nm. Blue fluorescence emission by a cell demonstrates specific cell surface proteolytic activity. Reproduced from ref.12.
Fig. 2.
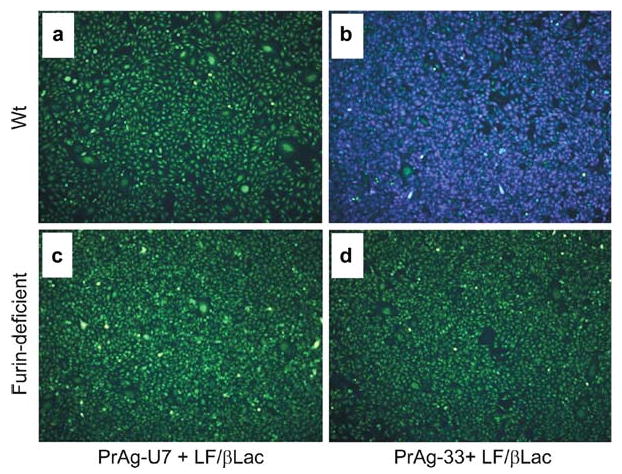
Specific imaging of endogenous cell surface furin proteolytic activity in single living cells. Wild-type (a and b) or furin-deficient (c and d) Chinese hamster ovary cells were incubated with 90 nM LF/β-Lac and 26 nM (2 μg/mL) PrAg-U7 (a and c), or 90 nM LF/β-Lac and 26 nM PrAg-33 (b and d) for 60 min. Thereafter, 1.5 μM CCF2/AM was added to the cells for 60 min at room temperature. The CCF2/AM remaining in the medium was removed by washing, and the cells were incubated for 60 min at room temperature to allow for CCF2/AM hydrolysis. The cells then were subjected to fluorescence microscopy using an excitation wavelength of 405 nm and emission filters of 530 nm (green light) and 460 nm (blue light). Specific imaging of furin proteolytic activity is demonstrated by the bright blue fluorescence of wild-type cells (b), but not furin-deficient cells (d), treated with LF/β-Lac and PrAg-33, or wild-type and furin-deficient cells treated with LF/β-Lac and PrAg-U7 (a and c). Reproduced in part from ref.12.
Fig. 3.
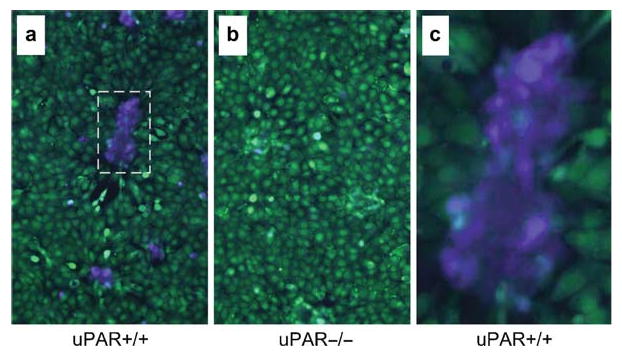
Specific imaging of endogenous cell surface uPA activity in mixed tumor cell stromal cell cultures. Mixed cultures of HN-26 oral squamous carcinoma cells and wild type (uPAR+/+, a and c) or urokinase plasminogen activator receptor (uPAR) knockout (uPAR-/-, b) fibroblasts were treated with 90 nM LF/β-Lac (5 μg/mL), 26 nM (2 μg/mL) PrAg-U2, and 11 nM (1 μg/mL) plasminogen for 60 min. Thereafter, 1.5 μM CCF2/AM was added to the cells for 60 min at room temperature. The CCF2/AM remaining in the medium was removed by washing, and the cells were incubated for 60 min at room temperature to allow for CCF2/AM hydrolysis. The cells then were subjected to fluorescence microscopy using an excitation wavelength of 405 nm and emission filters of 530 (green light) and 460 nm (blue light). Cell surface uPA activity is exclusively observed in nontransformed fibroblasts (a and c) and is dependent on the expression of uPAR, as shown by the strong blue fluorescence of islands of uPAR+/+ (a), but not in uPAR−/− (b) fibroblasts, and by green fluorescence of the tumor cells. (c) is a high magnification of the boxed area in (a) illustrating the confinement of uPA activity to fibroblasts. Reproduced in part from ref.12.
