

Abstract
IκB kinase β (IKKβ), a major kinase downstream of various proinflammatory signals, mediates multiple cellular functions through phosphorylation and regulation of its substrates. On the basis of protein sequence analysis, we identified arrest-defective protein 1 (ARD1), a protein involved in apoptosis and cell proliferation processes in many human cancer cells, as a new IKKβ substrate. We provided evidence showing that ARD1 is indeed a bona fide substrate of IKKβ. IKKβ physically associated with ARD1 and phosphorylated it at Ser209. Phosphorylation by IKKβ destabilized ARD1 and induced its proteasome-mediated degradation. Impaired growth suppression was observed in ARD1 phosphorylation-mimic mutant (S209E)-transfected cells as compared with ARD1 non-phosphorylatable mutant (S209A)-transfected cells. Our findings of molecular interactions between ARD1 and IKKβ may enable further understanding of the upstream regulation mechanisms of ARD1 and of the diverse functions of IKKβ.
Keywords: Phosphorylation, Arrest-defective protein 1, IκB kinase β, Destabilization, Degradation
Introduction
Arrest-defective protein 1 (ARD1), first identified in yeast, is the catalytic subunit of NatA acetyltransferase, responsible for N-terminal α-acetylation [1]. Mutation of Ard1 in yeast leads to defective entry into the stationary phase and sporulation in response to nutrient deprivation or mating pheromone α-factor [2; 3]. In mammalian cells, ARD1 possesses both N-terminal α-protein and ε-protein acetylation activities, thus representing a novel kind of acetyltransferase [4; 5]. ARD1 has been reported to mediate hypoxia-inducible factor 1α (HIF-1α) ubiquitination and degradation through Lys532 acetylation [5]; however, several groups were unable to replicate this observation [6; 7; 8]. Another ε-acetylation substrate of ARD1 is β-catenin, which was shown to mediate the cell proliferation effect of ARD1 in lung cancer cells [9]. In addition to cell growth control, ARD1 is also involved in DNA damage-induced apoptosis [10]. Although ARD1 plays a critical role in regulating cell proliferation and apoptosis, the molecular mechanisms regulating ARD1 stability and functions remain largely unclear.
IκB kinase β (IKKβ) is a component of the IKK complex, which contains IKKα, IKKβ, and a regulatory subunit, IKKγ. When activated by proinflammatory signals such as tumor necrosis factor α (TNFα) and lipopolysaccharide (LPS), IKKβ triggers the degradation of IκBα through phosphorylation, which in turn releases and mediates the nuclear translocation of nuclear factor κB (NF-κB). NF-κB then activates gene expression by binding to the target DNA sequence and thus contributing to diverse functions. Although first identified as the kinase for IκBα, IKKβ was subsequently shown to have other substrates as well. By identification of these non-IκBα downstream substrates, more cellular functions independent of IκBα have been found. For example, IKKβ is able to phosphorylate insulin receptor substrate 1 (IRS1) to suppress insulin signaling [11]. IKKβ also affects mitogen-activated protein kinase (MAPK) pathway by repressing DOK1 via phosphorylation-dependent manner and therefore increases cell migration [12; 13]. Additionally, IKKβ has been shown to promote breast cancer development through phosphorylation-mediated inhibition of two tumor suppressors, forkhead box O3a (FOXO3a) and tuberous sclerosis complex 1 (TSC1). IKKβ triggers the degradation of FOXO3a and TSC1, thereby exerting anti-apoptosis effects [14] and promoting angiogenesis [15]. All these findings suggest that IKKβ might have versatile roles in participating in physiological functions.
In the current study, we identified ARD1 as a substrate of IKKβ. IKKβ associated with and phosphorylated ARD1 at Ser209 in vitro and in vivo. Phosphorylation of ARD1 by IKKβ decreased its stability and led to the proteasome-mediated degradation of ARD1. IKKβ reduced the growth suppression effect of ARD1 through phosphorylation. We conclude that IKKβ down-regulates ARD1 through phosphorylation and destabilization.
