

Abstract
CDC25 phosphatases are not only rate-limiting activators of cyclin-dependent kinases (CDKs) but also important targets of the CHK1/CHK2-mediated checkpoint pathway. Each isoform of the mammalian CDC25 famiy seems to exert unique biological functions. CDC25A is a critical regulator for both G1-S and G2-M transitions and essential for embryonic cell proliferation after the blastocyst stage. CDC25B is dispensable for embryogenesis but required for meiotic progression of oocytes in a manner analogous to Drosophila Twine or C. elegans cdc-25.1. Moreover, CDC25A and CDC25B appear to regulate different events or stages of mitosis. CDC25B may mediate the activation of CDK1/Cyclin B at the centrosome during prophase, while CDC25A may be required for the subsequent full activation of nuclear CDK1/Cyclin B. CDC25C is dispensable for both mitotic and meiotic divisions, although it is highly regulated during the processes. Excessive levels of CDC25A and CDC25B are often observed in various human cancer tissues. Deregulated expression of these phosphatases allows cells to overcome DNA damage-induced checkpoint, leading to genomic instability. Studies using mouse models demonstrated that deregulated expression of CDC25A significantly promotes RAS- or NEU-induced mammary tumor development with chromosomal aberrations, whereas decreased CDC25A expression in heterozygous knockout mice delays tumorigenesis. These biological properties of CDC25 phosphatases provide significant insight into the pathobiology of cancer and scientific foundation for anti-CDC25 therapeutic intervention.
Keywords: cell cycle, mitosis, meiosis, development, checkpoint, transgenic mice, knockout mice
INTRODUCTION
The cell division cycle of eukaryotic cells is regulated by temporal activation of cyclin-dependent kinases (CDKs), highly conserved serine/threonine protein kinases that are activated by association with the regulatory cyclin subunits [1]. Mammalian cells entering the cell cycle from quiescence first exhibit the activation of CDK3/Cyclin C during the G0/G1 transition, followed by sequential activation of CDK4(6)/Cyclin D and CDK2/Cyclin E during mid to late G1 phase [2, 3]. The activities of these G1-specific CDK complexes are highly influenced by extracellular signals, e.g., growth factors, hormones, cytokines, nutrients, cell-cell contact, and anchorage attachment [4]. Immediately after cells enter S phase, the activity of CDK2/Cyclin A progressively accumulates, which is essential for DNA replication. During late G2, CDK1/Cyclin A and CDK1/Cyclin B are activated, which play central roles in the initiation and completion of mitosis (M phase). Cells progressing during S, G2 or M phase are generally less sensitive to the extracellular signals than cells in G1 are. Nonetheless, CDK activity in these phases must be tightly controlled for the fidelity of DNA replication and the sophisticated coordination of mitotic events.
CDK activity is under the control by multiple layers of regulatory mechanisms. CDK inhibitors, such as the KIP/CIP family proteins (p21, p27 and p57) and the INK4 family proteins (p16, p15, p18 and p19), can inhibit CDK/cyclin complexes by physical association [5]. In addition, the CDK catalytic subunit undergoes activating and inactivating phosphorylation. Phosphorylation of a threonine residue on the T-loop domain of each CDK protein, such as Thr161 of CDK1, Thr160 of CDK2 and Thr172 of CDK4, is required for full activation. The CDK activating kinases (CAKs), including CDK7/Cyclin H and CAK1, mediate the T-loop phosphorylation [6]. In contrast, phosphorylation of a tyrosine residue within the ATP binding domain, e.g., Tyr15 of CDK1 and CDK2 and Tyr17 of CDK4, is inhibitory on the CDK activity. These tyrosine residues, together with adjacent threonine residues (i.e., Thr14 of CDK1 and CDK2), are phosphorylated by Wee1/Mik1/Myt1 protein kinases following cyclin assembly, providing a fine tuning mechanism for proper timing of CDK activation [7]. The inhibitory tyrosine and threonine phosphorylation on CDK1 and CDK2 forms a major target of checkpoint signaling, which delays or halts cell cycle progression in response to cellular damages. The CDC25 family dual specificity phosphatases play key roles in activating tyrosine-phosphorylated CDKs by dephosphorylating the tyrosine and threonine residues at the ATP binding sites [8, 9]. In this review the biological roles of the CDC25 family members are discussed with emphasis on development and oncogenesis.
Developmental roles for CDC25 phosphatases in the worm, fly and mouse
Like most other cell cycle-regulatory proteins, CDC25 phosphatases are conserved in all eukaryotic cells. The fission yeast cdc25+ was originally identified as an essential gene for entry into mitosis [10, 11], and the budding yeast also has an ortholog of the cdc25+ gene [12]. Interestingly, Caenorhabditis elegans has four cdc25 genes, cdc-25.1 to cdc-25.4 [13]. cdc-25.1 is required for germline proliferation and the regulation of meiosis [14, 15], while this gene has been demonstrated to possess an oncogenic function by a gain-of-function mutation [16]. Drosophila melanogaster has two cdc25 genes, string and twine. string is required for mitotic divisions after the onset of organogenesis (patterning) during Drosophila embryogenesis [17–20]. In contrast, twine is required for meiosis in germline [21, 22]. These observations in the worm and fly suggest evolutional development of differential roles for multiple CDC25 phosphatases in the control of mitosis and meiosis.
