

Summary
Human glutamate carboxypeptidase II (GCPII) is involved in neuronal signal transduction and intestinal folate absorption by means of the hydrolysis of its two natural substrates, N-acetyl-aspartyl-glutamate (NAAG) and folyl-poly-γ-glutamates, respectively. During the past years, tremendous efforts have been made towards the structural analysis of GCPII. Crystal structures of GCPII in complex with various ligands have provided insight into the binding of these ligands, particularly to the S1′ site of the enzyme. In this paper, we have extended structural characterization of GCPII to its S1 site by using dipeptide-based inhibitors that interact with both S1 and S1′ sites of the enzyme. To this end, we have determined crystal structures of human GCPII in complex with phosphapeptide analogs of folyl-γ-glutamate, aspartyl-glutamate and γ-glutamyl-glutamate, reined at resolution of 1.50 Å, 1.60 Å and 1.67 Å, respectively. The S1 pocket of GCPII could be accurately defined and analyzed for the first time, and the data indicate the importance of Asn519, Arg463, Arg534, and Arg536 for recognition of the penultimate (i.e., P1) substrate residues. Direct interactions between the positively charged guanidinium groups of Arg534 and Arg536 and a P1 moiety of a substrate/inhibitor provide mechanistic explanation of GCPII preference for acidic dipeptides. Additionally, observed conformational flexibility of the Arg463 and Arg536 side chains likely regulates GCPII affinity towards different inhibitors and modulates GCPII substrate specificity. The biochemical experiments assessing the hydrolysis of several GCPII substrate derivatives modified at the P1 position, also included in this report, further complement and extend conclusions derived from the structural analysis. The data described here form an excellent foundation for the structurally aided design of novel low-molecular weight GCPII inhibitors and imaging agents.
Keywords: prostate-specific membrane antigen, metallopeptidase, folate hydrolase, NAALADase, phosphapeptide
Introduction
In humans, the nervous system, small intestine, kidney and prostate are major sites of expression of glutamate carboxypeptidase II (GCPII, E.C. 3.17.21), a zinc-dependent metallopeptidase.1-3 Within the nervous system and small intestine, GCPII hydrolyses its two recognized natural substrates, N-acetyl-aspartyl-glutamate (NAAG) and folyl-poly-γ-glutamates, participating directly in signal transmission via neural pathways and intestinal folate absorption, respectively.4,5 In the kidney and prostate, the physiological function remains unknown.
As NAAG hydrolysis by GCPII in the nervous system leads to the increase in extracellular glutamate levels, the enzyme represents a therapeutic target for treatment of pathologies associated with dysregulated glutamatergic transmission.6,7 Highly selective and potent GCPII inhibitors were reported in the past8-12, and these showed efficacy in a variety of experimental models of neurological disorders, including neuropathic and inflammatory pain, stroke, diabetic neuropathy, amyotrophic lateral sclerosis, and schizophrenia (see refs.13,14 and references therein). Moreover, GCPII is an excellent target for prostate cancer imaging and therapy because of its membrane localization and highly upregulated expression in prostate tumors and metastases.15 Several reports have demonstrated the feasibility of imaging of GCPII-positive cells in experimental models of prostate cancer in vitro and in vivo, using low molecular weight GCPII ligands.16-18 Optimization of such therapeutic and diagnostic strategies could greatly benefit from structural studies on GCPII.
Recently published X-ray structures of GCPII offered the first insight into the substrate binding cavity of the protein. A series of three GCPII structures reported by Mesters et al19 highlighted the preferences of GCPII for substrates containing the C-terminal glutamate, and led to the hypothesis of an induced-fit mechanism of substrate recognition. The subsequent structural study addressing GCPII inhibition by the glutamate mimetics/derivatives illustrated a flexibility of the S1′ pocket of the enzyme and helped to define the structural features required for potent GCPII inhibition.20
In contrast, little is understood about the interaction of the penultimate residue of GCPII substrates with the S1 site of the enzyme, despite the important role of this residue in substrate recognition.21-23 To address this issue, we utilized three phosphapeptide analogs of GCPII substrates in which the planar scissile peptide bond is substituted by a phosphinate moiety. These compounds mimic unstable tetrahedral transition state formed during the course of substrate hydrolysis24 and potently inhibit GCPII by interacting with both S1 and S1′ sites of the enzyme (Figure 1). Structures of GCPII in complex with these inhibitors should shed light on structural features of P1-S1 interactions as well as their role in binding of the inhibitors to the enzyme. The relevance of this research is further supported by the fact that covalent conjugates of one of the inhibitors described in this study (EPE) were successfully used in preclinical studies as contrast agents for image-guided surgery and radiotherapy.17,25 Consequently, our data on the nature of enzyme-substrate/inhibitor interactions should aid the development of novel low molecular weight GCPII inhibitors and imaging agents.
Figure 1. Chemical formulas of GCPII substrates (Panel A) and phosphinate transition-state analogs thereof (Panel B).

Panel A, folyl-γ-glu, a natural GCPII substrate present in the small intestine; γ-glu-glu; γ-glutamyl-glutamate; NAAG, N-acetyl-aspartyl-glutamate, a dipeptidic natural GCPII substrate in the nervous system. Panel B, MPE, 2-[(3-{4-[(2-amino-4-hydroxy-pteridin-6-ylmethyl)-amino]-benzoylamino}-3-carboxy-propyl)-hydroxy-phosphinoylmethyl]-pentanedioic acid; EPE, (2S,3′S)-{[(3′-Amino-3′-carboxy-propyl)-hydroxyphosphinoyl]methyl}-pentanedioic acid; SPE, (2S)-2-{[(2-carboxy-ethyl)-hydroxy-phosphinoyl]methyl}-pentanedioic acid. Chiral centers are indicated by asterisks, numbers 1 and 1′ mark P1 and P1′ chiral centers, respectively.
Results
Phosphapeptide analogs of GCPII substrates
Unstable tetrahedral transition state formed during metallopeptidase-catalyzed peptide hydrolysis can be mimicked by phosphinate analogs of respective substrates, and such compounds have been shown to be effective in inhibiting various enzymes including GCPII.8,26 Phosphinates used in this study include 2-[(3-{4-[(2-amino-4-hydroxy-pteridin-6-ylmethyl)-amino]-benzoylamino}-3-carboxy-propyl)-hydroxy-phosphinoylmethyl]-pentanedioic acid (MPE), (2S,3′S)-{[(3′-Amino-3′-carboxy-propyl)-hydroxyphosphinoyl]methyl}-pentanedioic acid (EPE), and (2S)-2-{[(2-carboxy-ethyl)-hydroxy-phosphinoyl]methyl}-pentanedioic acid (SPE). MPE, EPE, and SPE could be viewed as analogs of GCPII substrates folyl-γ-glutamate, γ-glutamyl-glutamate, and aspartyl-glutamate, respectively (Figure 1).