Materials and methods
Constructs
The FLAG-IKKβ and FLAG-nIKKβ plasmids were generated as previously described [15]. The Myc-ARD1 and HA-ARD1 plasmids were constructed by inserting the cDNA of hARD1 into the pcDNA6 and pCMV5 vectors containing the Myc and HA tags, respectively. We constructed the GST-ARD1 plasmid by subcloning the ARD1 fragment into the pGEX6P-1 GST vector. All constructs of ARD1 mutants were generated as follows. The PCR reaction was performed in a total volume of 25 μl pfu reaction buffer containing 30 ng DNA, 0.2 mM dNTPs, 0.4 pmol of each primer, 1.5 μl DMSO and 1 μl pfu polymerase. Cycling conditions were 94°C (5 minutes) for one cycle; 94°C (1 minute), 55°C (1 minute), and 68°C (14 minutes) for 20 cycles; and a final extension of 68°C (10 minutes). 1 μl DpnI was then added and incubated at 37°C for 1.5 hours to remove the methylated DNA. The sequences were confirmed by DNA sequencing.
Experimental reagents
We used antibodies to FLAG (F3165, Sigma, St. Louis, MO), HA (11666606001, Roche, Switzerland), Myc (11667203001, Roche), ARD1 (15-288-22667, GenWay, San Diego, CA), acetyl lysine (05-515 and 06-933, Upstate, Billerica, MA), and α-tubulin (T-5168, Sigma). Cycloheximide and MG132 were purchased from Sigma.
Cell culture
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 medium supplemented with 10% fetal bovine serum (FBS). For transient transfection, cells at 50-60% confluence were transfected with DNA by SN liposome (DNA:SN=1μg:1μl) [16]. Six hours after transfection, the DNA-liposome mixture was removed and the fresh medium was added. Cells were harvested for analysis 2 days after transfection.
Immunoprecipitation and immunoblotting assays
Immunoprecipitation and immunoblotting assays were performed as described previously [14]. Briefly, radioimmunoprecipitation assay-B (RIPA-B) buffer (1% Triton X-100, 150 mM NaCl, 20 mM Na2PO4, 1 mM PMSF, 3 μg/ml aprotinin, 750 μg/ml benzamidine, 2 mM Na3VO4, 5 mM NaF, pH 7.4) was used as lysis and immunoprecipitation buffers. For immunoprecipitation, specific antibodies were incubated with cell lysates at 4°C for 16-18 hours. Protein A or Protein G was then added for another 3 hours. Immunoprecipitates were washed five times using RIPA-B buffer. Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) sampling buffer was added, and the samples were boiled for 10 minutes. Beads were spun down, and the supernatants were loaded onto an SDS-PAGE. For immunoblotting, proteins subjected to SDS-PAGE were transferred onto polyvinylidene fluoride (PVDF) membrane pretreated with methanol. Membranes were then blocked with 5% skim milk or 1% bovine serum albumin (BSA) in Tris-buffered saline (TBS) buffer (10 mM Tris, 150 mM NaCl, pH 7.9) with 0.05% Tween 20. The indicated proteins and phosphorylation levels were analyzed by using specific antibodies. Horseradish peroxidase (HRP)-conjugated secondary antibodies and Enhanced Chemiluminescence (ECL) kit were used for detection.
In vitro kinase assays
IKKβ kinase assays were performed as described previously [14; 17]. Briefly, FLAG-IKKβ or FLAG-nIKKβ transfected-cells were lysed and immunoprecipitated with anti-FLAG antibodies. IKKβ kinase activity was analyzed by in vitro kinase assay using purified GST-ARD1 as the substrate and GST and GST-IκBα were served as negative and positive controls, respectively. Reactions were performed at 30°C for 30 minutes in a final volume of 50 μl consisting of kinase buffer (20 mM Tris, 7.5 mM MgCl2, 30 μM ATP, 10 μCi γ-[32P]-ATP) and were stopped by adding 20 μl of SDS-PAGE sampling buffer and boiling for 10 minutes. Reaction products were loaded on an SDS-PAGE and analyzed by autoradiography.
Identification of phosphorylation sites by mass spectrometry analysis
Cell lysate from FLAG-IKKβ- and Myc-ARD1-cotransfected HEK293T cells was immunoprecipitated by anti-Myc antibodies. After separation on SDS-PAGE, protein bands corresponding to ARD1 were identified, excised from the gel, and then subjected to digestion by trypsin or other proteases. After isolation by immobilized metal affinity chromatography, the enriched phosphopeptides were analyzed by using mass spectrometry analysis.