Fig. 4.
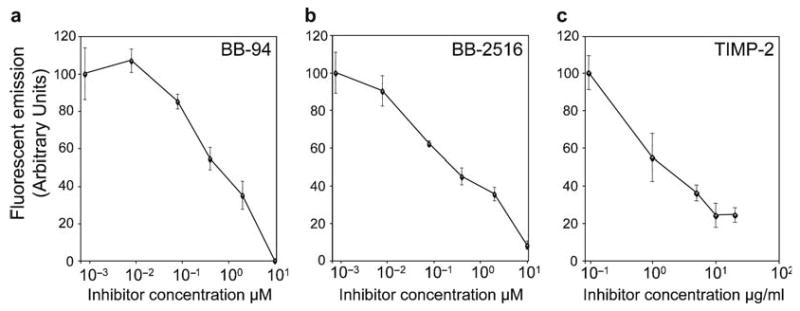
Quantitative analysis of the inhibition of cell surface metalloproteinase activity in human tumor cells. HT-1080 fibrosarcoma cells were seeded in a 96-well microtiter plate and treated for 60 min with 90 nM (5 μg/mL) LF/β-Lac and 26 nM (2 μg/mL) PrAg-L1 in the presence of increasing concentrations of the metalloprotease inhibitors BB-94 (a), BB-2516 (b), and TIMP-2 (c). Thereafter, 1.5 μM CCF2/AM was added to the cells for 60 min at room temperature. The CCF2/AM remaining in the medium was removed by washing, and the cells were incubated for 60 min at room temperature to allow for CCF2/AM hydrolysis. Fluorescence emission was recorded with a plate reader using 405 nm excitation and 460/25 nm bandpass for blue fluorescence and 535/25 nm bandpass for green fluorescence. The data are expressed as mean ± standard error of the mean of triplicate determinations. Reproduced in part from ref.12.
3.1. Imaging Cell Surface Proteolytic Activity by Fluorescence Microscopy Analysis
Grow cells to be imaged in phenol red-free DMEM supplemented with 10% fetal bovine serum (FBS), L-glutamine, and gentamicin reagent solution (see Note 1). Avoid using penicillin and other β-lactam-based antibiotics that are substrates for β-lactamase.
Treat eight-well chamber slides with 250 μL/well 0.1 mg/mL poly-D-lysine in PBS at 37°C in a tissue culture incubator for 60 min to facilitate cell adhesion. Other treatments of chamber slides can be used as is expedient for the adhesion of the cell line or primary cells employed (see Note 2).
Seed cells on the eight-well chamber slides the day prior to imaging in phenol red-free DMEM supplemented with 10% FBS, L, and gentamicin reagent solution. The seeding of 2 × 105 cells/well is appropriate for most cells (see Note 3). Incubate overnight in a standard 37°C tissue culture incubator.
Gently wash the cells twice in 0.5 mL 37°C phenol red-free DMEM.
Add either PrAg-33, PrAg-U2, or PrAg-L1 to 26 nM (2 μg/mL) final concentration together with LF/β-Lac fusion protein to 90 nM (5 μg/mL) final concentration in 0.25 mL 37°C phenol red-free DMEM. When imaging uPA cell surface proteolytic activity, include 1 μg/mL Glu-plasminogen with or without 100 ng/mL pro-uPA (see Note 4). As a negative control, substitute PrAg-33, PrAg-U2, or PrAg-L1 with PrAg-U7. As a positive control, substitute PrAg-33, PrAg-U2, or PrAg-L1 with wild-type PrAg (see Note 5).
Incubate the PrAg and LF/β-Lac-treated cells for 60 min at 37°C in a tissue culture incubator.
Wash cells twice with room temperature phenol red-free DMEM. Transfer plate to room temperature (see Note 6).