The synthesis of MPE has been described previously27 and the compound has been provided as a mixture of four diastereomers. Meanwhile, EPE and SPE are provided as single stereoisomers with stereochemistry corresponding to L-γ-Glu-L-Glu and aspartyl-L-Glu, respectively (Figure 1). We determined that all three compounds are potent inhibitors of GCPII with IC50 values of 0.12 nM, 14 nM and 0.7 nM for MPE, EPE and SPE, respectively (Table I).
Table I.
Inhibition of rhGCPII by phosphapeptide analogs of transition state reaction intermediates
| Inhibitor | IC50 (nM) |
|---|---|
| MPE | 0.12 ± 0.03* |
| EPE | 14.0 ± 4.2 |
| SPE | 0.70 ± 0.26 |
Data taken from22
An entrance to the GCPII substrate-binding cavity could be closed by the flexible Trp541-Gly548 loop
The overall fold of the three rhGCPII complexes is similar as demonstrated by r.m.s. deviations of 0.75 Å and 0.74 Å between the rhGCPII/MPE complex and the rhGCPII/EPE or rhGCPII/SPE complexes, respectively. The only notable exception is the fragment Trp541-Gly548 (the ‘entrance lid’), for which the maximum displacement between equivalent Cα-atoms reaches 9.4 Å (for rhGCPII/MPE and rhGCPII/SPE). In both rhGCPII/SPE and rhGCPII/EPE complexes, the ‘entrance lid’ accommodates closed conformation, effectively shielding the active-site bound ligands from the external space. In the rhGCPII/MPE complex, the ‘entrance lid’ adopts an open conformation, due to a steric barrier presented by the bulky distal (pteroyl) part of the inhibitor (Figure 2 and Supplementary Figure S1). Motion of the ‘entrance lid’ is realized by the flipping of the peptide bond between Asn540 and Trp541 at one hinge and flexibility of Gly548 at the other hinge. The side chain of Tyr549, adjacent to the second hinge (Gly548), is inserted into a hydrophobic pocket shaped by Ile386, Asp387, Glu457, Tyr552, Glu557, Leu561, and Tyr566, and evidently plays a role of an anchor at the C-terminal side of the ‘entrance lid’.
Figure 2. The substrate binding cavity of GCPII can be closed by the ‘entrance lid’.

Panel A, Superposition of the ‘entrance lids’ from rhGCPII/MPE and rhGCPII/SPE complexes. The model of rhGCPII/MPE is shown in cartoon representation and colored gray. The ‘entrance lid’, formed by the amino acids Trp541-Gly548, is painted in red and blue for the open (observed in the rhGCPII/MPE complex) and closed (taken from the superimposed structure of the rhGCPII/SPE complex) conformations, respectively. The active site-bound SPE inhibitor is represented by sticks and the active-site Zn2+ ions by magenta spheres. Panels B and C, A close-up view of the ‘entrance lid’ in open/closed conformation. The protein is represented by its molecular surface with the ‘entrance lid’ colored in red or in blue for the open (rhGCPII/MPE, Panel B) and closed (rhGCPII/SPE, Panel C) conformations, respectively. The molecule of MPE, visible in the current projection, is represented by sticks (Panel B), while a molecule of SPE is buried by the ‘entrance lid’ (Panel C). A surface defined by the active-site zinc ions is colored in magenta.
Enzyme-inhibitor interactions within the S1′ pocket and at the vicinity of Zn2+ ions
MPE, one of the phosphapeptides co-crystallized with rhGCPII, features 1′-(R,S)-methylglutarate as the C-terminal moiety (Figure 1). In agreement with the reported preferences of GCPII for L-amino acids at the P1′ (i.e., C-terminal) position4,23, only the (1′-S) diastereomer of MPE was identified to be bound in the rhGCPII active-site. The arrangement of the C-terminal part of all inhibitors within the S1′ pocket is virtually identical to the binding mode of a free glutamate reported previously, with all principal enzyme-inhibitor interactions preserved19 (Figure 3 and Supplementary Figure S2).
Figure 3. The electron density maps of the substrate binding cavity of human GCPII in complex with SPE (Panel A) and EPE (Panel B).

The protein residues are shown in ball-and-stick representation, while Zn2+ and Cl- ions are depicted as blue and yellow spheres, respectively. The Fo-Fc electron density omit maps around inhibitor molecules are contoured at the 3σ level (green) and the 2Fo-Fc electron density maps at the 1σ level (blue). The picture was generated using Molscript41, Bobscript42 and rendered with PovRay.
The phosphinate group of each inhibitor mimics the gem-diolate of a tetrahedral transition state of the reaction, and this functionality contributes prominently to the high affinity of the inhibitor towards GCPII.8 The phosphinate group interacts with the enzyme via contacts with both Zn2+ ions, with the interatomic distances of 2.0 Å (O1...Zn1) and 1.9 Å (O2...Zn2; distances are taken from the rhGCPII/MPE complex). The O1 oxygen atom forms additional hydrogen bonds with the side chains of Tyr552 (2.7 Å), His553 (3.0 Å), and Asp387 (3.2 Å), while the second oxygen atom from the phosphinate group (O2) interacts with the side chains of His377 (3.1 Å), Asp387 (3.1 Å), Asp453 (3.1 Å), and Glu425 (3.2 Å).
Additionally, the O2 atom forms a hydrogen bond (2.8 Å) with the γ-carboxylate of Glu424, which is positioned at the distance of 3.2 Å from a carbon atom substituting the peptide-bond amide of a ‘real’ dipeptidic substrate (Supplementary Figure S2). The side chain of Glu424 can thus participate in proton transfer from the activated water molecule to the ultimate nitrogen of the newly formed N-terminus of a hypothetical reaction product. This data support an earlier prediction suggesting that the Glu424 residue acts as a proton shuttle during hydrolysis by GCPII.28
The S1 pocket of GCPII and positional flexibility of Arg536 and Arg463
Principal interactions between rhGCPII and a carboxylate group of a P1 moiety of a substrate are mediated via side chains of Asn519, Arg534, and Arg536. Arginine residues 534, 536, and 463 form a positively charged patch on the wall of the S1 pocket that determines a preference of GCPII for acidic dipeptides (Figure 4). An important feature of the S1 arginines (notably Arg536 and to a lesser extent Arg463) is their discrete flexibility. Arg536 can adopt two distinct conformations, referred here as the ‘stacking’ conformation and the ‘binding’ conformation. In the ‘stacking’ conformation, Arg536 is not available for substrate binding and its guanidinium group is wedged between the guanidinium groups of Arg534 and Arg463. This conformation is further stabilized by an ion-pair interaction with the Asp465 carboxylate and a hydrogen bond with the Arg463 carbonyl oxygen. The transition into a ‘binding’ conformation is associated with a 4.5 Å shift (measured by the displacement of Cζ atom) of the guanidinium group. Here, stabilization as well as the charge neutralization are provided primarily by the S1-bound chloride anion (Nη1...Cl-, 3.5 Å) and the Glu457 carboxylate (Nη2...Oε1, 2.7 Å; Figure 5A).