Mammals have three CDC25 genes, CDC25A, CDC25B and CDC25C [23–25]. Studies on the expression of CDC25 proteins in mouse tissues showed that the three family members have distinct patterns of expression in embryos, as well as in adult tissues [26, 27]. CDC25A is not expressed in the pre-implantation mouse embryo until the late blastocyst stage. The initiation of CDC25A expression correlates with establishment of a typical G1 phase of the embryonic cell cycle. During later stages of embryogenesis CDC25A is expressed widely in most developing tissues. In contrast, CDC25B expression is observed in mouse embryos as early as the 1-cell stage, apparently from maternal transcripts. CDC25B levels go down at the 2-cell stage and then up again at the 4-cell stage, corresponding to the maternal to zygotic transition (MZT). Similar degradation of maternal CDC25C transcripts is observed in porcine embryos [28]. The dynamic control of CDC25B and CDC25C during MZT of mammalian embryos is analogous to the zygotic degradation of maternal Twine and String transcripts in Drosophila [29]. These observations suggest that CDC25B and CDC25C regulate cell cycle progression in pre-implantation embryos. In adult animals, CDC25B and CDC25C are expressed most highly in testes and ovaries. CDC25B is expressed also in the spleen, lung, heart, and intestine at lower levels [30]. CDC25C expression is observed in various tissues including the thymus, spleen and intestine of adult rodents. Thus, the CDC25 family members are expressed during development and adulthood in distinct but overlapping manners.
Studies on knockout mice deficient for CDC25 phosphatases further revealed developmental functions of this gene family, as summarized in Table (1). Cdc25A-null mice are lethal during the peri-implantation period (embryonic day 5–7), consistently with its expression pattern in embryogenesis [31]. Cdc25A-null blastocysts exhibit a defect in hatching, suggesting that the inner cell mass cannot proliferate without CDC25A (S. Shevtsov and H. Kiyokawa, unpublished observations). In sharp contrast, Cdc25B-null mice and Cdc25C-null mice are viable, suggesting these phosphatases are dispensable for embryogenesis. While most adult tissues also develop normally in the absence of CDC25B or CDC25C, female Cdc25B-null mice are sterile with a meiotic defect in the ovary [32], which is consistent with high CDC25B expression in wild-type gonads. In vertebrates, oocytes are arrested at prophase of meiosis I at birth. During puberty, oocytes in dominant follicles resume meiosis in prior to ovulation, and progress until metaphase of meiosis II [33]. This resumption of meiosis is associated with an increase of Cyclin B-CDK1 activity, a.k.a., MPF (maturation promoting factor). Cdc25B-null oocytes are defective in activating Cyclin B-CDK1 at the pre-ovulatory stage; thus, they are unable to resume meiotic progression and permanently arrested in meiosis (prophase) I. These observations imply that mammalian CDC25B is functionally homologous to C. elegans cdc-25.1 and Drosophila Twine. Cdc25C-null mice display no appreciable phenotype with normal fertility in both males and females [34]. Interestingly, Cdc25B, Cdc25C-double null mice essentially phenocopy Cdc25B-null mice without any additional defect [35]. These data imply that CDC25A is able to compensate for the absence of CDC25B and CDC25C in development, although the expression of CDC25A is not upregulated in embryonic fibroblasts from Cdc25B, Cdc25C-double null mice. Taken together, these studies using mutant mice highlight the functional differences among the three CDC25 phosphatases.
Fig. 1.
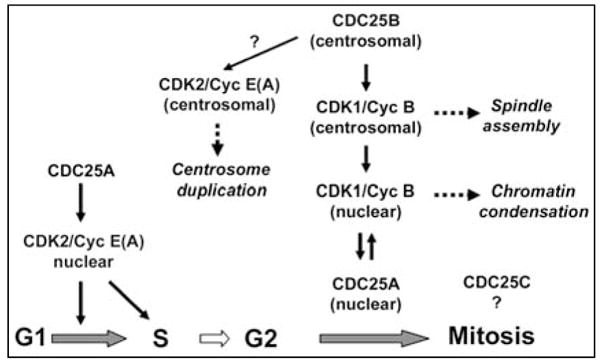
Specific roles for CDC25 phosphatases in the regulation of mammalian cell cycle progression.