Add 40 μL of 6× “Standard substrate loading solution” or “Alternative substrate loading solution” (see Subheading 2.3) to each chamber well containing 210 μL of room temperature phenol red-free DMEM. The use of standard or alternative loading solutions is cell type-dependent (see Note 7).
Cover the plate with aluminum foil and incubate for 60 min at room temperature to allow the loading of cells with CCF2/AM (see Note 8).
Wash the cells three times with room temperature phenol red-free DMEM and incubate for additional 60 min at room temperature to allow hydrolysis of CCF2/AM by the cytoplasmically-translocated LF/β-Lac (see Note 9).
Inspect cells using an inverted fluorescence microscope such as a Zeiss Axioplan (Carl Zeiss, Jena, Germany). For acquisition of blue fluorescence with this microscope, use excitation filter HQ405/20 nm bandpass, dichroic 425DCXR, and emitter filter HQ460/40 nm bandpass. For acquisition of green fluorescence with this microscope, use HQ405/20 nm bandpass, dichroic 425DCXR, and emitter filter HQ530/30 nm bandpass from Chroma Technology (Rockingham, VT) (see Note 10).
3.2. Imaging Cell Surface Proteolytic Activity with a Fluorescence Plate Reader
Grow cells to be imaged in phenol red-free DMEM (Gibco-BRL, Gaithersburg, MD) supplemented with 10% FBS, glutamine, and gentamicin reagent solution (see Note 1). Avoid using penicillin and other β-lactam-based antibiotics that are substrates for β-lactamase.
Treat 96-well black wall plates with a clear bottom with 150 μL/well 0.5 mg/mL poly-D-lysine in PBS at room temperature for 60 min to facilitate cell adhesion (see Note 2).
Seed cells 24 h before the assay at a density of 2 × 104 cells/well in 150 μL phenol red-free DMEM supplemented with 10% fetal bovine serum (FBS), glutamine, and gentamicin reagent solution (see Note 3). Add 150 μL medium without cells to six wells (see step 14).
Incubate plates over-night in a standard 37°C tissue culture incubator.
Gently wash the wells twice with 150 μL 37°C phenol red-free DMEM.
Add 26 nM (2 μg/mL) PrAg variant and 90 nM (5 μg/mL LF/β-Lac) fusion protein in 120 μL 37°C phenol red-free DMEM to each well. When imaging cell surface uPA proteolytic activity include 1 μg/mL Glu-plasminogen with or without 100 ng/mL pro-uPA (see Note 4). As a negative control, substitute PrAg-33, PrAg-U2, or PrAg-L1 with PrAg-U7. As a positive control, substitute PrAg-33, PrAg-U2, or PrAg-L1 with wild type PrAg (see Note 5).
Incubate the PrAg and LF/β-Lac-treated plate for 60 min at 37°C in a tissue culture incubator.
Wash cells twice with 150 μL room temperature phenol red-free DMEM. Transfer the plate to room temperature (see Note 6).
Add 20 μL of 6× “Standard substrate loading solution” or “Alternative substrate loading solution” (see Subheading 2.3) in 100 μL room temperature phenol red-free DMEM to each well. The use of standard or alternative loading solutions is cell type-dependent (see Note 7).
Cover the plate with aluminum foil to protect CCF2/AM from the light and incubate for 60 min at room temperature to allow the loading of cells with CCF2/AM (see Note 8).
Wash the cells three times with 150 μL room temperature phenol red-free DMEM containing Probenicid and incubate for an additional 60 min at room temperature to allow hydrolysis of CCF2/AM by the cytoplasmically-translocated LF/β-Lac (see Note 9).
Remove dust from the bottom of the plate.
Collect data using a fluorescence plate reader, such as a VICTOR plate reader from Perkin Elmer, Wellesley, MA, set in the bottom-read mode. Use the following filters in dual read mode: excitation 405/10 nm bandpass, emission filters 460/25 nm bandpass for blue fluorescence, and 535/25 nm bandpass for green fluorescence (see Note 10).
Determine “average blue background” and “average green background” from the wells without cells.
Subtract the “average blue background” and the “average green background” from all wells with cells to obtain “net blue signal” and “net green signal” for each well.
