

Abstract
The specificity of human glutathione transferase (GST) A1-1 is drastically altered to favor alkenal substrates in the GIMFhelix mutant designed to mimic first-sphere interactions utilized by GSTA4-4. This redesign serves as a model to further understand the structural determinants that contribute to the distinct specificities of alpha class GSTs. Herein we report the first crystal structures of GIMFhelix, both in complex with GSH and in apo form at 1.98 and 2.38 Ǻ resolution. In contrast to the pre-organized hydrophobic binding pocket that accommodates alkenals in GSTA4-4, GSTA1-1 includes a dynamic α9-helix that undergoes a ligand-dependent localization to complete the active site. Comparisons of the GIMFhelix structures with previously reported structures show a striking similarity with the GSTA4-4 active site obtained within an essentially GSTA1-1 scaffold and reveal the α9-helix assumes a similar localized structure regardless of active site occupancy in a manner resembling GSTA4-4. However, all the structural elements important in GSTA4-4 are not entirely accounted for within the mutant's active site. The contribution of Phe10 to the Tyr212-Phe10-Phe220 network prevents complete C-terminal closure and demonstrates that the presence of Phe10 within the context of a GSTA4-4-like active site may ultimately hinder Phe220, a key C-terminal residue, from effectively contributing to the active site. In total these results illustrate the remaining structural differences presumably reflected in the previously reported catalytic efficiencies of GIMFhelix and GSTA4-4 and emphasize the F10P mutation as necessary to completely accomplish the transformation to a highly specific GST from the more promiscuous GSTA1-1 enzyme.
The cytosolic glutathione transferases (GSTs)1 comprise a family of multifunctional enzymes that share similar overall topologies, dimer interactions, and glutathione- (GSH) binding sites, yet retain the capacity to detoxify a broad spectrum of electrophiles (1-3). All members are functionally dimeric proteins that contain one active site per subunit that consists of a GSH- and substrate-binding site, referred to as the G-site and H-site, respectively. The conserved G-site is located in the N-terminal domain while the H-site is located between the bundle of α–helices in the C-terminal domain, variations of which allow GSTs to accommodate structurally diverse electrophiles.
The alpha class GSTs, despite strong intra-class sequence similarity, are highly differentiated by their substrate specificities. While GSTA1-1 is regarded as a highly promiscuous enzyme with catalytic activity towards structurally diverse substrates, GSTA4-4 is recognized for its specificity toward alkenals such as 4-hydroxynonenal (HNE), an electrophile that has been described as one of the most toxic end products of oxidative stress (4-7). Both of these alpha class GSTs utilize three substrate recognition modules distributed throughout the primary structure to construct the H-site in the folded protein. The β1-α1 region, the end of the α4-helix, and the C-terminus all contribute to some extent to the H-site. However, the unique characteristics of the C-terminal α9-helix, a distinguishing feature of alpha class GSTs, have been the subject of numerous studies (5, 6, 8-20) and play a central role in the different specificities achieved within the constraints of a conserved scaffold (5, 6, 21, 22).
While the position of the C-terminus of GSTA1-1 is highly heterogeneous, and converts to a more ordered helix that completes the H-site in a ligand-dependent manner, the well-defined α9-helix of GSTA4-4 creates a pre-existing H-site pocket shaped to accommodate alkenals (5). Aromatic-aromatic interactions can contribute to protein stabilization (23) and the human GSTA4-4 (hGSTA4-4) enzyme contains an edge-to-face aromatic-aromatic interaction between Phe111 in the α4-helix and Tyr217 in the α9-helix that is absent within the H-site region of hGSTA1-1. This interaction was previously hypothesized to contribute to a closer association between the α4-α5 helix-turn-helix “tower” and C-terminal regions of hGSTA4-4 that helps to pre-organize the C-terminus for alkenal binding (6). Incorporation of this interaction into hGSTA1-1 resulted in a tower mutant with a less dynamic C-terminus and increased selectivity for HNE, as well as stereoselectivity of product formation more reminiscent of hGSTA4-4 (24). This result also demonstrated that the conformational heterogeneity is highly coupled throughout the structure of the promiscuous hGSTA1-1 enzyme (6).
Although the dynamics have yet to be explicitly characterized, a more extensive redesign of hGSTA1-1, made to mimic first-sphere interactions utilized by hGSTA4-4, was found to drastically alter the specificity of hGSTA1-1 from aromatic substitution reactions to Michael-like additions in the GIMFhelix mutant described by Nilsson et al. (22). While it is Tyr212 in the α9-helix that is proposed to interact with the aldehyde of alkenals, this area is occupied by the Cβ of Ala12 in hGSTA1-1 and the lack of a side chain at residue 12 is known to be required for high catalytic efficiency with alkenals (5, 22, 25). Residues lining the hydrophobic binding cavity in hGSTA4-4 were also selected for incorporation in the redesign of hGSTA1-1 (22). These include Ile107, Met108, and Phe111 in the α4-helix as well as Val216, Tyr217, and the aforementioned Tyr212. Since a number of the targeted residues comprise the C-terminus, which is known to be static and positioned to contribute the H-site in hGSTA4-4, the entire C-terminus (residues 208-222) was exchanged with that of hGSTA4-4. As a result, the final GIMFhelix mutant contains both residues necessary to potentially establish the tower region interaction, in addition to several other substitutions aimed to confer catalytic activity like that of hGSTA4-4. In fact the GIMFhelix mutant provides an excellent example of rational enzyme design and a model to further understand the structural and dynamic basis for the functional differences between hGSTA1-1 and hGSTA4-4.