Cell cycle regulation by mammalian CDC25 phosphatases
The mitotic control in mammalian somatic cells apparently involves the collaboration of all three CDC25 phosphatases, which determines the timing of CDK1/Cyclin B activation and the initiation of mitosis. Although an undefined level of functional redundancy exists among the three CDC25 phosphatases, a recent study using siRNA and time-lapse microscopy elegantly showed that CDC25A and CDC25B cooperate for mitotic entry presumably by exerting distinct functions [36][see Fig. (1)]. CDC25B may specifically activate CDK1/Cyclin B at the centrosome, the microtubule organizing center for mitotic spindles. The activation of centrosomal CDK1/Cyclin B1 is an essential process not only for spindle assembly but also for other mitotic events. Active CDK1/Cyclin B first appears on centrosomes in prophase, and subsequently is imported into the nucleus [37]. Thus, CDC25B is a critical initiator of mitosis, triggering the first ignition of CDK1/Cyclin B activation. In addition, CDC25B may be involved in activation of CDK2/Cyclin E or A at the centrosome, which is required for the centrosome duplication cycle [38–41]. This hypothesis is supported by a recent study showing that overexpression of CDC25B results in centrosome overduplication and forced expression of CDC25B targeted on the centrosomes causes centrosome amplification and aberrant microtubule organization [42]. At this time it is not well understood how CDC25B is activated at the centrosomes. Aurora-A has been shown to phosphorylate Ser339 of centrosomal CDC25B, possibly activating the phosphatase [43]. During interphase, the checkpoint kinase Chk1 negatively regulates CDC25B and prevents premature mitotic entry [44, 45]. On the other hand, CDC25A may be involved directly in promoting chromatin condensation. CDK1/Cyclin B imported into the nucleus stabilizes CDC25A by direct phosphorylation [46], which in turn sustains CDK1/Cyclin B activity high enough for chromatin condensation. It is still controversial whether CDC25C plays a unique or essential role for mitotic initiation, although it has been shown that microinjection of CDC25C protein could induce premature mitosis [47].
In contrast to the G2-M regulation involving multiple CDC25 proteins, the G1-S progression is controlled predominantly by CDC25A. In human cell cultures synchronized with serum starvation, CDC25A is activated prior to the S phase initiation [48]. Forced expression of CDC25A accelerates the G1-S transition with an increase in CDK2/Cyclin E activity [49], while microinjection of cells with anit-CDC25A antibody has been shown to result in G1 arrest [48]. These data suggest a role for CDC25A in the control of the G1-S transition, as shown in Fig. (1). The transcription of CDC25A is activated by the E2F family transcription factors during late G1, and sustained high during S and G2 [50]. Furthermore, an additional layer of regulation exists at the level of protein degradation [51, 52]. The anaphase promoting complex or cyclosome (APC/C) in complex with the activating subunit Cdh1 ubiquitinates CDC25A during mitosis through G1, targeting the phosphatase to proteasomal degradation [53]. In late G1, E2F transactivates Emi1, a major inhibitor of APC/C-Cdh1 [54], leading to a decrease in CDC25A degradation. These mechanisms allow cells to upregulate CDC25A at the G1-S boundary. During S phase, CDC25A continuously activates CDK2/Cyclin A as a driving force of DNA replication. Consistent with the critical G1/S function of CDC25A, inactivation of this phosphatases is one of the mechanisms of transforming growth factor-β (TGF-β)-induced G1 arrest. While TGF-β signals repress the CDC25A promoter via the E2F-responsive element [55, 56], TGF-β signals also inactivate CDC25A at the post-translational level. TGF-β promotes ubiquitination of CDC25A mediated by the β-TrCP ubiquitin ligase complex [57]. TGF-β also upregulates ROCK1 kinase, which phosphorylates and inhibits the phosphatase activity of CDC25A [58]. Thus, CDC25A activity constitutes a rate-limiting mechanism for G1-S progression, which is highly regulated by extracellular signals. Interestingly, a previous study suggested that CDC25C was involved in S phase transition [59], awaiting follow-up investigations.
The meiosis-promoting function of CDC25 phosphatases has been well established in lower eukaryotes. The initiation of meiosis requires the activation of CDC25 phosphatase(s) in prior to CDK1 activation. At meiotic initiation of Xenopus eggs, CDC25C is activated by a series of phosphorylation at the N-terminal regulatory domain, mediated by p42MAPK (the Xenopus orthologue of ERK2), CDK1/Cyclin B, and Pix1 (the orthologue of polo-like kinase 1 or PLK1)[60–62]. Progesterone, a meiosis-promoting hormone, triggers the MOS-MEK-MAPK kinase cascade, leading to CDC25C phosphorylation. CDK1- and Pix1-mediated phosphorylation forms a positive feed-back loop to further activate CDC25C. Phosphorylation of the Polo-box motif in CDC25C by CDK1 facilitates the association with Pix1 and subsequent phosphorylation of CDC25C, which is analogous to the regulation of other Plk1 substrates. CDC25 phosphatases in mammalian oocytes seem to undergo a similar activation process [61]. Purified ERK2 can phosphorylate and activate all three human CDC25 proteins in vitro, and CDC25C is found in complex with ERK2 in human cells. Although the mouse knockout studies indicate that CDC25B plays an essential role in meiosis [32], the detail in the regulation and action of CDC25B during meiotic initiation remains to be fully understood.