Results and discussion
IKKβ physically interacts with ARD1
Since IKKβ functions as an oncoprotein and ARD1 might have a role in suppression of tumor progression, it is tempting to know whether IKKβ-mediated tumor development via regulating ARD1. To examine the physical association between IKKβ and ARD1, we first performed exogenous protein reciprocal coimmunoprecipitation assays. We cotransfected FLAG-IKKβ and HA-ARD1 into HEK293T cells, and found the presence of HA-ARD1 in FLAG-IKKβ immunoprecipitates (Fig. 1A). Consistently, FLAG-IKKβ was detected in HA-ARD1 immunoprecipitates (Fig. 1A). This interaction was also observed with endogenous IKKβ and ARD1 using specific antibodies to IKKβ and ARD1 (Fig. 1B). Together, these results demonstrated the association between ARD1 and IKKβ.
Fig. 1.
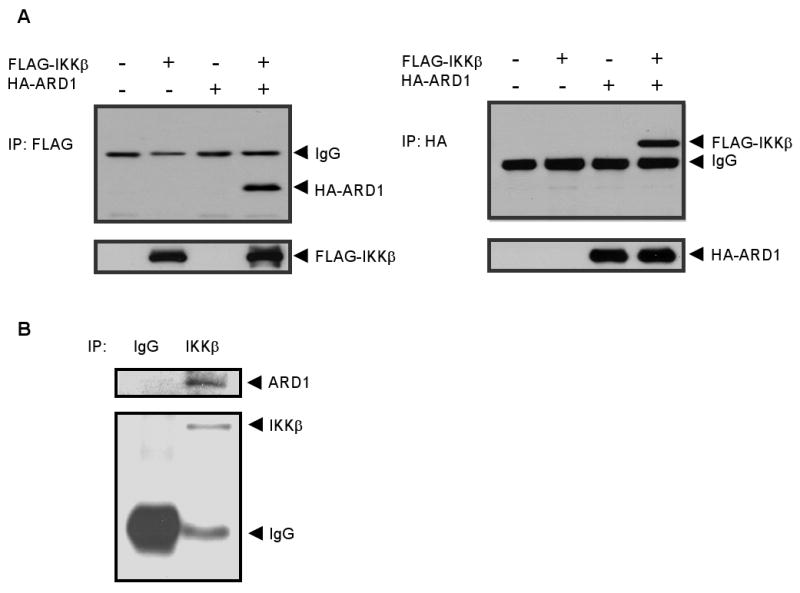
ARD1 physically interacted with IKKβ. (A) Exogenous interaction of ARD1 and IKKβ. HEK293T cells were cotransfected with HA-ARD1 and FLAG-IKKβ, and cell lysates were immunoprecipitated with anti-FLAG or anti-HA antibodies. The association between HA-ARD1 and FLAG-IKKβ was analyzed by reciprocal coimmunoprecipitation and immunoblotting assays. (B) Endogenous interaction of ARD1 and IKKβ in HEK293T cells. Cell lysates were immunoprecipitated with specific antibodies to ARD1 and IKKβ to identify the association of endogenous proteins.
IKKβ phosphorylates ARD1
Analysis of the amino acid sequence revealed an IKKβ consensus motif (DSψXXS/T) on ARD1 (Fig. 2A), which is present in many substrates of IKKβ, for example, IκBα, FOXO3a, and TSC1. This sequence (DSKDLS) is evolutionarily conserved from human to mouse (Fig. 2B), suggesting the importance of this region. The observation that ARD1 contains a potential IKKβ phosphorylation motif prompted us to investigate whether ARD1 is a substrate of IKKβ. As shown in Fig. 2C, in vitro kinase assay demonstrated that GST-ARD1 but not GST protein is efficiently phosphorylated by FLAG-IKKβ (lanes 1 and 2, left panel). In contrast, no phosphorylation of ARD1 was observed with kinase dead FLAG-nIKKβ control (lane 4, left panel). This result demonstrated that IKKβ phosphorylates ARD1 in vitro. Because ARD1 is an acetyltransferase, we next questioned whether ARD1 could induce ε-acetylation of IKKβ. HEK293T cells were cotransfected with ARD1 and IKKβ and treated with sodium butyrate to prevent protein deacetylation. The acetylated IKKβ was detected by anti-acetyl lysine antibodies. We were unable to detect any ε-acetylation of IKKβ (data not shown), even after using two different antibodies. In summary, our results suggest that ARD1 is a physiological substrate of IKKβ.