This redesign of hGSTA1-1, with only 6% of the residues mutated to key residues present in hGSTA4-4, resulted in 20- and > 300-fold increases in catalytic efficiency with HNE and nonenal, respectively (21, 22). An 11-fold decrease in the catalytic efficiency was observed for the aromatic substitution reaction involving 1-chloro-2,4-dinitrobenzene (CDNB) favored by hGSTA1-1, indicating the changes were also correlated with a general shift in specificity toward Michael-like additions similar to hGSTA4-4 and illustrating the importance of first-sphere interactions for altered specificity.
Herein we describe the first crystal structures reported for GIMFhelix that offer a crystallographic look at the β1-α1 loop, α4-helix, and C-terminal region targeted to convey high alkenal activity to an essentially hGSTA1-1 protein scaffold. Comparisons of crystal structures also provide an opportunity to dissect interactions exploited by hGSTA1-1 and A4-4 that led to a unique situation inadvertently created by the Tyr212-Phe10-Phe220 network within the mutant's active site. Overall these structures help to elucidate the structural and conformational basis underlying the specificity and catalytic efficiency emerged in GIMFhelix.
Experimental Procedures
Protein Expression and Purification
The GIMFhelix mutant was constructed as described previously (22) and expressed in Escherichia coli followed by purification via GSH-agarose affinity chromatography (4). The protein was exchanged into 10 mM HEPES, pH 7.0 for crystallization screens and the subunit concentration was determined using the extinction coefficient, ε276 = 22,800 M-1 cm-1, for GIMFhelix (22).
Crystallization and X-ray Diffraction Data Collection
All crystallization screens were carried out using the sitting drop vapor-diffusion method at room temperature under conditions based on previously published protocols (5, 26, 27) as well as using the Hampton crystallization screen kits, HR2-110 and HR2-112, with protein concentrations of 10 mg/ml in 10 mM HEPES, pH 7.0. Data sets were collected for samples crystallized from: 2 μl apo GIMFhelix mixed with 2 μl reservoir solution containing 18% PEG 4000, 0.1 M Tris-Cl, pH 7.5; and 2 μl GIMFhelix with a 5-fold molar excess of GSH per subunit and 10 mM dithiothreitol mixed with 2 μl reservoir solution containing 18% PEG 4000, 0.1 M Tris-Cl, pH 7.5 and 10 mM dithiothreitol. Paraffin oil was used as a cryoprotectant and crystals and X-ray diffraction data were collected at − 180°C using a Rigaku RA-Micro 7 Desktop Rotating Anode Generator (λ = 1.5418 Ǻ) and an R-AXIS IV++ image-plate detector and processed using HKL2000 (28). Data collection and processing statistics are summarized in Table 1.
TABLE 1.
Data collection and processing statistics.
| GIMFheilx Structure | GSH Complex | Apo |
|---|---|---|
| Space Group | P21 | P21 |
| Unit-cell Dimensions (Ǻ,°) | 96.78, 114.67, 97.74 90.0, 117.9, 90.0 | 96.90, 114.68, 97.54 90.0, 117.8, 90.0 |
| Resolution Limit (Ǻ) | 1.98 | 2.30 |
| Reflections | 128626 | 83015 |
| Completeness (%) | 98.0 | 98.9 |
| Redundancy | 5.1 | 4.0 |
| Rmergea | 0.096 | 0.12 |
| <I> / <σ(I)> | 15.0 | 10.1 |
Rmerge = Σ|(I - <I>)| / ΣI
Molecular Replacement and Refinement
The crystal structure of GIMFhelix in complex with GSH was solved using the molecular replacement (MR) pipeline, BALBES (29), with a hGSTA1-1 structure (PDB code 1K3Y) selected as the search model. The MR solution was used to build a model for the GIMFhelix mutant that was refined with REFMAC5 (30) in the CCP4 program suite (31) using rigid body refinement followed by restrained refinement. The entire model was checked and adjusted in Xfit (32) after each cycle of refinement and the GSH ligand as well as water molecules were fit to the difference density. Over the course of the refinement, hydrogen atoms were added, weights were adjusted and Babinet scaling and TLS (Translation/Libration/Screw) refinement of the individual subunits were included when appropriate. The final GSH-bound GIMFhelix structure without the ligand and solvent was used to initiate the refinement of the apo GIMFhelix model. Omit maps were generated to aid in interpretation in the case of Phe10 and the structures were evaluated by Molprobity (33). Refinement and model statistics are summarized in Table 2. Coordinates for the apo and GSH-bound GIMFhelix models have been deposited in the Protein Data Bank, PDB identifiers 3I69 and 3I6A, respectively.
TABLE 2.
Model and refinement statistics.
| GIMFheilx Structure | GSH Complex | Apo |
|---|---|---|
| PDB code | 3I6A | 3I69 |
| Resolution Limit (Ǻ) | 1.98 | 2.38 |
| R valuea | 0.206 | 0.228 |
| Rfreeb | 0.244 | 0.289 |
| r.m.s.d. bonds (Ǻ) | 0.016 | 0.010 |
| r.m.s.d. angles (°) | 1.47 | 1.16 |
| No. Atoms: | ||
| Protein | 14269 | 14264 |
| Solvent | 553 | 239 |
| Other | 160 | 40 |
| Average B values (Ǻ2): | ||
| Protein | 37.7 | 57.6 |
| Solvent | 35.4 | 43.7 |
| Ligand | 44.0 | 75.7 |
| Model | A,C,D,E,G,H:2-220, B:2-222, F:4-220, 8GSH | A,C,D,E,G,H:2-220, B:2-222, F:4-219, 2GSH (B, D) |
R = Σ‖Fo| - |Fc‖ / Σ|Fo|
Rfree is R over the 5% of the data not included in model refinement.