Figure 4. The active site of human GCPII with the EPE inhibitor bound.

The dissected substrate-binding cavity of GCPII is represented by its molecular surface and the active site-bound EPE inhibitor is shown in stick representation. The atoms are colored blue (nitrogen), red (oxygen), green (phosphorus), yellow (Zn2+ ions), and gray (carbon atoms). Note the patch of positively charged arginines 463, 534 and 536 contributing towards the preference of GCPII for acidic residues in the P1 position of a substrate. The figure has been generated using Pymol.
Figure 5. The side chain of Arg536 adopts two alternate conformations.

Panel A, Arrangement of S1 arginines in the rhGCPII/SPE complex. Arginine 536 can adopt two distinct conformations referred to as the ‘stacking’ (R536s) and ‘binding’ (R536b) conformation, respectively. Transition between the two conformations is associated with a 4.5 Å shift of the guanidinium group. The atoms are colored blue (nitrogen), red (oxygen), gray (carbon) and yellow (the chloride ion). Polar interactions stabilizing individual conformations of Arg536 are shown as dashed lines. The SPE inhibitor and water molecules have been omited for clarity. Panel B, Superposition of SPE and EPE inhibitors in the substrate-binding cavity of GCPII. The rhGCPII/EPE and rhGCPII/SPE complexes were superimposed based on corresponding Cα atoms (only GCPII atoms from the rhGCPII/SPE complex are shown). The S1 residues and the active-site bound inhibitors are shown in stick representation, Zn2+ and Cl- ions as blue and yellow spheres, respectively. The atoms are colored blue (nitrogen), red (oxygen), gray (rhGCPII/SPE carbons), cyan (EPE carbons), and green (phosphorus). Direct polar interactions between inhibitors and the protein in the S1 pocket are shown as dashed lines.
GCPII-phosphapeptide interactions in the S1 site
Only two direct contacts between GCPII and SPE are observed in the S1 pocket of the enzyme. They are represented by hydrogen bonds between one of the carboxylate oxygen atoms of SPE and the side chains of Asn519 (2.8 Å) and Arg534 (2.8 Å). The remaining four indirect interactions between GCPII and SPE are mediated by the S1-bound water molecules. It should be noted that in the rhGCPII/SPE complex, the Arg536 adopts both ‘stacking’ and ‘binding’ conformations, with the ‘stacking’ conformation being more populated (about 70%). The arrangement of the Arg536 side chain is similar to that in the ligand-free GCPII structure29, implying that the SPE binding is not associated with significant conformational changes within the S1 site. Similar arrangement of the S1 site and comparable set of enzyme-ligand interactions are observed in the rhGCPII/EPE complex (Figure 5B).
Analysis of the S1 site in the rhGCPII/MPE complex reveals considerable rearrangements of the S1 arginines. Here, the guanidinium group of Arg463 forms a hydrogen bond with the carbonyl oxygen of the benzoyl group (3.2 Å) of the inhibitor and this interaction is assisted by the 2.0 Å shift of the Arg463 side chain. The alternate position of Arg463 is stabilized by electrostatic interactions between its guanidinium group and carboxylate groups of Glu457 and Asp465. Repositioning of Arg463 places it closer to the side chain of Arg534 (4.9 Å), sterically preventing Arg536 from adopting the ‘stacking’ conformation. Consequently, in the rhGCPII/MPE complex only the ‘binding’ conformation is observed for the Arg536 side chain contributing two additional hydrogen bonds formed with the γ-carboxylate of the P1 glutamate (3.1 Å and 2.7 Å; Figure 6). The absence of contacts between the distal (i.e., pteroyl) part of MPE and the enzyme suggests that the observed rearrangement of the S1 arginines is responsible for the 100-fold increase in affinity of GCPII toward MPE, as compared to EPE (Table I). Finally, it was somewhat unexpected to find that the absolute configuration at the P1 glutamate of MPE, unambiguously clear from the electron density map, corresponds to the (1-R) diastereomer, i.e., configuration equivalent to D-glutamate, even though the (1-RS) diastereomers were used in the study (Figure 1).
Figure 6. MPE binding leads to rearrangement of the S1 arginines of GCPII.

Repositioning of Arg463 by 2.0 Å prevents Arg536 from adopting the ‘stacking’ (R536s) conformation. As a result, only the ‘binding’ (R536b) conformation of Arg536 is observed in the rhGCPII/MPE complex. The rhGCPII/MPE and rhGCPII/EPE complexes were superimposed using corresponding Cα atoms. A protein part of the rhGCPII/MPE complex is shown in cartoon representation and selected amino acid residues from both complexes and the active-site bound MPE are shown in stick representation. Zn2+ and Cl- ions are depicted as blue and yellow spheres, respectively. Atoms are colored blue (nitrogen), red (oxygen), gray (rhGCPII/MPE carbons), cyan (rhGCPII/EPE carbons) and green (phosphorus). Direct H-bonding interactions between MPE and the protein in the S1 pocket are shown as dashed lines.
Effects of P1 modifications on substrate hydrolysis
To corroborate and extend conclusions drawn from the presented crystal structures, we designed and synthesized GCPII substrates modified at the P1 moiety and analyzed their hydrolysis by rhGCPII. Molecular formulas of the substrates together with the corresponding kinetic parameters, as determined by an HPLC assay (see Materials and Methods), are summarized in Table II. In agreement with structural analysis of the rhGCPII/MPE complex, we found that Ac-γ-D-Glu-L-Glu binds to GCPII slightly better than the Ac-γ-L-Glu-L-Glu form. On the other hand, the 1-D enantiomers of the remaining dipeptides tested (i.e., NAAG, β-NAAG, and Ac-D-Glu-L-Glu) are hydrolyzed by rhGCPII less efficiently than corresponding dipeptides with the 1-L stereochemistry. An observed 10 to 100-fold decrease in the catalytic efficiency of rhGCPII towards 1-D enantiomers is attributable to both the increase in substrate binding constants and lower turnover numbers.