Fig. 2.

IKKβ phosphorylated ARD1 in vitro. (A) ARD1 contains a putative IKKβ phosphorylation motif (DSψXXS/T). D, aspartic acid; S, serine; ψ, hydrophobic amino acid; X, any amino acid. (B) IKKβ consensus motif (DSKDLS) on ARD1 is conserved from human to mouse. (C) ARD1 was phosphorylated by IKKβ. FLAG-IKKβ or FLAG-nIKKβ was transfected into HEK293T cells and immunoprecipitated for in vitro kinase assay using GST-ARD1 as the substrate. GST and GST-IκBα proteins were used as negative and positive controls, respectively.
IKKβ phosphorylation site is identified on ARD1
To further identify the IKKβ phosphorylation site on ARD1, we cotransfected Myc-ARD1 and FLAG-IKKβ plasmids into HEK293T cells and purified ARD1 protein for mass spectrometry analysis. The results demonstrated in vivo phosphorylation of ARD1 by IKKβ at Ser209 (Fig. 3A). To demonstrate the phosphorylation is dependent on IKKβ but not other IKKβ-regulated Ser/Thr kinases, we performed in vitro kinase assays using immunoprecipitated IKKβ or purified IKKβ. Substitution of Ala for Ser209 (S209A) abolished the phosphorylation of ARD1 by IKKβ immunocomplex (Fig. 3B) or purified IKKβ (Fig. 3C). Together, these results from kinase assays and mass spectrometry analysis indicate that IKKβ phosphorylates ARD1 at Ser209 in vitro and in vivo.
Fig. 3.

Identification of IKKβ phosphorylation site on ARD1. (A) HEK293T cells were transfected with FLAG-IKKβ and Myc-ARD1. After separation by SDS-PAGE electrophoresis, the band representing ARD1 protein was isolated and analyzed by mass spectrometry. (B) The IKKβ phosphorylation site on ARD1, Ser209, was identified by in vitro kinase assays. Immunoprecipitated FLAG-IKKβ was used in the kinase assay and kinase dead FLAG-nIKKβ was served as a negative control. (C) Purified IKKβ protein was incubated with GST-ARD1 (WT) and GST-ARD1 (S209A) in the kinase assay.
Phosphorylation by IKKβ decreases the stability of ARD1
We next generated ARD1 (S209E) mutant to mimic the phosphorylation by IKKβ and studied the mechanisms of ARD1 regulation by IKKβ. Since lower expression level of ARD1 (S209E) protein was observed, we first clarified whether the phosphorylation of ARD1 affects its stability. Treatment with cycloheximide to inhibit protein translation showed the decreased stability of ARD1 (S209E) (Fig. 4A) compared with that of wild-type (WT) ARD1 or ARD1 (S209A). The protein of ARD1 (S209E) was restored to a level similar to that of ARD1 (WT) or ARD1 (S209A) after MG132 treatment (Fig. 4B), suggesting that phosphorylation of ARD1 enhances its proteasome-mediated degradation. Together, these results demonstrated that phosphorylation of ARD1 by IKKβ contributes to its destabilization and degradation.
Fig. 4.