Results
Overall Structure
The GSH-bound GIMFhelix mutant structure was solved at 1.98 Ǻ. The final model of the complex is composed of four dimers identified as chains AB, CD, EF, and GH in the asymmetric unit, with a GSH molecule bound to each of the corresponding eight G-sites. All eight subunits are located in unique crystal packing arrangements in the crystal with the dimers related through non-crystallographic pseudo two-fold rotation axes that give an average root mean square deviation (r.m.s.d.) value of 0.41 Ǻ for the Cα atoms between the pairs of subunits in the four dimers.
Each GIMFhelix subunit exhibits the canonical fold consisting of the N-terminal domain that exists as a mixed four-strand β–sheet situated between α–helices followed by the C-terminal domain composed entirely of α–helices (1). The Ramachandran plot identified nine outliers in the eight subunits in the asymmetric unit. Eight of them are functionally significant and belong to Gln67 in each of the subunits. Gln67 lies in the highly conserved ββα motif that recognizes the γ-glutamyl portion of GSH. Consistent with other known GST structures, this residue is positioned to participate in a hydrogen bond with the amino group of GSH and adopts these dihedral angles in order to permit the binding of a solvent molecule between the Gln67 amide nitrogen and the γ-glutamyl carboxylate group of GSH (34). The remaining outlier, Arg13 in subunit F, is only slightly outside the generally accepted limits and well-defined in electron density. A highly conserved proline residue is also observed at position 56 in the less favored cis conformation known to be important in the “cis-Pro loop” that helps to conserve the overall fold and is also part of the ββα motif mentioned above (3). Similar results were obtained for the apo GIMFhelix crystal structure solved at 2.38 Ǻ. It should be noted that weak difference density resembling GSH in two of the eight subunits (B and D) was observed in the apo structure and is likely the result of residual GSH used in the purification procedure. The statistics for data collection, processing, and refinement of both structures are summarized in Tables 1 and 2.
Subunit Comparisons
Structural superpositions of the different subunits in the asymmetric unit revealed two consistent differences. The first involves Gln53, a residue conserved between hGSTA1-1 and hGSTA4-4 that is hydrogen-bonded through a bridging water molecule to the carbonyl oxygen of residue 45 and also interacts with the carbonyl oxygen of residue 50 via its amide nitrogen (34). The side chain orientation of Gln53 in the AB and CD dimers alternates between the aforementioned position and one which points towards the G-site and allows contact with Arg131 across the dimer interface. However, the altered conformation of Gln53 did not disrupt GSH binding. Instead, the position was found to reflect dimer-dimer interactions between the amide nitrogen of the Gln53 side chain and the carbonyl of Lys141 from a separate dimer as a result of crystal packing.
The second difference encountered between the subunits relates to the orientation of Tyr166 of the α6-helix in the AB, CD, and EF dimers of the GSH-bound structure and the CD and EF dimers of the apo structure. Although this residue is not conserved in hGSTA4-4, which has an alanine at position 166, in hGSTA1-1 it has been observed to form a intra-domain hydrogen bond to Glu162 that contributes to the creation of a solvent cavity between the two domains of a single subunit (35). Inspection of the subunits shows that Tyr166 alternates between interacting with Glu162 and residing between Leu170 of the α6-helix and Ile106 of the α4-helix. The conformations of surrounding residues are not affected; however, the placement of Tyr166 between Leu170 and Ile106 is associated with a small but consistent increase in the distance between the α6- and α4-helices from 7.9 to 9.1 Ǻ, as measured between the corresponding Cα atoms of the backbone. The reason for the alternating positions is unclear and there are no confounding factors from crystal packing when dimer-dimer interactions are taken into consideration. However, given that Ile106 resides behind the H-site, just prior to three of the mutations (L107I, L108M and V111F) made in the GIMFhelix mutant, the small shift in this portion of the α4-helix towards the H-site may allow enough room for Tyr166 to occupy multiple positions, possibly giving rise to a more dynamic α6-helix than expected in this region.
Comparisons of GST Structures and Binding Sites
The GIMFhelix mutant shares 94% and 59% amino acid sequence identity with hGSTA1-1 and hGSTA4-4, respectively, which share 53% identity with each other and hence, only minimal deviations are observed for the superposition of all Cα atoms in the polypeptide backbone (0.59 and 0.87 Ǻ r.m.s.d with hGSTA1-1 and hGSTA4-4, respectively). In addition to the resemblance in overall topology, the G-site is also well-conserved among GSTs. The GSH molecule is bound in an extended conformation and participates in a variety of hydrogen bonds and salt links with the main and side chain functional groups lining the G-site as observed previously for other GSTs (1, 5, 26, 35). While some of the subsequently described GIMFhelix H-site features reflect the design of hGSTA4-4, the positions of the GSH sulfur and glycyl moiety within the G-site more closely resemble wild-type hGSTA1-1 (26). Although each enzyme has the Tyr9 hydroxyl group in proximity to the GSH sulfur, the distance is increased from 3.1 Ǻ in hGSTA1-1 (1PKW) and 3.3 Ǻ in GIMFhelix, to 5.2 Ǻ with an intervening water molecule that is discernible in the hGSTA4-4 structure (1GUL). The salt bridge formed with Arg45 also creates a noticeable difference in the position of the glycyl moiety when compared with Gln45, which allows for a greater extension of the tripeptide in the hGSTA4-4 G-site (5).