CDC25 inactivation as a major mechanism of p53-independent checkpoint
Cell cycle checkpoint is a major mechanism of tumor suppression, delaying or halting cell cycle progression in response to a variety of damage on the genome, such as ionizing irradiation, ultraviolet light, oxidative stress, perturbed DNA replication, and other genotoxic agents [63]. It is well known that the critical tumor suppressor p53 plays a key role in cell cycle checkpoint, by transactivating the CDK inhibitor p21 (CDKN1A) and a CDK1 inhibitor, 14-3-3σ. While the p53-dependent checkpoint is important in essentially all cell types, p53-deficient cells are still capable of arresting in response to DNA damage, with mechanisms leading to inactivation of CDC25 phosphatases [8, 64–66]. DNA damage generally triggers activation of ATM (ataxia-telangiectasia-mutated) and ATR (ATM-and Rad3-related) kinases. These proximal checkpoint kinases in turn activate the downstream effector kinases, Chk1 and Chk2. A number of studies indicated that CDC25 phosphatases are critical substrates of Chk1 and Chk2. Chk1 phosphorylates CDC25A on Ser76, Ser124, Ser178, Ser279, Ser 293 and Thr507, while Chk2 can phosphorylate Ser124, Ser178, Ser279 and Ser 293. Of these sites, Ser76 phosphorylation functions as a priming event, promoting subsequent phosphorylation of Ser82 and Ser88 by undefined kinase(s). The sequence including Ser82/Ser88 constitutes a phospho-degron, which is recognized by the β-TrCP ubiquitin ligase complex, SCFFbw1(β-TrCP) [67, 68]. Polyubiquitinated CDC25A is recruited to proteasome-mediated degradation. Therefore, Ser76 phosphorylation initiates the process of destabilizing CDC25A protein. Identification of the Ser82/Ser88 kinase(s) should provide a significant insight into the checkpoint pathway. As the entire process from Chk1 activation through CDC25A degradation does not require new protein synthesis, it constitutes a fast checkpoint response in comparison with the p53-dependent checkpoint requiring transcription of effector genes. In addition, Chk1/Chk2-mediated phosphorylation results in inhibition of CDC25A phosphatase activity. The sequence surrounding Ser178 contains a 14-3-3 docking site, and Ser178 phosphorylation facilitates association with 14-3-3 scaffold proteins. 14-3-3 binding has been shown to inactivate the enzymatic activity of CDC25A. The significance of CDC25A as a checkpoint target is further supported by the data that Cdc25A-heterozygous mouse embryonic fibroblasts (MEFs) show a decrease in the basal level of CDC25A expression and modestly enhanced G2 checkpoint response to ionizing irradiation [31]. Besides Chk1, two other kinases have been identified to phosphorylate Ser76 of CDC25A and destabilize the protein. MAPKAP kinase-2, an effector kinase of the p38MAPK pathway, phosphorylates Ser76 and Ser124, leading to CDC25A ubiquitination and degradation. This p38MAPK-dependent control has been implicated for cell cycle arrest of lymphocytes in response to cytokine (IL-3 or IL-7) withdrawal [69] and arrest by osmotic stress [70]. The other kinase that can phosphorylates Ser76 is GSK-3β [71]. Interestingly, Ser76 phosphorylation by GSK-3β requires priming phosphorylation of Thr80 by polo-like kinase-3 (PLK3). The regulation of CDC25A degradation by GSK-3β implicates that CDC25A protein may be stabilized in cancer cells that have mutations in the PTEN-PI3K-AKT pathway and consequently lower GSK-3β activity.
CDC25B and CDC25C also undergo similar phosphorylation by Chk1/Chk2 and MAPKAP kinase-2 [66, 72–76]. However, the consequence of phosphorylation is not ubiquitination or degradation. Chk2-mediated phosphorylation of CDC25C at Ser216 facilitates binding with 14-3-3, which sequesters CDC25C in the cytoplasm. MAPKAP kinase-2-mediated phosphorylation of CDC25B at Ser309 and Ser361 promotes similar cytoplasmic sequestration with 14-3-3. Interestingly, CDC25B, but not CDC25A or CDC25C, is required along with PLK1 for recovery from DNA damage-induced G2 arrest and subsequent mitotic entry [77]. Taken together, damage-induced phosphorylation and inactivation of CDC25 phosphatases are a central mechanism of p53-independent checkpoint. Cells are equipped with multiple modes of CDC25 inactivation, all of which are rapidly responsive to damage. Such nature of the regulation makes these phosphatases attractive targets of therapeutic intervention.