Phosphorylation by IKKβ destabilized ARD1 and reduced the growth suppression effect of ARD1. (A) Phosphorylation of ARD1 by IKKβ decreased its stability as determined by treatment with cycloheximide (100 μg/ml). The graph shows the relative intensity of various ARD1 proteins at different time points (standardized to 1 for the cycloheximide-pretreated [CHX 0 hours] sample). W, WT; A, S209A; E, S209E. (B) Treatment of MG132 restored the protein expression of ARD1 (S209E) mutant. HEK293T cells were transfected with various ARD1 constructs and treated with MG132 for 6 hours before analysis. W, WT; A, S209A; E, S209E. (C) Constructs of vector control, ARD1 WT and various ARD1 mutants were transfected into HEK293T cells. Cells (1×106) were plated 2 days after transfection and the number of cells was counted at different time points. V, vector control; W, WT; A, S209A; E, S209E. (D) A model by which phosphorylation of ARD1 by IKKβ induces ARD1 degradation and decreases its growth suppression effect.
Phosphorylation of ARD1 by IKKβ reduced its growth suppression effect
On the basis of our observation that IKKβ phosphorylates and destabilizes ARD1, we next asked whether phosphorylation by IKKβ affects the biological function of ARD1. We found ARD1 (WT)-transfected cells grow much slowly than vector control-transfected cells. In addition, expression of ARD1 non-phosphorylatable mutant (S209A) significantly inhibited cell growth of HEK293T cells as compared with ARD1 phosphorylation-mimic mutant (S209E) (Fig. 4C), suggesting phosphorylation by IKKβ decreases the growth suppression function of ARD1.
Whether ARD1's function in tumorigenesis is as an oncoprotein or a tumor suppressor has remained a matter of controversy in the literature [10; 18; 19]. It has been reported that protein post-translational modifications may change its function, for example, IKKα phosphorylation of CREB-binding protein (CBP) determines its associated partners and the oncoprotein/tumor suppressor role [20]. In the current studies, we identified IKKβ as a kinase of ARD1 that mediated its phosphorylation on Ser209, thereby resulting in the degradation of ARD1. Considering that IKKβ has been shown an association with oncogenic activity through negatively regulating its downstream substrates, for example, TSC1 and FOXO3a [14; 15], the destabilization of ARD1 by IKKβ seems to favor a tumor suppressor role for ARD1. Indeed, our results showed expression of ARD1 in HEK293T cells suppresses cell growth.
In summary, we identified an upstream kinase, IKKβ, which associated with and phosphorylated ARD1, resulting in its destabilization (Fig. 4D). The phosphorylation of ARD1 by IKKβ reduced the growth suppression effect of ARD1. Further investigation into the biological effect of this post-translational modification may advance our knowledge of IKKβ functions and may clarify the controversial role of ARD1 in the development of cancer.
Acknowledgments
We thank the Department of Scientific Publications at The University of Texas M. D. Anderson Cancer Center for editing this manuscript. This work was partially supported by National Institutes of Health (NIH) grants R01 CA109311, CCSG CA16672, and P01 CA099031, The University of Texas M. D. Anderson SPORE grants (P50 CA116199 for breast cancer, and P50 CA83639 for ovarian cancer), MDACC/CMUH Sister Institution Fund, Patel Memorial Breast Cancer Endowment Fund, and grants from the Kadoorie Charitable Foundations and the National Breast Cancer Foundation, Inc., to M.-C.H.; a grant from Taiwan National Science Council (NSC-96-3111-B) to L.-Y.L. and M.-C.H.; a predoctoral fellowship from the U.S. Army Breast Cancer Research Program (grant W81XWH-08-1-0397) and the Andrew Sowell-Wade Huggins Scholarship from The University of Texas Graduate School of Biomedical Sciences at Houston to H.-P.K.; a predoctoral fellowship from the U.S. Army Breast Cancer Research Program (grant W81XWH-05-1-0252) and the T.C. Hsu Endowed Memorial Scholarship, Andrew Sowell-Wade Huggins Scholarship and Presidents' Research Scholarship from The University of Texas Graduate School of Biomedical Sciences at Houston to D.-F.L.; and grants from National Health Research Institutes (NHRI-EX98-9603BC) and Department of Health (DOH98-TD-I-111-TN002) to L.-Y.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mullen JR, Kayne PS, Moerschell RP, Tsunasawa S, Gribskov M, Colavito-Shepanski M, Grunstein M, Sherman F, Sternglanz R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989;8:2067–2075. doi: 10.1002/j.1460-2075.1989.tb03615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whiteway M, Szostak JW. The ARD1 gene of yeast functions in the switch between the mitotic cell cycle and alternative developmental pathways. Cell. 1985;43:483–492. doi: 10.1016/0092-8674(85)90178-3. [DOI] [PubMed] [Google Scholar]