Despite all the G-site similarities with wild-type hGSTA1-1, the structural ramifications of the redesign of hGSTA1-1 were readily apparent upon inspection of the three hydrophobic substrate-binding modules targeted. The hGSTA4-4 crystal structure-based model as well as mutagenesis indicate that the simultaneous presence of Tyr212 in the α9-helix and Gly12 in the β1-α1 loop is critical for binding and activating alkenals (5, 22, 25). Both of the GIMFhelix crystal structures reported here illustrate the proper placement of Tyr212 when incorporated into the H-site with the simultaneous introduction of Gly12 to avert interference by a side chain at this position, thus confirming the importance of a sterically negligible residue here.
The remaining regions targeted in the construction of the GIMFhelix mutant reside in the α4- and α9-helices. Previous results from our lab have demonstrated one of the most critical interactions implicated in these regions is the aromatic-aromatic interaction between Phe111 and Tyr217 that increases core packing and stability in hGSTA4-4 (6). There is no corresponding interaction in hGSTA1-1; however, examination of the GIMFhelix crystal structures demonstrates the edge-to-face aromatic-aromatic interaction was indeed established between these two residues (Figure 1), with the Phe111-CD and Tyr217-CZ atoms separated by an average distance of 3.9 and 4.0 Ǻ, in the apo and GSH-bound structures, respectively. As expected, the C-terminal region is defined in electron density in the GSH-bound GIMFhelix crystal structure with the exception of the last two residues. Although the apo GIMFhelix structure reported here does have weak difference density corresponding to GSH in two of the eight subunits in the asymmetric unit, in contrast to other wild-type apo GSTA1-1 structures (34, 36) it also presents a C-terminus defined in electron density extending to residue 219-220 for all subunits.
FIGURE 1.
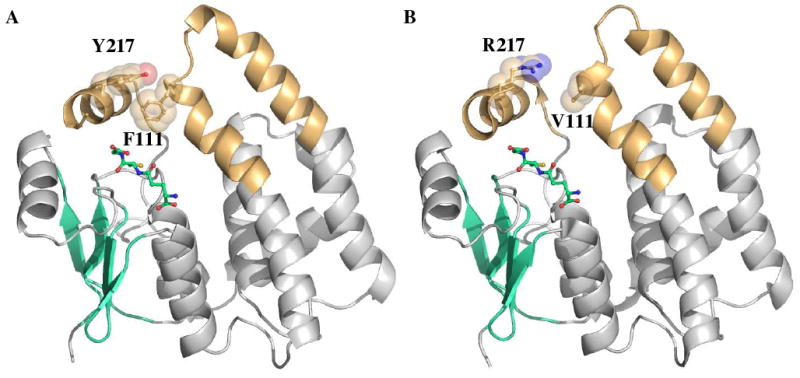
C-terminal domain interactions in the GIMFhelix mutant. Ribbon diagrams depicting the three-dimensional structures of GST subunits viewed perpendicular to the two-fold axis of symmetry for the dimer. The α9-helix and the tower region are highlighted in orange, β-strands are colored cyan, and GSH ligands are colored green. (A) Incorporation of the aromatic-aromatic interaction between Y217 in the α9-helix and F111 in the α4-turn-α5 tower region. (B) This interaction in not present in hGSTA1-1 (1PKW).
When viewed perpendicular to the two-fold axis, one easily recognizable difference in quaternary structure between hGSTA1-1 and hGSTA4-4 is the size of the crevice formed between the two subunits. From the backbone Cα atoms at the tip of the tower region, this crevice measures 16.7 and 26.1 Ǻ in the apo hGSTA1-1 (1PKZ) and apo hGSTA4-4 (1GUM) structures, respectively, while the apo GIMFhelix structure is intermediate with an average dimer crevice of 20.1 Ǻ (Figure 2). The increase in the size of this crevice presumably reflects the closer association between the α4-turn-α5 and the α9-helix regions, with the α9-helix anchored to the core by Tyr212.
FIGURE 2.
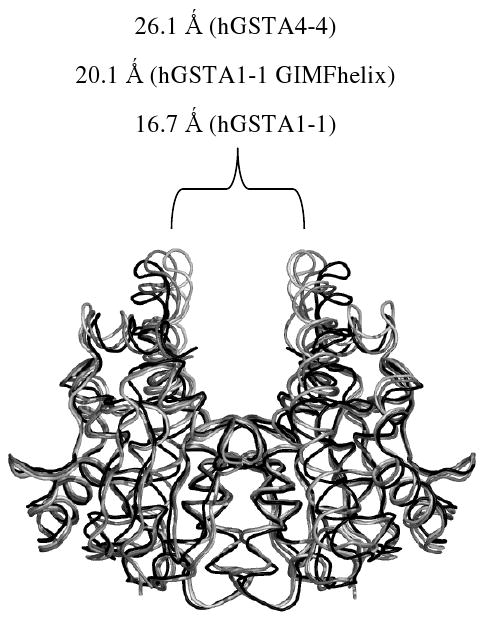
Comparison of A-class GST quaternary structure. The view is aligned perpendicular to the two-fold axis of symmetry for the dimer. The size of the crevice formed between the two subunits is shown for the apo hGSTA1-1 (light gray, 1PKZ), apo hGSTA1-1 GIMFhelix (gray) and apo hGSTA4-4 (black, 1GUM) structures.