Table II.
rhGCPII-catalyzed hydrolysis of acidic dipeptides modified at the P1 position
| Substrate | Molecular Structure | KM (μM) | kcat (s-1) | kcat/KM (103s-1M-1) |
|---|---|---|---|---|
| N-Ac-L-Asp-L-Glu (NAAG) | 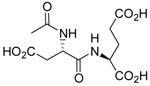 |
1.2 ± 0.5# | 1.1 ± 0.2 | 917 |
| N-Ac-D-Asp-L-Glu | 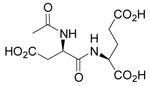 |
3.1 ± 0.6 | 0.27 ± 0.01 | 87 |
| N-Ac-β-L-Asp-L-Glu (β-NAAG) | 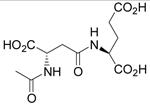 |
5.0 ± 1.2 | 0.14 ± 0.01 | 28 |
| N-Ac-β-D-Asp-L-Glu | 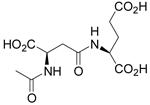 |
ND | < 0.02& | ND |
| N-Ac-L-Glu-L-Glu | 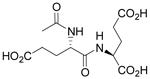 |
2.0 ± 0.9 | 1.0 ± 0.1 | 500 |
| N-Ac-D-Glu-L-Glu | 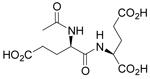 |
5.0 ± 1.0 | 0.026 ± 0.001 | 5 |
| N-Ac-γ-L-Glu-L-Glu | 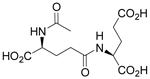 |
4.6 ± 0.7 | 1.6 ± 0.1 | 348 |
| N-Ac-γ-D-Glu-L-Glu | 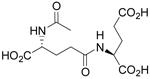 |
3.55 ± 0.90 | 1.95 ± 0.10 | 549 |
| N-Malonyl-L-Glu | 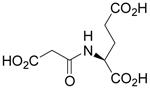 |
1.53 ± 0.7 | 0.049 ± 0.005 | 32 |
| N-Succinyl-L-Glu | 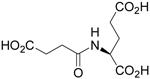 |
< 1.0$ | 0.22 ± 0.02 | > 220 |
| N-Glutaryl-L-Glu | 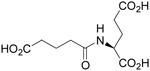 |
2.45 ± 0.90 | 0.97 ± 0.09 | 396 |
| N-Adipyl-L-Glu | 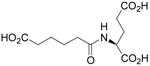 |
2.89 ± 0.92 | 0.14 ± 0.02 | 48 |
substrate hydrolysis below the detection limit
the exact value of the Michaelis constant could not be calculated due to sensitivity limits of the assay
data taken from22
To assess the importance of N-acetylation and a size of P1 residues for substrate hydrolysis by GCPII, we synthesized a series of dipeptides lacking the N-terminal acetylated amino group and differing in the length of a P1 side chain (from carboxymethyl to carboxybutyl; Table II). Comparison of kinetic parameters with corresponding data on N-acetylated dipeptides reveals that N-acetylation does not contribute significantly to the binding affinity of a substrate. In addition, we show that succinyl-L-Glu and glutaryl-L-Glu, analogous to NAAG and Ac-Glu-L-Glu, respectively, are the best substrates within this series, even though all of the substrates within this series bind rhGCPII with the comparable affinity in the low micromolar range.
Discussion
The main objective of this work was to enhance our understanding of the interactions governing substrate specificity of human GCPII, with particular emphasis on the S1 pocket. Our results provide a mechanistic insight into GCPII preferences for dipeptides with acidic side chains and can also be used for the structurally aided design of novel therapeutics. For example, the EPE phosphapeptide used in this study serves as a modular ligand for the GCPII active site with the amino group utilized for an incorporation of various functional groups, including fluorescent dyes or radiolabels.17,25 Such derivatives could be exploited as GCPII inhibitor-based imaging agents or therapeutics for prostate cancer and/or neurodegenerative diseases and provide an alternative to conventional antibody-based applications.
The examination of the rhGCPII structure in complex with MPE reveals that only the glutamate residues and the benzoyl carbonyl oxygen interact with the enzyme, while the pteridine ring is located outside of the GCPII substrate binding pocket. Contrary to the C-terminal part of the inhibitor, which is well defined in the electron density map, the absence of the electron density for the pteridine ring indicates its high flexibility. This observation supports previous findings that a wide variety of functional groups could be attached to the N-terminal amino group of EPE without an appreciable loss of an inhibitor affinity.17,30,31 This trend is also supported by the study of Luthi-Carter et al who showed that there is no significant difference in GCPII affinity towards non-substituted poly-γ-glutamates and their pteroyl or aminobenzyl derivatives. Furthermore, the degree of poly-γ-glutamylation did not affect binding affinity of a substrate as long as it possessed at least two glutamyl residues.30 These results agree well with our structural observations and strengthen the conclusions drawn for EPE and its derivatives.
Although MPE was provided as a mixture of four diastereomers, the crystal structure shows only a single diastereomer corresponding to γ-D-Glu-L-Glu bound to GCPII. This finding is not necessarily surprising given that both Ac-γ-D-Glu-L-Glu and Ac-γ-L-Glu-L-Glu are equally potent substrates for rhGCPII (with the D-form even slightly better; Table II). While there is convincing experimental evidence that the L-stereochemistry is much preferred at the C-terminus of GCPII substrates/inhibitors4,20,23,32, the enzyme is evidently more tolerant to varying stereochemistry at the P1 position. However, the stereochemical preference of GCPII at the P1 position is clearly regioselective and likely depends on the nature/chemistry of a functionality attached to the P1 moiety (ref.33 and data presented here). For example, while Ac-γ-L-Glu-L-Glu and Ac-γ-D-Glu-L-Glu show similar KM and kcat values, the kinetic constants for Ac-α-L-Glu-L-Glu and Ac-α-D-Glu-L-Glu exhibit significant difference with the L-stereoisomer being clearly preferred. Overall, these observations suggest that specificity towards P1 modified ligands would be dictated by the combination of stereochemistry and regiochemistry of the peptide bond (or its mimetics) as well as the physicochemical characteristics of a P1 substituent.
Comparison of rhGCPII/MPE and rhGCPII/SPE(EPE) complexes indicates substantial differences in positioning of the Trp541-Gly548 amino acid segment (the ‘entrance lid’) that can shield the substrate binding cavity of GCPII from an extracellular environment. It is interesting to note that in the high-resolution structures of GCPII complexes and ligand-free GCPII published previously, a portion of the ‘entrance lid’ was not modeled, due to the lack of corresponding electron density, implying high positional flexibility of this loop.20,29 Our observations suggest that the closed conformation of the ‘entrance lid’ might be stabilized by ligand binding in the active-site of GCPII (i.e., the occupancy of the S1 pocket by a glutamate/aspartate moiety of the substrate/inhibitor), despite the lack of direct contacts between a ligand and the ‘entrance lid’. Additionally, when larger substrates such as folyl-poly-γ-glutamates are processed by GCPII, the ‘entrance lid’ probably remains open during the course of hydrolysis. Taken together, an access to the substrate binding cavity of GCPII could be blocked upon substrate binding, but the closure of the ‘entrance lid’ is not required for substrate hydrolysis to occur.