Divide “net blue signal” with “net green signal” for each well to obtain the “response ratio” (see Note 11).
3.3. Imaging Cell Surface Proteolytic Activity in Individual Cells by Flow Cytometry
Grow cells to be imaged in phenol red-free DMEM supplemented with 10% FBS, glutamine, and gentamicin reagent solution (see Note 1). Avoid using penicillin and other β-lactam ring-based antibiotics that are substrates for β-lactamase.
Treat 10 cm standard tissue culture dishes with 2 mL 0.1 mg/mL poly-D-lysine in PBS at 37°C in a tissue culture incubator for 60 min to facilitate cell adhesion (see Note 2).
Seed cells in the 10 cm tissue culture dishes 24 h before at a density of 1 × 106 cells/plate in phenol red-free DMEM supplemented with 10% FBS, glutamine, and gentamicin reagent solution (see Note 3).
Incubate overnight in a standard 37°C tissue culture incubator.
Gently wash the plates twice with 3 mL 37°C phenol red-free DMEM.
Add 26 nM (2 μg/mL) PrAg variant and 90 nM (5 μg/mL LF/β-Lac) fusion protein in 4 mL 37°C phenol red-free DMEM. When imaging cell surface uPA proteolytic activity include 1 μg/mL Glu-plasminogen with or without 100 ng/mL pro-uPA (see Note 4). As a negative control, substitute PrAg-33, PrAg-U2, or PrAg-L1 with PrAg-U7. As a positive control, substitute PrAg-33, PrAg-U2, or PrAg-L1 with wild-type PrAg (see Note 5).
Incubate the PrAg and LF/β-Lac-treated plates for 60 min at 37°C in a tissue culture incubator.
Wash cells twice with 3 mL room temperature phenol red-free DMEM. Transfer plate to room temperature (see Note 6).
Add 4 mL of 6× “Standard substrate loading solution” or 6× “Alternative substrate loading solution” (see Subheading 2.3) in 4 mL room temperature phenol red-free DMEM to each well. The use of standard or alternative loading solutions is cell type-dependent (see Note 7).
Cover the plates with aluminum foil to protect CCF2/AM from the light and incubate for 60 min at room temperature to allow the loading of cells with CCF2/AM (see Note 8).
Wash the plates three times with 3 mL room temperature phenol red-free DMEM and incubate for additional 60 min at room temperature to allow hydrolysis of CCF2/AM by the cytoplasmically-translocated LF/β-Lac (see Note 9).
Remove the cells from the plate using 1 mL trypsin-EDTA solution. Neutralize trypsin with 1 mL phenol red-free DMEM with 10% FBS. Add to a 10 mL tube with 8 mL ice-cold Hanks' balanced salt solution containing 2 mM probenecid (see Note 12).
Centrifuge the cells for 2 min at 700 × g at 4°C. Discard medium.
Gently resuspend cells in 10 mL ice-cold Hanks' balanced salt solution containing 2 mM probenicid.
Centrifuge the cells for 2 min at 700 × g at 4°C. Discard medium. Gently resuspend cells in ice-cold Hanks' balanced salt solution containing 2 mM probenecid at a concentration of 1 × 106 cells/mL.
Analyze cells using a flow cytometer such as a BD LSRII flow cytometer from Beckton Dickenson, San Jose, CA, equipped with a standard 488 nm laser and a 405 nm violet laser. Exclude dead cells, cell debris, and aggregates using side scatter versus forward scatter.
Use a 405 nm excitation filter, 525/50 nm filter for green light emission, and 440/40 nm filter for blue light emission with a 505 LP dichroic mirror to determine blue and green fluorescence signals from each living cell.
Determine the relative cell surface proteolytic activity of each cell by dividing the blue signal with the green signal for each individual cell (see Note 13). Plot cell number versus blue/green signal as a histogram.
Acknowledgments
We thank Drs. Silvio Gutkind and Mary Jo Danton for critically reviewing this manuscript, and Drs. Kevin L. Holmes and David Stephany for expert assistance with flow cytometry. This work was supported by the NIH Intramural Research Program, by NIAID Support of Intramural Biodefense Research from ICs other than NIAID and by the Department of Defense (DAMD-17-02-1-0693) to Dr. Thomas H. Bugge. For imaging reagents, contact S. H. Leppla (Leppla@nih.gov) or T. H. Bugge (thomas. bugge@nih.gov).