Although the H-site of both GSTA1-1 and A4-4 are hydrophobic, the GIMFhelix mutations outlined above result in a putative H-site that is more compact with C-terminal characteristics more reminiscent of hGSTA4-4 even in the apo subunits. In addition to the hGSTA4-4-derived residues, Phe10 also appears to influence the orientation of residues in the targeted region. Previously described GSTA1-1 structures demonstrated that the side chain position of Phe10 differs between the apo and ligand-bound forms with Phe10 occupying the position adopted by Phe220 in the ligand-bound complex (26, 34, 36). However, position 10 was not included in the mutations chosen in the construction of GIMFhelix. As a consequence, Phe10 of GIMFhelix was generally found to encroach upon the position previously occupied by Phe220, regardless of whether GSH was bound, due to the presence of Tyr212 near the alternate Phe10 location (Figure 3). Interestingly, the electron density verified by omit maps, particularly in subunit C of the structures, suggests that Phe10 can still alternate and occupy a second conformation that allows Phe220 to shift back towards the active site, but only at the expense of displacing Try212 from its proper position with respect to that surmised for optimal binding and catalysis. This is likely to have important functional consequences as discussed below.
FIGURE 3.
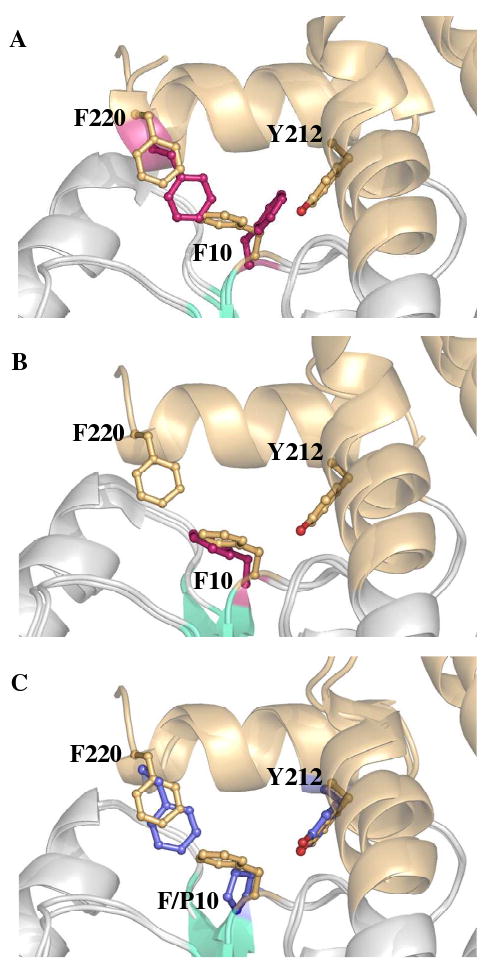
Position of residues 10 and 220 within alpha-class GST active sites. Structural superpositions contrasting the side chain of F10 and F220 in GSH-bound GIMFhelix with that of (A) ligand-bound hGSTA1-1 (1K3Y), (B) apo hGSTA1-1 (1K3O), which does not have an ordered C-terminus modeled in the crystal structure, and (C) P10 in hGSTA4-4 (1GUL). The ball and stick representation of residues from hGSTA1-1 and hGSTA4-4 are colored pink and blue, respectively. The ligands are not shown for clarity.
Comparison of the seven residues (Gly14, Ile107, Met108, Phe111, Tyr212, Val216 and Tyr217) lining the sides of this cavity in hGSTA4-4 and the GIMFhelix mutant also reveal an unanticipated location of Met108 in the α4-helix. Although Met108 lies along the side of the hydrophobic cavity in hGSTA4-4 and GIMFhelix, its side chain is oriented away from the H-site in both the apo (1GUM) and ligand-bound hGSTA4-4 structures (1GUL). In contrast, the Met108 side chain points into the putative H-site in the GIMFhelix structures reported herein. This residue appears to be displaced from its previous location in hGSTA4-4 by Lys120 remaining from the hGSTA1-1 sequence that extends towards the same space occupied by Met108 in hGSTA4-4 (Figure 4). Figure 4 also illustrates the relative position of the S-(2-iodobenzyl)-GSH (IBG) ligand from the hGSTA4-4 crystal structure (1GUL) within the context of the GIMFhelix active site. This superposition demonstrates that the tip of the benzyl ring would come as close as 1.2 Ǻ to the sulfur of Met108 in GIMFhelix.
FIGURE 4.