CDC25A and CDC25B as oncogenes critical for tumor initiation and progression
Accumulating evidence indicates that CDC25 phosphatases, especially CDC25A and CDC25B, are oncogenes (reviewed in [8, 65]). CDC25A overexpression has been reported in a variety of human malignancies, such as liver, breast, ovarian, thyroid, colorectal, laryngeal and esophageal cancers and non-Hodgkin lymphomas. CDC25B overexpression also has been observed in all these types of malignancies, plus gastric, endometrial and prostate cancers and gliomas. In most cases, overexpression of CDC25A and CDC25B correlates with poor prognosis of patients. As gene amplification has not been identified, precise mechanisms of cancer-associated overexpression of these proteins remain to be elucidated. Since CDC25 phosphatases are critical targets of the checkpoint pathway, deregulated expression of the proteins could allow aberrant cell cycle progression even in the presence of DNA damage, possibly leading to genomic instability. A previous report described that ectopic expression of CDC25A or CDC25B cooperates with RAS activation or RB loss to transform rodent fibroblasts in culture [78], suggesting that these phosphatases are involved in tumor initiation. In vivo studies using MMTV-CDC25A [79] and MMTV-CDC25B [80, 81] transgenic mice demonstrated that ectopic expression of CDC25A or CDC25B alone results in alveolar hyperplasia of mammary glands, but is insufficient for spontaneous tumorigenesis [Table (1)]. However, CDC25A overexpression in MMTV-H-ras transgenic mice significantly shortens the latency of mammary tumorigenesis. Ectopic expression of CDC25A in MMTV-c-neu transgenic mice minimally affects tumor latency, but significantly accelerates tumor growth with mis-coordinated cell cycle progression and various chromosomal aberrations. Furthermore, it has been demonstrated that decreased expression of CDC25A is inhibitory to in vivo tumorigenesis [31]. Mammary tumor initiation of MMTV-H-ras or MMTV-c-neu mice is markedly delayed in the Cdc25A-heterozygous knockout background. These studies suggest that CDC25A plays a rate-limiting role in tumorigenesis, cooperating with other tumor initiating oncogenes such as RAS [82]. It is noteworthy that Chk1 heterozygous mice exhibit increased susceptibility to Wnt-induced mammary tumorigenesis [83], and Chk1 heterozygous mammary epithelial cells overexpress CDC25A [84]. There is also some experimental evidence for the in vivo oncogenic action of CDC25B. Ectopic expression of CDC25B increases the susceptibility of murine mammary tissues to tumorigenesis induced by the chemical carcinogen DMBA [81]. Finally, it has been obscure whether CDC25C is involved in tumorigenesis. A recent study showed that MEFs lacking the Fez1/Lzts1 tumor suppressor display enhanced mitotic degradation of CDC25C, impaired CDK1 activation, accelerated mitotic progression and chromosomal instability [85]. Importantly, Fez1/Lzts1-null mice are predisposed to tumorigenesis. Thus, perturbation of the fine tuning mechanism for CDC25C regulation, unlike CDC25A or CDC25B overexpression, may lead to tumor predisposition. It will be informative to examine the susceptibility of Cdc25B-and Cdc25C-null mice to tumorigenesis induced by various oncogenes or carcinogens.
Conclusion
The specific biological functions of CDC25A and CDC25B implicate that deregulation of these two proteins may promote tumorigenesis in distinct manners. Since CDC25 phosphatases are therapeutic targets with high potential, further investigations are necessary to fully characterize the biological impacts of ablation or inhibition of each phosphatase in vivo. The mechanisms of cancer-associated overexpression of CDC25A and CDC25B are yet to be clarified. In addition, the developmental and oncogenic roles for CDC25C remain to be defined. The biological functions of each CDC25 phosphatase should be carefully considered to refine the strategy of anti-CDC25 therapeutic intervention.
Table 1.
Mouse models for altered expression of CDC25 phosphatases
| Mouse strain | Phenotype | Reference |
|---|---|---|
| Cdc25A−/− | Embryonic lethal at the peri-implantation stage (E5–E7) | [31] |
| Defective hatching of null blastocysts | ||
| Cdc25A+/− | Viable | [31] |
| No developmental defect observed | ||
| Delayed mammary tumorigenesis induced by MMTV-H-ras or c-neu | ||
| MEFs: Normal cell cycle progression but earlier replicative senescence. | ||
| Partial resistance to Ras-mediated transformation | ||
| Cdc25B−/− | Viable | [32] |
| Female sterility with oocytes permanently arrested in meiosis I. | ||
| Normal male fertility | ||
| Cdc25C−/− | Viable | [34] |
| No developmental defect observed | ||
| MEFs: Normal cell cycle progression. Normal G1, S and G2 checkpoint | ||
| T- and B-lymphocytes: Normal development and proliferation | ||
| Cdc25B−/−; | Viable | [35] |
| Cdc25C−/− | Female sterility similar to Cdc25B−/− mice | |
| MEFs: Normal cell cycle progression. Normal G1, S and G2 checkpoint | ||
| T-lymphocytes: Normal development and checkpoint response to in vivo Ionizing irradiation | ||
| MMTV-CDC25A | Alveolar hyperplasia of mammary glands | [79] |
| Rare spontaneous tumorigenesis (<5%) | ||
| Shortened latency of mammary tumorigenesis and more invasiveness in MMTV-H-ras transgenic mice | ||
| Accelerated growth of mammary tumors with more invasiveness and Genomic instability in MMTV-c-neu transgenic mice | ||
| MMTV-CDC25B | Alveolar hyperplasia and retarded involution of mammary glands | [80, 81] |
| No spontaneous tumorigenesis but higher susceptibility to DMBA-induced mammary tumorigenesis |
Acknowledgments
We thank current and former members of the laboratory, Helen Piwnica-Worms, Maddalena Donzelli, Sophia Tsai, and all collaborators for helpful discussions. Work in the laboratory has been supported partly by funds to H.K. from the National Institutes of Health (R01-CA100204, CA112282, and HD38085), the Department of Defense (DAMD 17-02-1-0413), the Searle Leadership Fund, the Zell Scholar Fund, and the Robert H. Lurie Comprehensive Cancer Center.