- 3.Whiteway M, Freedman R, Van Arsdell S, Szostak JW, Thorner J. The yeast ARD1 gene product is required for repression of cryptic mating-type information at the HML locus. Mol Cell Biol. 1987;7:3713–3722. doi: 10.1128/mcb.7.10.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnesen T, Anderson D, Baldersheim C, Lanotte M, Varhaug JE, Lillehaug JR. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem J. 2005;386:433–443. doi: 10.1042/BJ20041071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 6.Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR. Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett. 2005;579:6428–6432. doi: 10.1016/j.febslet.2005.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murray-Rust TA, Oldham NJ, Hewitson KS, Schofield CJ. Purified recombinant hARD1 does not catalyse acetylation of Lys532 of HIF-1alpha fragments in vitro. FEBS Lett. 2006;580:1911–1918. doi: 10.1016/j.febslet.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Bilton R, Mazure N, Trottier E, Hattab M, Dery MA, Richard DE, Pouyssegur J, Brahimi-Horn MC. Arrest-defective-1 protein, an acetyltransferase, does not alter stability of hypoxia-inducible factor (HIF)-1alpha and is not induced by hypoxia or HIF. J Biol Chem. 2005;280:31132–31140. doi: 10.1074/jbc.M504482200. [DOI] [PubMed] [Google Scholar]
- 9.Lim JH, Park JW, Chun YS. Human arrest defective 1 acetylates and activates beta-catenin, promoting lung cancer cell proliferation. Cancer Res. 2006;66:10677–10682. doi: 10.1158/0008-5472.CAN-06-3171. [DOI] [PubMed] [Google Scholar]
- 10.Yi CH, Sogah DK, Boyce M, Degterev A, Christofferson DE, Yuan J. A genome-wide RNAi screen reveals multiple regulators of caspase activation. J Cell Biol. 2007;179:619–626. doi: 10.1083/jcb.200708090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, Ye J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277:48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- 12.Lee S, Andrieu C, Saltel F, Destaing O, Auclair J, Pouchkine V, Michelon J, Salaun B, Kobayashi R, Jurdic P, Kieff ED, Sylla BS. IkappaB kinase beta phosphorylates Dok1 serines in response to TNF, IL-1, or gamma radiation. Proc Natl Acad Sci U S A. 2004;101:17416–17421. doi: 10.1073/pnas.0408061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Cristofano A, Niki M, Zhao M, Karnell FG, Clarkson B, Pear WS, Van Aelst L, Pandolfi PP. p62(dok), a negative regulator of Ras and mitogen-activated protein kinase (MAPK) activity, opposes leukemogenesis by p210(bcr-abl) J Exp Med. 2001;194:275–284. doi: 10.1084/jem.194.3.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 15.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, He X, Hung JY, Lai CC, Ding Q, Su JL, Yang JY, Sahin AA, Hortobagyi GN, Tsai FJ, Tsai CH, Hung MC. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 16.Zou Y, Peng H, Zhou B, Wen Y, Wang SC, Tsai EM, Hung MC. Systemic tumor suppression by the proapoptotic gene bik. Cancer Res. 2002;62:8–12. [PubMed] [Google Scholar]
- 17.Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, Li Y, Lin SY, Hung MC. beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell. 2002;2:323–334. doi: 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- 18.Arnesen T, Gromyko D, Pendino F, Ryningen A, Varhaug JE, Lillehaug JR. Induction of apoptosis in human cells by RNAi-mediated knockdown of hARD1 and NATH, components of the protein N-alpha-acetyltransferase complex. Oncogene. 2006;25:4350–4360. doi: 10.1038/sj.onc.1209469. [DOI] [PubMed] [Google Scholar]
- 19.Fisher TS, Etages SD, Hayes L, Crimin K, Li B. Analysis of ARD1 function in hypoxia response using retroviral RNA interference. J Biol Chem. 2005;280:17749–17757. doi: 10.1074/jbc.M412055200. [DOI] [PubMed] [Google Scholar]
- 20.Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26:75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]