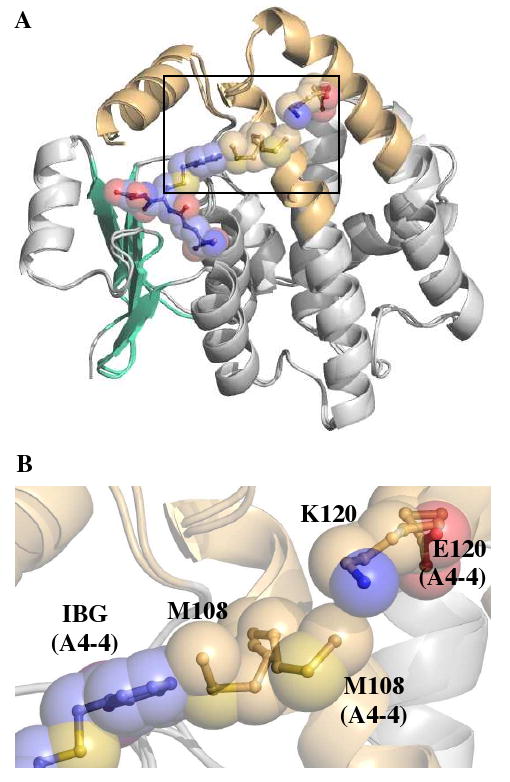
Side chain of M108. (A) Structural superposition of GSH-bound GIMFhelix and hGSTA4-4 (1GUL) contrasting the position of M108 and illustrating the relative position of the IBG ligand (blue) within the context of the GIMFhelix active site. Only the IBG ligand is shown for clarity. (B) Expansion of the denoted region in (A) showing the incompatibility of the GIMFhelix M108 conformation with the H-site ligand of hGSTA4-4.
Discussion
Prior characterizations of apo GSTA1-1 show a great deal of variation with respect to the dynamics and ordering of the α9-helix. In addition to apo hGSTA1-1 crystal structures such as 1K3O, which contains a highly disordered C-terminus that converts to a more ordered helix upon S-hexyl-GSH binding in 1K3Y (34), two of the apo structures reported by Grahn et al. (26) captured a range of conformations for the partial to complete α-helical C-terminus in some of the subunits. While it is not as closely packed to the rest of the protein as in the ligand-bound form, the degree of order was correlated with the solvation state of Tyr9 in the active site. In contrast to the heterogeneity observed for hGSTA1-1, the C-terminus of hGSTA4-4 exists as a well-defined α9-helix in both apo and ligand-bound forms that provides pre-existing ligand complementarity (5). Likewise, both the apo and GSH-bound GIMFhelix subunits reported herein present an α9-helix defined in electron density and indicate that the important elements in the GIMFhelix mutant are pre-organized in a manner similar to wild-type hGSTA4-4. The stability and orientation of the C-terminus presumably reflects the tighter packing between the tower region and the α9-helix predominately established through the Phe111-Tyr217 interaction, with the α9-helix fastened to the core by Tyr212 as detailed in the results section. Therefore, in addition to mimicking first-sphere interactions with a substrate in hGSTA4-4, these mutations likely contribute to a reduction in local dynamics that are highly coupled to other parts of the structure and can be correlated with reduced promiscuity, similar to that reported for the hGSTA1-1 tower mutant (6) and consistent with the change in substrate specificity already reported for the GIMFhelix mutant (21, 22).
Despite the successful redesign of GIMFhelix, this mutant does not quite achieve the catalytic efficiency of wild-type hGSTA4-4 with the lipid peroxidation product HNE (22). Although the GIMFhelix mutant has a three-fold lower KM with HNE, it also has a ten-fold lower kcat. It was initially suggested that the discrepancy with HNE could be related to an inability of the mutant to accommodate both configurations of the carbon 4 hydroxyl group (22). However, results from our laboratory indicate both enantiomers are indeed good substrates. Results from an extensive follow-up study also found the GIMFhelix mutant falls just short of maximal catalytic efficiency with nonenaland lacks complete functional identity with less active substrates as well (21). Collectively these results suggest that although Tyr212 is properly positioned to interact with the aldehyde while the H-site provides a hydrophobic groove that accommodates alkenals, some minor structural elements important in hGSTA4-4 may not be entirely accounted for in the GIMFhelix mutant active site.
Examination of the GIMFhelix crystal structures suggests that these discrepancies are the result of the hGSTA1-1-derived Phe10 participating in the hGSTA4-4-like H-site. Interestingly, Phe10 has formerly been correlated with the ionization of Tyr9 and the open and closed conformational forms of GSTA1-1 (10). As illustrated in Figure 3, the side chain of Phe10 alternates between apo and ligand-bound forms of previously described GSTA1-1 structures (26, 34, 36). In the apo form it is found to occupy the region where Phe220 resides in the ligand-bound complex, thereby sterically hindering C-terminal closure. Upon ligand binding Phe10 moves to accommodate complete C-terminal closure over the active site and allows Phe220, a key C-terminal residue conserved in both hGSTA1-1 and A4-4, to effectively interact with the Cys methylene of GSH as well as contribute to the H-site (8, 9, 14).
The identity and positioning of key residues in the α9-helix of hGSTA4-4 are known to confer specificity toward alkenals and take over the function of the N-terminal region that makes a larger contribution to the GSTA1-1 H-site (5). It was initially thought that the proper placement of Tyr212 in GIMFhelix would prevent the remaining Phe10 residue from contributing to the H-site (21). However, the GIMFhelix structures reveal the position of Tyr212 ultimately resulted in Phe220 being displaced further toward the periphery of the H-site by the intervening Phe10 residue in this network. Although the C-terminal α9-helix is discernible in electron density and relatively ordered in a position that contributes to the H-site, the “apo” position of Phe10 within the unique Tyr212-Phe10-Phe220 network in GIMFhelix is not entirely optimal for catalysis since it obstructs the complete C-terminal pre-organization of hGSTA4-4, which averts this with the incorporation of a proline at position 10.