Thermal parameters of the P1′ residues of inhibitors are substantially lower when compared to the P1 counterparts. While C-termini of MPE, EPE, and SPE have an average B-factor of 18.3, 21.2, and 17.8 Å2, respectively, the corresponding P1 residues are characterized by the B-factors’ values of 26.0, 34.0, and 27.0 Å2, respectively. The decreased mobility of atoms at the C-termini of the ligands suggests tighter binding of this fragment to the enzyme. This fact is in good agreement with available biochemical evidence. Site-directed mutagenesis of GCPII residues participating in the substrate/inhibitor binding has led to the suggestion that amino acids defining the S1′ pocket are required for the high-affinity interactions with substrates/inhibitors, while the residues within S1 pocket might be more important for the ‘fine-tuning’ of the GCPII substrate specificity.34 Furthermore, structure-activity relationship (SAR) analyses revealed that although a wide variety of substitutions are tolerated at the P1 position of phosphinate/urea-based inhibitors without a significant loss of potency, the P1′ moiety is very sensitive to structural changes and is primarily responsible for tight GCPII binding.8,9,11
Several mammalian homologs and orthologs of GCPII have been described in literature.35 Alignment of the available amino acid sequences reveals that, with the exception of Asn519, all of the S1 residues are conserved among NAAG-peptidases (i.e., GCPII and GCPIII), while the only S1 residue shared between human GCPII and mammalian NAALADases L is Arg463. This observation suggests that the architecture of the S1 site of human GCPII and its mammalian GCPII/GCPIII homologs/orthologs is similar and that comparable enzyme-substrate interactions could exist within the S1 site. On the other hand, the observed variability of the S1 residues in NAALADases L is consistent with distinct (yet unknown) substrate specificities of these enzymes and their inability to hydrolyze NAAG.36
Conclusions
The data presented here allow for the first time a detailed definition and analysis of the S1 site of GCPII. In particular, the structural observations provide mechanistic insight into GCPII-substrate interactions and help to understand the substrate specificity of the enzyme. Furthermore, structures of rhGCPII/inhibitor complexes can assist the rational design and development of novel diagnostic and therapeutic agents targeting GCPII.
Experimental Section
rhGCPII expression and purification
Recombinant human GCPII (rhGCPII) comprising amino acids 44 to 750 of the full-length enzyme was expressed and purified as described previously.21 Briefly, the extracellular part of human GCPII was overexpressed in the Drosophila Schneider’s S2 cells and the protein was purified from the conditioned medium by the combination of ion-exchange (Q-Sepharose and SP-Sepharose; GE Healthcare), affinity (Lentil-Lectin Sepharose; GE Healthcare), and size-exclusion (Superdex HR200, GE Healthcare) chromatography steps. The final protein preparation was >98% pure (as determined by silver-stained SDS-PAGE, data not shown). The rhGCPII stock solution (10 mg/ml, determined using Bio-Rad Protein Assay kit with bovine serum albumin as a standard) in 20 mM Tris-HCl, 100 mM NaCl, pH 8.0, was frozen in liquid nitrogen and stored at -80°C until further use.
Synthesis of phosphapeptide inhibitors
The synthesis of MPE has been described previously.27 The syntheses of SPE and EPE are described in the Supplementary Material (Scheme 1, 2). For crystallization experiments, SPE and EPE were dissolved in distilled water to a final concentration of 50 mM. MPE was dissolved in distilled water (50 mM) and pH of the solution adjusted to 7.0 with NaOH. For kinetic measurements, the inhibitors were dissolved in 20 mM MOPS, 20 mM NaCl, pH 7.4.
Crystallization and X-ray data collection
The rhGCPII stock solution was mixed with 1/10 volume of the individual phosphinate substrate analog, and the crystallization droplets were prepared by combining 2 μl of the rhGCPII-analog mixture and 2 μl of the reservoir solution containing 33% pentaerythriol propoxylate (PO/OH 5/4; Hampton Research), 1% PEG3350, and 100 mM Tris-HCl, pH 8.0. Crystals were grown by the hanging drop vapor diffusion method at 293 K. Pyramidal crystals, of the dimensions of approximately 0.5 × 0.4 × 0.1 mm, belonging to the I222 space group with one rhGCPII molecule per the asymmetric unit, grew typically within one week. The crystals were flash-frozen in liquid nitrogen directly from the reservoir solution and the diffraction data were collected at 100 K, using synchrotron radiation at the SER-CAT sector 22 beamlines of the Advanced Photon Source (Argonne, IL, USA) at the X-ray wavelength of 1.0 Å (rhGCPII/MPE and rhGCPII/EPE complexes) or 0.976 Å (the rhGCPII/SPE complex). In all cases, the diffraction intensities were collected from a single crystal, recorded on a CCD detector and processed using the HKL2000 software package.37
Structure determination and refinement
Since the crystals of the rhGCPII complexes were isomorphous with the crystals of ligand-free rhGCPII29 (PDB code 2OOT), the latter was used as the starting model for the structure determination using difference Fourier methods. The initial structure determinations, carried out at a resolution of 2.5 Å, were followed by the refinement of individual atomic coordinates and B-factors, with gradual extension of the resolution to the limits of the experimental data. Calculations were performed with the program Refmac 5.138, and the refinement protocol was interspersed with manual corrections to the model, employing the programs O39 and Xtalview.40 In the final stage of the refinement, the anisotropic model of the displacement parameters (B-factors) was applied to all non-hydrogen atoms in the rhGCPII/MPE complex, and to “heavy atoms” (i.e., S, Zn2+, Ca2+, and Cl-) of rhGCPII/EPE and rhGCPII/SPE complexes. During the refinement process, ∼ 1% of the randomly selected reflections were kept aside for cross-validation (Rfree). The data collection and refinement statistics are shown in Table III.
Table III.