Footnotes
Other phenol red-free media compositions, sera, and antibiotic combinations can be used for cultivation as required for each individual cell line/primary cell type.
Cell types that adhere well to untreated tissue culture plastic or glass during normal culturing conditions may dislodge from an uncoated surface during the imaging procedure. The need for coating should be determined empirically.
Loading efficiency of cells with CCF2/AM depends on cell density and has been reported by Invitrogen to be most efficient at 60–80% confluence. However, we have successfully imaged cell surface protease activity in highly confluent cultures.
Addition of plasminogen is essential to convert pro-uPA to active two-chain uPA if the assay is performed in serum-free medium. Add pro-uPA when imaging the capacity of cells to convert exogenous pro-uPA to active uPA on the cell surface. Do not add pro-uPA when imaging endogenous uPA activity. Note that pro-uPA and uPA binding to the urokinase plasminogen activator receptor (uPAR) is species-specific. For example, human uPA will not bind to mouse uPAR and mouse uPA will not bind to human uPAR.
PrAg-U7 is a noncleavable PrAg variant that binds to cells but cannot support the translocation of LF/β-Lac to the cytoplasm. The combination of PrAg-U7 and LF/β-Lac is the optimal negative control. Wild-type PrAg is cleaved by furin as well as a number of furin-like proprotein convertases. The combination of wild-type PrAg and LF/β-Lac is the optimal positive control.
The uptake of CCF2/AM into cells is faster at 37°C than at room temperature, but the outwards transport of CCF2/AM is also higher. The net result of incubation of cells at 37°C is decreased loading of cells with CCF2/AM.
In our experience, the alternative substrate loading solution method from Invitrogen works best with cell lines, and the standard loading solution works best with primary cell cultures.
Loading with CCF2/AM for 60 min suffices for most cells. The loading time can be extended for higher sensitivity.
The substrate hydrolysis time can be extended to increase sensitivity.
Light of 409 nm wavelength causes maximum excitation of the coumarin group in CCF2/AM, leading to the emission of green light by the fluorescein group with maximum intensity at 520 nm due to fluorescent resonance energy transfer before hydrolysis by β-lactamase, and blue light with maximum intensity at 450 nm after hydrolysis by β-lactamase. The excitation and emission filters used here are those recommended by Invitrogen for the imaging of CCF2/AM. Other filters with similar properties can be used.
A ratiometric readout compensates for well-to-well variation in terms of cell number and CCF2/AM uptake and provides the most consistent data.
Probenecid is an anion transport inhibitor that reduces the outwards transport of CCF2/AM, CCF2, and probably hydrolyzed CCF2 from cells and increases the sensitivity of the assay.
A ratiometric readout compensates for variations in terms of cell size and CCF2/AM uptake and provides the most accurate data.