Although there is no crystal structure reported with HNE bound, the model for hGSTA4-4 in complex with the inhibitor IBG suggests that the H-site will favor the placement of the alkyl chain tucked in a hydrophobic binding cavity that extends up past Phe111 in the α4-helix (5). Speculatively, additional binding modes, accessible depending on the location of the entire C-terminus, would prevent the GIMFhelix mutant from consistently binding and maintaining the substrates in the optimal position and could explain the remaining structural differences reflected in the catalytic efficiencies of GIMFhelix and hGSTA4-4.
An altered conformation of the Met108 side chain, mediated by an interaction involving Lys120 remote from the active site, was also observed and may alter or hinder binding in the H-site region. Human GSTA4-4 has a glutamic acid at residue 120, which not only has a shorter side chain, but can be drawn even farther away from the same area occupied by Lys120 in hGSTA1-1 through electrostatic interactions with charged residues present nearby only in hGSTA4-4. Although Met108 may simply be able to occupy alternate locations to accommodate H-site ligands, since the presence of Lys120 appears responsible for the Met108 side chain conformation in the mutant, the additional mutation of residue 120 would ensure that Met108 has room to occupy the conformation observed in hGSTA4-4 (1GUM and 1GUL).
In conclusion, we have described the first crystal structures of GIMFhelix, both with and without subunits bound to GSH. Previous studies have been based on functional characterizations of the recombinant enzyme, but the structural basis for the altered activities was merely inferred by comparison with the hGSTA4-4 model. The novel structures provide the proper correlates between structural changes and functional consequences. Analysis of the β1-α1 loop and the α4- and α9-helix regions targeted to install high alkenal activity into the hGSTA1-1 framework illustrate a striking similarity with the wild-type hGSTA4-4 active site obtained in spite of 94% sequence identity with wild-type hGSTA1-1. However, the structures also reveal a unique situation encountered by the hGSTA1-1-derived Phe10 residue within the context of an hGSTA4-4-like active site that indicates all the structural elements important in hGSTA4-4 are not entirely accounted for in the mutant's active site. To this end, the incorporation of a F10P mutation into the rational design that drastically altered specificity could ultimately complete the transformation to an essentially hGSTA1-1 protein scaffold that detoxifies alkenals with the same catalytic efficiency achieved by wild-type hGSTA4-4.
Acknowledgments
This work was supported by NIHGM 62284 (WMA)
Footnotes
Abbreviations: GST, glutathione transferase; GSH, glutathione; HNE, 4-hydroxynonenal; CDNB, 1-chloro-2,4-dinitrobenzene; MR, molecular replacement; TLS, Translation/Libration/Screw; IBG, S-(2-iodobenzyl)-GSH.
References
- 1.Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 2.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 3.Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Board PG. Identification of cDNAs encoding two human alpha class glutathione transferases (GSTA3 and GSTA4) and the heterologous expression of GSTA4-4. Biochem J. 1998;330:827–831. doi: 10.1042/bj3300827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, Tainer JA. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]
- 6.Hou L, Honaker MT, Shireman LM, Balogh LM, Roberts AG, Ng KC, Nath A, Atkins WM. Functional promiscuity correlates with conformational heterogeneity in A-class glutathione S-transferases. J Biol Chem. 2007;282:23264–23274. doi: 10.1074/jbc.M700868200. [DOI] [PubMed] [Google Scholar]
- 7.Hubatsch I, Ridderstrom M, Mannervik B. Human glutathione transferase A4-4: an alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem J. 1998;330:175–179. doi: 10.1042/bj3300175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gustafsson A, Etahadieh M, Jemth P, Mannervik B. The C-terminal region of human glutathione transferase A1-1 affects the rate of glutathione binding and the ionization of the active-site Tyr9. Biochemistry. 1999;38:16268–16275. doi: 10.1021/bi991482y. [DOI] [PubMed] [Google Scholar]
- 9.Ibarra C, Nieslanik BS, Atkins WM. Contribution of aromatic-aromatic interactions to the anomalous pK(a) of tyrosine-9 and the C-terminal dynamics of glutathione S-transferase A1-1. Biochemistry. 2001;40:10614–10624. doi: 10.1021/bi010672h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibarra CA, Chowdhury P, Petrich JW, Atkins WM. The anomalous pKa of Tyr-9 in glutathione S-transferase A1-1 catalyzes product release. J Biol Chem. 2003;278:19257–19265. doi: 10.1074/jbc.M301566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mosebi S, Sayed Y, Burke J, Dirr HW. Residue 219 impacts on the dynamics of the C-terminal region in glutathione transferase A1-1: implications for stability and catalytic and ligandin functions. Biochemistry. 2003;42:15326–15332. doi: 10.1021/bi035671z. [DOI] [PubMed] [Google Scholar]
- 12.Nieslanik BS, Dabrowski MJ, Lyon RP, Atkins WM. Stopped-flow kinetic analysis of the ligand-induced coil-helix transition in glutathione S-transferase A1-1: evidence for a persistent denatured state. Biochemistry. 1999;38:6971–6980. doi: 10.1021/bi9829130. [DOI] [PubMed] [Google Scholar]