Data collection and refinement statistics
| Data collection statistics | |||
|---|---|---|---|
| rhGCPII/MPE | rhGCPII/EPE | rhGCPII/SPE | |
| Wavelength (Å) | 1.000 | 1.000 | 0.976 |
| Temperature (K) | 100 | 100 | 100 |
| Space group | I222 | I222 | I222 |
| Unit-cell parameters a, b, c (Å) | 102.1, 130.5, 159.5 | 102.4, 130.8, 159.1 | 101.5, 130.3, 159.2 |
| Resolution limits (Å) | 30.0 - 1.50 (1.55 - 1.50)* | 50.0 - 1.67 (1.73 - 1.67) | 30.0-1.60 (1.66-1.60) |
| Number of unique reflections | 169,309 (16,622) | 119,268 (9,667) | 135,823 (13,157) |
| Redundancy | 10.0 (6.6) | 10.9 (6.1) | 3.9 (3.9) |
| Completeness (%) | 99.5 (98.2) | 97.4 (79.1) | 97.9 (95.6) |
| I/σI | 19.0 (2.2) | 23.4 (2.1) | 20.7 (4.3) |
| Rmerge | 0.085 (0.62) | 0.091 (0.52) | 0.042 (0.29) |
| Refinement statistics | |||
| Resolution limits (Å) | 15.0 - 1.50 (1.54 - 1.50) | 15.0 - 1.67 (1.72 - 1.67) | 15.0 - 1.60 (1.64 - 1.60) |
| Total number of reflections | 166,133 (11,987) | 117,320 (6,680) | 134,043 (9,606) |
| Number of reflection in working set | 164,450 (11,872) | 115,534 (6,577) | 132,404 (9,491) |
| Number of reflection in test set | 1,683 (115) | 1,786 (103) | 1,639 (115) |
| R / Rfree | 0.148 (0.201)/0.169 (0.274) | 0.184 (0.314)/0.211 (0.366) | 0.181 (0.263)/0.201 (0.31) |
| Total number of non-H atoms | 6,643 | 6,572 | 6,601 |
| Number of ligand atoms | 43 | 20 | 18 |
| Number of ions | 4 | 4 | 4 |
| Number of water molecules | 715 | 644 | 661 |
| Average B-factor (Å2) | |||
| Protein atoms | 25.1 | 27.7 | 24.6 |
| Waters | 39.4 | 39.0 | 36.9 |
| Inhibitor | 36.6 | 25.6 | 20.8 |
| R.m.s deviations: | |||
| Bond lengths (Å) | 0.020 | 0.021 | 0.020 |
| Bond angles (°) | 1.77 | 1.78 | 1.88 |
| Planarity (Å) | 0.011 | 0.010 | 0.011 |
| Chiral-centers (Å3) | 0.13 | 0.13 | 0.13 |
| Ramachandran Plot (%) | |||
| Most favored | 90.7 | 89.7 | 89.8 |
| Additionally allowed | 8.8 | 9.7 | 9.5 |
| Generously allowed | 0.3 | 0.5 | 0.5 |
| Disallowed | 0.2 (Lys207) | 0.2 (Lys207) | 0.2 (Lys207) |
| Gaps in the structure | 44-54, 654-655 | 44-54, 541, 654-655 | 44-54, 654-655 |
Values in parentheses correspond to the highest resolution shells.
Synthesis of peptidic substrates
Peptide substrates were synthetized using an ABI 433A Peptide Synthesizer (Applied Biosystems, Foster City, CA, USA) and a standard Fmoc chemistry method.
Determination of kinetic constants
Purified rhGCPII was mixed with various concentrations (2-400 μM) of a substrate in solution containing 20 mM MOPS (pH 7.4) and 20 mM NaCl to a final volume of 120 μl. Following 20 min incubation at 37 °C, the reaction was terminated by the addition of 60 μl of solution containing 33 mM EDTA and 66 mM sodium borate (pH 9.2). Released glutamate was derivatized using 20 μl of an AccQ-Fluor reagent (Waters, Milford, MA), dissolved in acetonitrile. The resulting mixture was resolved on a Luna C18 column 250 × 4.6 mm, 5 μm (Phenomenex, Torrance, CA) mounted to a Waters Alliance 2795 system equipped with a Waters 2475 fluorescence detector using a 20%-35% linear gradient of B (56 mM sodium acetate, 6.8 mM triethanolamine, 60% acetonitrile, pH 6.6) in A (140 mM sodium acetate, 17 mM triethanolamine pH 5.05) over 9 mins. Glutamate quantification was performed using a calibration curve constructed from known concentrations of a glutamic acid standard. The KM and kcat values were determined from reaction rate vs. substrate concentration plots using the GraFit program (version 5.0.4, Erithacus Software Limited, Horley, UK).
Determination of inhibition constants
The inhibition constants of rhGCPII were determined by an enzymatic assay using 3H-NAAG (radiolabeled on the terminal glutamate) as described previously.22 rhGCPII (30 ng/ml) was preincubated with increasing concentrations of inhibitors in the solution containing 20 mM MOPS (pH 7.4) and 20 mM NaCl at 37 °C for 15 mins. The reaction was started by an addition of 20 μl of the mixture of 0.95 μM NAAG (Sigma) and 50 nM 3H-NAAG (50 Ci/mmol in Tris-HCl buffer, Perkin Elmer) to the total reaction volume of 200 μl. Following 20 min incubation, the reaction was terminated by the addition of 200 μl of 200 mM potassium phosphate (pH 7.4). Glutamate was separated from the reaction mixture by an ion exchange chromatography and quantified by a liquid scintillation. Each experimental point was measured in duplicate. The IC50 values were determined from plots of vi/v0 (ratio of individual reaction rate to rate of uninhibited reaction) vs. inhibitor concentration using the GraFit software.
Supplementary Material
Acknowledgement
We thank Jana Starkova for the excellent technical assistance, Zbigniew Dauter for help with data collection, and Drs. James K. Coward and Nadya Valiaeva (University of Michigan) for their generous gift of MPE. Diffraction data were collected at the South-East Regional Collaborative Access Team (SER-CAT) beamline 22-ID, at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng38. This project was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (J.L. and C.B.). J.K., M.R., and K.H. were in part supported by the Ministry of Education of the Czech Republic (Research Centre for New Antivirals and Antineoplastics, 1M0508).
Accession numbers:
Atomic coordinates of the present structures together with the experimental diffraction amplitudes have been deposited at the RCSB Protein Data Bank with accession numbers 3BI1 (the complex with MPE), 3BI0 (the complex with EPE), and 3BHX (the complex with SPE).