References
- 1.Werb Z. ECM and cell surface pro-teolysis: regulating cellular ecology. Cell. 1997;91(4):439–42. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 2.Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57(1):25–40. doi: 10.1007/s000180050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCawley LJ, Matrisian LM. Matrix metalloproteinases: multifunctional contributors to tumor progression. Mol Med Today. 2000;6(4):149–56. doi: 10.1016/s1357-4310(00)01686-5. [DOI] [PubMed] [Google Scholar]
- 4.Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta. 2000;1477(12):98–111. doi: 10.1016/s0167-4838(99)00263-0. [DOI] [PubMed] [Google Scholar]
- 5.Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291(2):113–35. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- 6.Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance, and regulation. Biochemistry. 1991;30(43):10363–70. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 7.Bugge TH. Proteolysis in carcinogenesis. In: Ensley JF, Gutkind JS, Jacob JR, Lippman SM, editors. Head and Neck Cancer. San Diego: Academic Press; 2003. pp. 137–49. [Google Scholar]
- 8.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6(10):764–75. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 9.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–92. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 10.McIntyre JO, Matrisian LM. Molecular imaging of proteolytic activity in cancer. J Cell Biochem. 2003;290(6):1087–97. doi: 10.1002/jcb.10713. [DOI] [PubMed] [Google Scholar]
- 11.Sloane BF, Sameni M, Podgorski I, Cavallo-Medved D, Moin K. Functional imaging of tumor proteolysis. Pharmacol Toxicol. 2006;46:301–15. doi: 10.1146/annurev.pharmtox.45.120403.095853. [DOI] [PubMed] [Google Scholar]
- 12.Hobson JP, Liu S, Rono B, Leppla SH, Bugge TH. Imaging specific cell-surface proteolytic activity in single living cells. Nat Meth. 2006;3(4):259–61. doi: 10.1038/nmeth862. [DOI] [PubMed] [Google Scholar]
- 13.Duesbery NS, Vande Woude GF. Anthrax toxins. Cell Mol Life Sci. 1999;55(12):1599–609. doi: 10.1007/s000180050399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect Immun. 1995;63(1):82–7. doi: 10.1128/iai.63.1.82-87.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adachi M, Kitamura K, Miyoshi T, et al. Activation of epithelial sodium channels by prostasin in Xenopus oocytes. J Am Soc Nephrol. 2001;12(6):1114–21. doi: 10.1681/ASN.V1261114. [DOI] [PubMed] [Google Scholar]
- 16.Liu S, Netzel-Arnett S, Birkedal-Hansen H, Leppla SH. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000;60(21):6061–7. [PubMed] [Google Scholar]
- 17.Liu S, Aaronson H, Mitola DJ, Leppla SH, Bugge TH. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc Natl Acad Sci USA. 2003;100(2):657–62. doi: 10.1073/pnas.0236849100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, Schubert RL, Bugge TH, Leppla SH. Anthrax toxin: structures, functions and tumour targeting. Expert Opin Biol Ther. 2003;3(5):843–53. doi: 10.1517/14712598.3.5.843. [DOI] [PubMed] [Google Scholar]
- 19.Leppla SH, Arora N, Varughese M. Anthrax toxin fusion proteins for intracellular delivery of macromolecules. J Appl Microbiol. 1999;187(2):284. doi: 10.1046/j.1365-2672.1999.00890.x. [DOI] [PubMed] [Google Scholar]
- 20.Arora N, Leppla SH. Residues 1–254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J Biol Chem. 1993;268(5):3334–41. [PubMed] [Google Scholar]
- 21.Zlokarnik G, Negulescu PA, Knapp TE, et al. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science. 1998;279(5347):84–8. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]
- 22.Liu S, Bugge TH, Leppla SH. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J Biol Chem. 2001;276(21):17976–84. doi: 10.1074/jbc.M011085200. [DOI] [PubMed] [Google Scholar]
- 23.Ke SH, Coombs GS, Tachias K, Navre M, Corey DR, Madison EL. Distinguishing the specificities of closely related proteases. Role of P3 in substrate and inhibitor discrimination between tissue-type plasminogen activator and urokinase. J Biol Chem. 1997;272(26):16603–9. doi: 10.1074/jbc.272.26.16603. [DOI] [PubMed] [Google Scholar]
- 24.Ke SH, Madison EL. Rapid and efficient site-directed mutagenesis by single-tube ‘megaprimer’ PCR method. Nucleic Acids Res. 1997;25(16):3371–2. doi: 10.1093/nar/25.16.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coombs GS, Bergstrom RC, Pellequer JL, et al. Substrate specificity of prostate-specific antigen (PSA) Chem Biol. 1998;5(9):475–88. doi: 10.1016/s1074-5521(98)90004-7. [DOI] [PubMed] [Google Scholar]
- 26.Inglese J, Johnson RL, Simeonov A, et al. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3(8):466–79. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 27.Arora N, Leppla SH. Fusions of anthrax toxin lethal factor with shiga toxin and diphtheria toxin enzymatic domains are toxic to mammalian cells. Infect Immun. 1994;62(11):4955–61. doi: 10.1128/iai.62.11.4955-4961.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]