- 13.Nieslanik BS, Ibarra C, Atkins WM. The C-terminus of glutathione S-transferase A1-1 is required for entropically-driven ligand binding. Biochemistry. 2001;40:3536–3543. doi: 10.1021/bi001869x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilsson LO, Edalat M, Pettersson PL, Mannervik B. Aromatic residues in the C-terminal region of glutathione transferase A1-1 influence rate-determining steps in the catalytic mechanism. Biochim Biophys Acta. 2002;1598:199–205. doi: 10.1016/s0167-4838(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 15.Zhan Y, Rule GS. Glutathione induces helical formation in the carboxy terminus of human glutathione transferase A1-1. Biochemistry. 2004;43:7244–7254. doi: 10.1021/bi0363329. [DOI] [PubMed] [Google Scholar]
- 16.Adman ET, Le Trong I, Stenkamp RE, Nieslanik BS, Dietze EC, Tai G, Ibarra C, Atkins WM. Localization of the C-terminus of rat glutathione S-transferase A1-1: crystal structure of mutants W21F and W21F/F220Y. Proteins. 2001;42:192–200. doi: 10.1002/1097-0134(20010201)42:2<192::aid-prot60>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Dirr HW, Little T, Kuhnert DC, Sayed Y. A conserved N-capping motif contributes significantly to the stabilization and dynamics of the C-terminal region of class Alpha glutathione S-transferases. J Biol Chem. 2005;280:19480–19487. doi: 10.1074/jbc.M413608200. [DOI] [PubMed] [Google Scholar]
- 18.Dirr HW, Wallace LA. Role of the C-terminal helix 9 in the stability and ligandin function of class alpha glutathione transferase A1-1. Biochemistry. 1999;38:15631–15640. doi: 10.1021/bi991179x. [DOI] [PubMed] [Google Scholar]
- 19.Kuhnert DC, Sayed Y, Mosebi S, Sayed M, Sewell T, Dirr HW. Tertiary interactions stabilise the C-terminal region of human glutathione transferase A1-1: a crystallographic and calorimetric study. J Mol Biol. 2005;349:825–838. doi: 10.1016/j.jmb.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 20.Nieslanik BS, Atkins WM. The catalytic Tyr-9 of glutathione S-transferase A1-1 controls the dynamics of the C terminus. J Biol Chem. 2000;275:17447–17451. doi: 10.1074/jbc.M002083200. [DOI] [PubMed] [Google Scholar]
- 21.Blikstad C, Shokeer A, Kurtovic S, Mannervik B. Emergence of a novel highly specific and catalytically efficient enzyme from a naturally promiscuous glutathione transferase. Biochim Biophys Acta. 2008;1780:1458–1463. doi: 10.1016/j.bbagen.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Nilsson LO, Gustafsson A, Mannervik B. Redesign of substrate-selectivity determining modules of glutathione transferase A1-1 installs high catalytic efficiency with toxic alkenal products of lipid peroxidation. Proc Natl Acad Sci U S A. 2000;97:9408–9412. doi: 10.1073/pnas.150084897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burley SK, Petsko GA. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 24.Balogh LM, Roberts AG, Shireman LM, Greene RJ, Atkins WM. The stereochemical course of 4-hydroxy-2-nonenal metabolism by glutathione S-transferases. J Biol Chem. 2008;283:16702–16710. doi: 10.1074/jbc.M801725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bjornestedt R, Tardioli S, Mannervik B. The high activity of rat glutathione transferase 8-8 with alkene substrates is dependent on a glycine residue in the active site. J Biol Chem. 1995;270:29705–29709. doi: 10.1074/jbc.270.50.29705. [DOI] [PubMed] [Google Scholar]
- 26.Grahn E, Novotny M, Jakobsson E, Gustafsson A, Grehn L, Olin B, Madsen D, Wahlberg M, Mannervik B, Kleywegt GJ. New crystal structures of human glutathione transferase A1-1 shed light on glutathione binding and the conformation of the C-terminal helix. Acta Crystallogr D Biol Crystallogr. 2006;62:197–207. doi: 10.1107/S0907444905039296. [DOI] [PubMed] [Google Scholar]
- 27.Xiao B, Singh SP, Nanduri B, Awasthi YC, Zimniak P, Ji X. Crystal structure of a murine glutathione S-transferase in complex with a glutathione conjugate of 4-hydroxynon-2-enal in one subunit and glutathione in the other: evidence of signaling across the dimer interface. Biochemistry. 1999;38:11887–11894. doi: 10.1021/bi990468i. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.McRee DE. A Visual Protein Crystallographic Software System for X11/Xview. J Mol Graphics. 1992;10:44–46. [Google Scholar]
- 33.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Trong I, Stenkamp RE, Ibarra C, Atkins WM, Adman ET. 1.3-A resolution structure of human glutathione S-transferase with S-hexyl glutathione bound reveals possible extended ligandin binding site. Proteins. 2002;48:618–627. doi: 10.1002/prot.10162. [DOI] [PubMed] [Google Scholar]
- 35.Sinning I, Kleywegt GJ, Cowan SW, Reinemer P, Dirr HW, Huber R, Gilliland GL, Armstrong RN, Ji X, Board PG, et al. Structure determination and refinement of human alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J Mol Biol. 1993;232:192–212. doi: 10.1006/jmbi.1993.1376. [DOI] [PubMed] [Google Scholar]
- 36.Cameron AD, Sinning I, L'Hermite G, Olin B, Board PG, Mannervik B, Jones TA. Structural analysis of human alpha-class glutathione transferase A1-1 in the apo-form and in complexes with ethacrynic acid and its glutathione conjugate. Structure. 1995;3:717–727. doi: 10.1016/s0969-2126(01)00206-4. [DOI] [PubMed] [Google Scholar]