Abbreviations
- rhGCPII
recombinant human glutamate carboxypeptidase II, extracellular domain (44-750)
- r.m.s.d.
root mean square deviation
- Fmoc
fluoren-9-yl-methoxycarbonyl
- HPLC
high-performance liquid chromatography
- MOPS
3-[N-morpholino]propanesulfonic acid
- EDTA
ethylenediaminetetraacetic acid
- AccQ
6-aminoquinolyl-N-hydroxysuccinimidyl carbamate
- Ac
acetyl
- MPE
2-[(3-{4-[(2-amino-4-hydroxy-pteridin-6-ylmethyl)-amino]-benzoylamino}-3-carboxy-propyl)-hydroxy-phosphinoylmethyl]-pentanedioic acid
- EPE
(2S,3′S)-{[(3′-Amino-3′-carboxy-propyl)-hydroxyphosphinoyl]methyl}-pentanedioic acid
- SPE
(2S)-2-{[(2-carboxy-ethyl)-hydroxy-phosphinoyl]methyl}-pentanedioic acid
- NAALADase
N-acetylated-α-linked-acidic dipeptidase
- SDS
sodium dodecylsulfate
- Tris
tris(hydroxymethyl)aminomethane
- NAAG
N-acetyl-L-aspartyl-L-glutamate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sacha P, Zamecnik J, Barinka C, Hlouchova K, Vicha A, Mlcochova P, Hilgert I, Eckschlager T, Konvalinka J. Expression of glutamate carboxypeptidase II in human brain. Neuroscience. 2007;144:1361–1372. doi: 10.1016/j.neuroscience.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 2.Troyer JK, Beckett ML, Wright GL., Jr. Detection and characterization of the prostate-specific membrane antigen (PSMA) in tissue extracts and body fluids. Int. J. Cancer. 1995;62:552–558. doi: 10.1002/ijc.2910620511. [DOI] [PubMed] [Google Scholar]
- 3.Rovenska M, Hlouchova K, Sacha P, Mlcochova P, Horak V, Zamecnik J, Barinka C, Konvalinka J. Tissue expression and enzymologic characterization of human prostate specific membrane antigen and its rat and pig orthologs. The Prostate. 2007 doi: 10.1002/pros.20676. DOI 10.1002/pros.20676. [DOI] [PubMed] [Google Scholar]
- 4.Robinson MB, Blakely RD, Couto R, Coyle JT. Hydrolysis of the brain dipeptide N-acetyl-L-aspartyl-L-glutamate. Identification and characterization of a novel N-acetylated alpha-linked acidic dipeptidase activity from rat brain. J. Biol. Chem. 1987;262:14498–14506. [PubMed] [Google Scholar]
- 5.Halsted CH, Ling EH, Luthi-Carter R, Villanueva JA, Gardner JM, Coyle JT. Folylpoly-gamma-glutamate carboxypeptidase from pig jejunum. Molecular characterization and relation to glutamate carboxypeptidase II. J. Biol. Chem. 1998;273:20417–20424. doi: 10.1074/jbc.273.32.20417. [DOI] [PubMed] [Google Scholar]
- 6.Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, Traystman RJ, Robinson MB, Britton P, Lu XC, Tortella FC, Wozniak KM, Yudkoff M, Potter BM, Jackson PF. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat. Med. 1999;5:1396–1402. doi: 10.1038/70971. [DOI] [PubMed] [Google Scholar]
- 7.Vornov JJ, Wozniak K, Lu M, Jackson P, Tsukamoto T, Wang E, Slusher B. Blockade of NAALADase: a novel neuroprotective strategy based on limiting glutamate and elevating NAAG. Ann. N. Y. Acad. Sci. 1999;890:400–405. doi: 10.1111/j.1749-6632.1999.tb08019.x. [DOI] [PubMed] [Google Scholar]
- 8.Jackson PF, Tays KL, Maclin KM, Ko YS, Li W, Vitharana D, Tsukamoto T, Stoermer D, Lu XC, Wozniak K, Slusher BS. Design and pharmacological activity of phosphinic acid-based NAALADase inhibitors. J. Med. Chem. 2001;44:4170–4175. doi: 10.1021/jm0001774. [DOI] [PubMed] [Google Scholar]
- 9.Kozikowski AP, Zhang J, Nan F, Petukhov PA, Grajkowska E, Wroblewski JT, Yamamoto T, Bzdega T, Wroblewska B, Neale JH. Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase II: efficacy as analgesic agents. J. Med. Chem. 2004;47:1729–1738. doi: 10.1021/jm0306226. [DOI] [PubMed] [Google Scholar]
- 10.Majer P, Hin B, Stoermer D, Adams J, Xu W, Duvall BR, Delahanty G, Liu Q, Stathis MJ, Wozniak KM, Slusher BS, Tsukamoto T. Structural optimization of thiol-based inhibitors of glutamate carboxypeptidase II by modification of the P1′ side chain. J. Med. Chem. 2006;49:2876–2885. doi: 10.1021/jm051019l. [DOI] [PubMed] [Google Scholar]
- 11.Kozikowski AP, Nan F, Conti P, Zhang J, Ramadan E, Bzdega T, Wroblewska B, Neale JH, Pshenichkin S, Wroblewski JT. Design of remarkably simple, yet potent urea-based inhibitors of glutamate carboxypeptidase II (NAALADase) J. Med. Chem. 2001;44:298–301. doi: 10.1021/jm000406m. [DOI] [PubMed] [Google Scholar]
- 12.Nan F, Bzdega T, Pshenichkin S, Wroblewski JT, Wroblewska B, Neale JH, Kozikowski AP. Dual function glutamate-related ligands: discovery of a novel, potent inhibitor of glutamate carboxypeptidase II possessing mGluR3 agonist activity. J. Med. Chem. 2000;43:772–774. doi: 10.1021/jm9905559. [DOI] [PubMed] [Google Scholar]
- 13.Zhou J, Neale JH, Pomper MG, Kozikowski AP. NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nat. Rev. Drug Discov. 2005;4:1015–1026. doi: 10.1038/nrd1903. [DOI] [PubMed] [Google Scholar]
- 14.Tsukamoto T, Wozniak KM, Slusher BS. Progress in the discovery and development of glutamate carboxypeptidase II inhibitors. Drug Discov. Today. 2007;12:767–776. doi: 10.1016/j.drudis.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Bostwick DG, Pacelli A, Blute M, Roche P, Murphy GP. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: a study of 184 cases. Cancer. 1998;82:2256–2261. doi: 10.1002/(sici)1097-0142(19980601)82:11<2256::aid-cncr22>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 16.Foss CA, Mease RC, Fan H, Wang Y, Ravert HT, Dannals RF, Olszewski RT, Heston WD, Kozikowski AP, Pomper MG. Radiolabeled small-molecule ligands for prostate-specific membrane antigen: in vivo imaging in experimental models of prostate cancer. Clin. Cancer Res. 2005;11:4022–4028. doi: 10.1158/1078-0432.CCR-04-2690. [DOI] [PubMed] [Google Scholar]
- 17.Humblet V, Lapidus R, Williams LR, Tsukamoto T, Rojas C, Majer P, Hin B, Ohnishi S, De Grand AM, Zaheer A, Renze JT, Nakayama A, Slusher BS, Frangioni JV. High-affinity near-infrared fluorescent small-molecule contrast agents for in vivo imaging of prostate-specific membrane antigen. Mol. Imaging. 2005;4:448–462. doi: 10.2310/7290.2005.05163. [DOI] [PubMed] [Google Scholar]