References
- 1.Nurse P. Cell. 2000;100:71–78. doi: 10.1016/s0092-8674(00)81684-0. [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- 3.Sage J. Dev Cell. 2004;6:607–8. doi: 10.1016/s1534-5807(04)00137-6. [DOI] [PubMed] [Google Scholar]
- 4.Pardee AB. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ, Roberts JM. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 6.Lolli G, Johnson LN. Cell Cycle. 2005;4:572–7. [PubMed] [Google Scholar]
- 7.Kellogg DR. J Cell Sci. 2003;116:4883–90. doi: 10.1242/jcs.00908. [DOI] [PubMed] [Google Scholar]
- 8.Boutros R, Lobjois V, Ducommun B. Nat Rev Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 9.Rudolph J. Biochemistry. 2007;46:3595–604. doi: 10.1021/bi700026j. [DOI] [PubMed] [Google Scholar]
- 10.Fantes P. Nature. 1979;279:428–30. doi: 10.1038/279428a0. [DOI] [PubMed] [Google Scholar]
- 11.Russell P, Nurse P. Cell. 1986;45:145–53. doi: 10.1016/0092-8674(86)90546-5. [DOI] [PubMed] [Google Scholar]
- 12.Popolo L, Alberghina L. Proc Natl Acad Sci U S A. 1984;81:120–4. doi: 10.1073/pnas.81.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashcroft NR, Kosinski ME, Wickramasinghe D, Donovan PJ, Golden A. Gene. 1998;214:59–66. doi: 10.1016/s0378-1119(98)00228-5. [DOI] [PubMed] [Google Scholar]
- 14.Ashcroft N, Golden A. Genesis. 2002;33:1–7. doi: 10.1002/gene.10083. [DOI] [PubMed] [Google Scholar]
- 15.Ashcroft NR, Srayko M, Kosinski ME, Mains PE, Golden A. Dev Biol. 1999;206:15–32. doi: 10.1006/dbio.1998.9135. [DOI] [PubMed] [Google Scholar]
- 16.Clucas C, Cabello J, Bussing I, Schnabel R, Johnstone IL. Embo J. 2002;21:665–74. doi: 10.1093/emboj/21.4.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edgar BA, O’Farrell PH. Cell. 1990;62:469–80. doi: 10.1016/0092-8674(90)90012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edgar BA, O’Farrell PH. Cell. 1989;57:177–87. doi: 10.1016/0092-8674(89)90183-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehner CF. Semin Cell Biol. 1991;2:223–31. [PubMed] [Google Scholar]
- 20.Jimenez J, Alphey L, Nurse P, Glover DM. Embo J. 1990;9:3565– 71. doi: 10.1002/j.1460-2075.1990.tb07567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alphey L, Jimenez J, White-Cooper H, Dawson I, Nurse P, Glover DM. Cell. 1992;69:977–88. doi: 10.1016/0092-8674(92)90616-k. [DOI] [PubMed] [Google Scholar]
- 22.Courtot C, Fankhauser C, Simanis V, Lehner CF. Development. 1992;116:405–16. doi: 10.1242/dev.116.2.405. [DOI] [PubMed] [Google Scholar]
- 23.Galaktionov K, Beach D. Cell. 1991;67:1181–1194. doi: 10.1016/0092-8674(91)90294-9. [DOI] [PubMed] [Google Scholar]
- 24.Sadhu K, Reed SI, Richardson H, Russell P. Proc Natl Acad Sci USA. 1990;87:5139–5143. doi: 10.1073/pnas.87.13.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagata A, Igarashi M, Jinno S, Suto K, Okayama H. New Biol. 1991;3:959–968. [PubMed] [Google Scholar]
- 26.Wu S, Wolgemuth DJ. Dev Biol. 1995;170:195–206. doi: 10.1006/dbio.1995.1207. [DOI] [PubMed] [Google Scholar]
- 27.Wickramasinghe D, Becker S, Ernst MK, Resnick JL, Centanni JM, Tessarollo L, Grabel LB, Donovan PJ. Development. 1995;121:2047–2056. doi: 10.1242/dev.121.7.2047. [DOI] [PubMed] [Google Scholar]
- 28.Anderson JE, Matteri RL, Abeydeera LR, Day BN, Prather RS. Mol Reprod Dev. 2001;60:181–8. doi: 10.1002/mrd.1075. [DOI] [PubMed] [Google Scholar]
- 29.Edgar BA, Datar SA. Genes Dev. 1996;10:1966–77. doi: 10.1101/gad.10.15.1966. [DOI] [PubMed] [Google Scholar]
- 30.Kakizuka A, Sebastian B, Borgmeyer U, Hermans-Borgmeyer I, Bolado J, Hunter T, Hoekstra MF, Evans RM. Genes Dev. 1992;6:578–90. doi: 10.1101/gad.6.4.578. [DOI] [PubMed] [Google Scholar]
- 31.Ray D, Terao Y, Nimbalkar D, Hirai H, Osmundson EC, Zou X, Franks R, Christov K, Kiyokawa H. Cancer Res. 2007;67:6605–11. doi: 10.1158/0008-5472.CAN-06-4815. [DOI] [PubMed] [Google Scholar]