- 18.Tang H, Brown M, Ye Y, Huang G, Zhang Y, Wang Y, Zhai H, Chen X, Shen TY, Tenniswood M. Prostate targeting ligands based on N-acetylated alpha-linked acidic dipeptidase. Biochem. Biophys. Res. Commun. 2003;307:8–14. doi: 10.1016/s0006-291x(03)01119-7. [DOI] [PubMed] [Google Scholar]
- 19.Mesters JR, Barinka C, Li W, Tsukamoto T, Majer P, Slusher BS, Konvalinka J, Hilgenfeld R. Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 2006;25:1375–1384. doi: 10.1038/sj.emboj.7600969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barinka C, Rovenska M, Mlcochova P, Hlouchova K, Plechanovova A, Majer P, Tsukamoto T, Slusher BS, Konvalinka J, Lubkowski J. Structural insight into the pharmacophore pocket of human glutamate carboxypeptidase II. J. Med. Chem. 2007;50:3267–3273. doi: 10.1021/jm070133w. [DOI] [PubMed] [Google Scholar]
- 21.Barinka C, Rinnova M, Sacha P, Rojas C, Majer P, Slusher BS, Konvalinka J. Substrate specificity, inhibition and enzymological analysis of recombinant human glutamate carboxypeptidase II. J. Neurochem. 2002;80:477–487. doi: 10.1046/j.0022-3042.2001.00715.x. [DOI] [PubMed] [Google Scholar]
- 22.Hlouchova K, Barinka C, Klusak V, Sacha P, Mlcochova P, Majer P, Rulisek L, Konvalinka J. Biochemical characterization of human glutamate carboxypeptidase III. J. Neurochem. 2007;101:682–696. doi: 10.1111/j.1471-4159.2006.04341.x. [DOI] [PubMed] [Google Scholar]
- 23.Mhaka A, Gady AM, Rosen DM, Lo KM, Gillies SD, Denmeade SR. Use of methotrexate-based peptide substrates to characterize the substrate specificity of prostate-specific membrane antigen (PSMA) Cancer Biol. Ther. 2004;3:551–558. doi: 10.4161/cbt.3.6.846. [DOI] [PubMed] [Google Scholar]
- 24.Holz RC, Bzymek KP, Swierczek SI. Co-catalytic metallopeptidases as pharmaceutical targets. Curr. Opin. Chem. Biol. 2003;7:197–206. doi: 10.1016/s1367-5931(03)00033-4. [DOI] [PubMed] [Google Scholar]
- 25.Humblet V, Misra P, Frangioni JV. An HPLC/mass spectrometry platform for the development of multimodality contrast agents and targeted therapeutics: prostate-specific membrane antigen small molecule derivatives. Contrast. Media Mol. Imaging. 2006;1:196–211. doi: 10.1002/cmmi.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsukamoto T, Majer P, Vitharana D, Ni C, Hin B, Lu XC, Thomas AG, Wozniak KM, Calvin DC, Wu Y, Slusher BS, Scarpetti D, Bonneville GW. Enantiospecificity of glutamate carboxypeptidase II inhibition. J. Med. Chem. 2005;48:2319–2324. doi: 10.1021/jm049258g. [DOI] [PubMed] [Google Scholar]
- 27.Valiaeva N, Bartley D, Konno T, Coward JK. Phosphinic acid pseudopeptides analogous to glutamyl-gamma-glutamate: Synthesis and coupling to pteroyl azides leads to potent inhibitors of folylpoly-gamma-glutamate synthetase. Journal of Organic Chemistry. 2001;66:5146–5154. doi: 10.1021/jo010283t. [DOI] [PubMed] [Google Scholar]
- 28.Rawlings ND, Barrett AJ. Structure of membrane glutamate carboxypeptidase. Biochim. Biophys. Acta. 1997;1339:247–252. doi: 10.1016/s0167-4838(97)00008-3. [DOI] [PubMed] [Google Scholar]
- 29.Barinka C, Starkova J, Konvalinka J, Lubkowski J. A high-resolution structure of ligand-free human glutamate carboxypeptidase II. Acta Crystallograph. Sect. F. Struct. Biol. Cryst. Commun. 2007;63:150–153. doi: 10.1107/S174430910700379X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luthi-Carter R, Barczak AK, Speno H, Coyle JT. Hydrolysis of the neuropeptide N-acetylaspartylglutamate (NAAG) by cloned human glutamate carboxypeptidase II. Brain Res. 1998;795:341–348. doi: 10.1016/s0006-8993(98)00244-3. [DOI] [PubMed] [Google Scholar]
- 31.Tsukamoto T, Flanary JM, Rojas C, Slusher BS, Valiaeva N, Coward JK. Phosphonate and phosphinate analogues of N-acylated gamma-glutamylglutamate: Potent inhibitors of glutamate carboxypeptidase II. Bioorganic & Medicinal Chemistry Letters. 2002;12:2189–2192. doi: 10.1016/s0960-894x(02)00360-8. [DOI] [PubMed] [Google Scholar]
- 32.Vitharana D, France JE, Scarpetti D, Bonneville GW, Majer P, Tsukamoto T. Syntesis and biological evaluation of (R)-and (S)-2-(phosphonomethyl)pentanedioic acids as inhibitors of glutamate carboxypeptidase II. Tetrahedron: Asymmetry. 2002;13:1609–1614. [Google Scholar]
- 33.Anderson MO, Wu LY, Santiago NM, Moser JM, Rowley JA, Bolstad ES, Berkman CE. Substrate specificity of prostate-specific membrane antigen. Bioorg. Med. Chem. 2007;15:6678–6686. doi: 10.1016/j.bmc.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mlcochova P, Plechanovova A, Barinka C, Majer P, Mahadevan D, Saldanha J, Rulisek L, Konvalinka J. Mapping of the active site of glutamate carboxypeptidase II by site-directed mutagenesis. FEBS Journal. 2007;274:4731–4741. doi: 10.1111/j.1742-4658.2007.06021.x. [DOI] [PubMed] [Google Scholar]
- 35.Lambert LA, Mitchell SL. Molecular evolution of the transferrin receptor/glutamate carboxypeptidase II family. J. Mol. Evol. 2007;64:113–128. doi: 10.1007/s00239-006-0137-4. [DOI] [PubMed] [Google Scholar]
- 36.Pangalos MN, Neefs JM, Somers M, Verhasselt P, Bekkers M, van der Helm L, Fraiponts E, Ashton D, Gordon RD. Isolation and expression of novel human glutamate carboxypeptidases with N-acetylated alpha-linked acidic dipeptidase and dipeptidyl peptidase IV activity. J. Biol. Chem. 1999;274:8470–8483. doi: 10.1074/jbc.274.13.8470. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr., Sweet RM, editors. Methods in Enzymology. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 38.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 39.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 41.Kraulis PE. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. App. Crystal. 1991;24:946–950. [Google Scholar]
- 42.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.