- 32.Lincoln AJ, Wickramasinghe D, Stein P, Schultz RM, Palko ME, De Miguel MP, Tessarollo L, Donovan PJ. Nat Genet. 2002;30:446–9. doi: 10.1038/ng856. [DOI] [PubMed] [Google Scholar]
- 33.Sagata N. Trends Cell Biol. 1996;6:22–8. doi: 10.1016/0962-8924(96)81034-8. [DOI] [PubMed] [Google Scholar]
- 34.Chen MS, Hurov J, White LS, Woodford-Thomas T, Piwnica-Worms H. Mol Cell Biol. 2001;21:3853–3861. doi: 10.1128/MCB.21.12.3853-3861.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferguson AM, White LS, Donovan PJ, Piwnica-Worms H. Mol Cell Biol. 2005;25:2853–60. doi: 10.1128/MCB.25.7.2853-2860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindqvist A, Kallstrom H, Lundgren A, Barsoum E, Rosenthal CK. J Cell Biol. 2005;171:35–45. doi: 10.1083/jcb.200503066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackman M, Lindon C, Nigg EA, Pines J. Nat Cell Biol. 2003;5:143–8. doi: 10.1038/ncb918. [DOI] [PubMed] [Google Scholar]
- 38.Lacey KR, Jackson PK, Stearns T. Proc Natl Acad Sci U S A. 1999;96:2817–22. doi: 10.1073/pnas.96.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumoto Y, Hayashi K, Nishida E. Curr Biol. 1999;9:429–32. doi: 10.1016/s0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- 40.Fukasawa K. Nat Rev Cancer. 2007;7:911–24. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- 41.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Cell. 2000;103:127–40. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 42.Boutros R, Lobjois V, Ducommun B. Cancer Res. 2007;67:11557–64. doi: 10.1158/0008-5472.CAN-07-2415. [DOI] [PubMed] [Google Scholar]
- 43.Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, Mirey G, Bouche JP, Theis-Febvre N, Schmitt E, Monsarrat B, Prigent C, Ducommun B. J Cell Sci. 2004;117:2523–31. doi: 10.1242/jcs.01108. [DOI] [PubMed] [Google Scholar]
- 44.Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J. Nat Cell Biol. 2004;6:884–91. doi: 10.1038/ncb1165. [DOI] [PubMed] [Google Scholar]
- 45.Loffler H, Rebacz B, Ho AD, Lukas J, Bartek J, Kramer A. Cell Cycle. 2006;5:2543–7. doi: 10.4161/cc.5.21.3435. [DOI] [PubMed] [Google Scholar]
- 46.Mailand N, Podtelejnikov AV, Groth A, Mann M, Bartek J, Lukas J. Embo J. 2002;21:5911–20. doi: 10.1093/emboj/cdf567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strausfeld U, Fernandez A, Capony JP, Girard F, Lautredou N, Derancourt J, Labbe JC, Lamb NJ. J Biol Chem. 1994;269:5989–6000. [PubMed] [Google Scholar]
- 48.Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, Okayama H. EMBO J. 1994;13:1549–1556. doi: 10.1002/j.1460-2075.1994.tb06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blomberg I, Hoffmann I. Mol Cell Biol. 1999;19:6183–6194. doi: 10.1128/mcb.19.9.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K. Mol Cell Biol. 1999;19:6379–6395. doi: 10.1128/mcb.19.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neely KE, Piwnica-Worms H. Cell Cycle. 2003;2:455–457. [PubMed] [Google Scholar]
- 52.Busino L, Chiesa M, Draetta GF, Donzelli M. Oncogene. 2004;23:2050–2056. doi: 10.1038/sj.onc.1207394. [DOI] [PubMed] [Google Scholar]
- 53.Donzelli M, Squatrito M, Ganoth D, Hershko A, Pagano M, Draetta GF. EMBO J. 2002;21:4875–4884. doi: 10.1093/emboj/cdf491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Cell. 2001;105:645–655. doi: 10.1016/s0092-8674(01)00361-0. [DOI] [PubMed] [Google Scholar]
- 55.Iavarone A, Massague J. Nature. 1997;387:417–422. doi: 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- 56.Iavarone A, Massague J. Mol Cell Biol. 1999;19:916–922. doi: 10.1128/mcb.19.1.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ray D, Terao Y, Nimbalkar D, Chu LH, Donzelli M, Tsutsui T, Zou X, Ghosh AK, Varga J, Draetta GF, Kiyokawa H. Mol Cell Biol. 2005;25:3338–47. doi: 10.1128/MCB.25.8.3338-3347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhowmick NA, Ghiassi M, Aakre M, Brown K, Singh V, Moses HL. Proc Natl Acad Sci USA. 2003;100:15548–15553. doi: 10.1073/pnas.2536483100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turowski P, Franckhauser C, Morris MC, Vaglio P, Fernandez A, Lamb NJ. Mol Biol Cell. 2003;14:2984–98. doi: 10.1091/mbc.E02-08-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perdiguero E, Nebreda AR. Cell Cycle. 2004;3:733–7. [PubMed] [Google Scholar]
- 61.Wang R, He G, Nelman-Gonzalez M, Ashorn CL, Gallick GE, Stukenberg PT, Kirschner MW, Kuang J. Cell. 2007;128:1119–32. doi: 10.1016/j.cell.2006.11.053. [DOI] [PubMed] [Google Scholar]
- 62.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 63.Dash BC, El-Deiry WS. Methods Mol Biol. 2004;280:99–161. doi: 10.1385/1-59259-788-2:099. [DOI] [PubMed] [Google Scholar]
- 64.Donzelli M, Draetta GF. EMBO Rep. 2003;4:671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kristjansdottir K, Rudolph J. Chem Biol. 2004;11:1043–51. doi: 10.1016/j.chembiol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 66.Karlsson-Rosenthal C, Millar JB. Trends Cell Biol. 2006;16:285–92. doi: 10.1016/j.tcb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 67.Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- 68.Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khaled AR, Bulavin DV, Kittipatarin C, Li WQ, Alvarez M, Kim K, Young HA, Fornace AJ, Durum SK. J Cell Biol. 2005;169:755–63. doi: 10.1083/jcb.200409099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goloudina A, Yamaguchi H, Chervyakova DB, Appella E, Fornace AJ, Jr, Bulavin DV. Cell Cycle. 2003;2:473–478. [PubMed] [Google Scholar]
- 71.Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, Piwnica-Worms H. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 73.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Science. 1997;277:1497–501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 74.Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ., Jr Nature. 2001;411:102–107. doi: 10.1038/35075107. [DOI] [PubMed] [Google Scholar]
- 75.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. Mol Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 76.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. Cancer Cell. 2007;11:175–89. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Vugt MA, Bras A, Medema RH. Mol Cell. 2004;15:799–811. doi: 10.1016/j.molcel.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 78.Galaktionov K, Lee AK, Eckstein J, Draetta G, Meckler J, Loda M, Beach D. Science. 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 79.Ray D, Terao Y, Fuhrken PG, Ma ZQ, DeMayo FJ, Christov K, Heerema NA, Franks R, Tsai SY, Papoutsakis ET, Kiyokawa H. Cancer Res. 2007;67:984–91. doi: 10.1158/0008-5472.CAN-06-3927. [DOI] [PubMed] [Google Scholar]
- 80.Ma ZQ, Chua SS, DeMayo FJ, Tsai SY. Oncogene. 1999;18:4564–4576. doi: 10.1038/sj.onc.1202809. [DOI] [PubMed] [Google Scholar]
- 81.Yao Y, Slosberg ED, Wang L, Hibshoosh H, Zhang YJ, Xing WQ, Santella RM, Weinstein IB. Oncogene. 1999;18:5159–66. doi: 10.1038/sj.onc.1202908. [DOI] [PubMed] [Google Scholar]
- 82.Ray D, Kiyokawa H. Cancer Res. 2008;68 doi: 10.1158/0008-5472.CAN-07-5983. in press. [DOI] [PubMed] [Google Scholar]
- 83.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 84.Lam MH, Liu Q, Elledge SJ, Rosen JM. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 85.Vecchione A, Baldassarre G, Ishii H, Nicoloso MS, Belletti B, Petrocca F, Zanesi N, Fong LY, Battista S, Guarnieri D, Baffa R, Alder H, Farber JL, Donovan PJ, Croce CM. Cancer Cell. 2007;11:275–89. doi: 10.1016/j.ccr.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]