

Abstract
Alveolar rhabdomyosarcoma (ARMS) is remarkably rare in adults over 45 years. Initial immunoprofiling of a small cell neoplasm of the head and neck region in an older adult may not include myogenic markers. A valuable diagnostic aid and important prognostic parameter in ARMS is the identification of PAX3-FOXO1 [t(2;13)(q35;q14)] or PAX7-FOXO1 [t(1;13)(p36;q14)] rearrangements. The purpose of this study was to document the clinicopathologic, immunophenotypic, and genetic features of head/neck ARMS in older adults. Prior isolated descriptions of three patients were included. Five patients were female and two male (median age 61 years). Each neoplasm was composed of undifferentiated, small round cells in a predominantly solid pattern. Initially ordered immunostains corresponded with early diagnostic impressions of a hematologic malignancy or neuroendocrine carcinoma. CD56 was positive in 5/5 tumors and synaptophysin in 1/6. Given the virtual absence of other lymphoid or epithelial markers, muscle immunostains were performed and these were positive. Definitive ARMS diagnoses were confirmed genetically. This study illustrates the diagnosis of head/neck ARMS in older adults is complicated by its rarity, lack of an alveolar pattern, and a potentially misleading immunoprofile (CD56 and synaptophysin immunoreactivity) if myogenic markers are not employed. Both PAX3- and PAX7-FOXO1 ARMSs were identified in these patients. In children, PAX7-FOXO1 ARMS is associated with a significantly longer event-free survival. In contrast, adult ARMS behaves more aggressively with a worse overall survival than pediatric ARMS. Further follow-up and additional cases are required to assess the prognostic relevance of these fusion transcripts in the context of advanced age.
Keywords: alveolar rhabdomyosarcoma, cytogenetics, FOXO1, PAX3, PAX7, RT-PCR
Introduction
Rhabdomyosarcoma (RMS) is a morphologically and clinically heterogeneous family of malignant soft tissue tumors related to myogenic lineage (1,2). Alveolar rhabdomyosarcoma (ARMS) and embryonal rhabdomyosarcoma (ERMS) represent the two main histologic patterns and must be differentiated from other small round cell tumors. RMS is the most common soft tissue sarcoma in the pediatric population, comprising approximately 5% of all childhood cancers and nearly 50% of soft tissue sarcomas arising in children 0–14 years of age (3,4). In contrast, RMS is remarkably uncommon in older adults representing merely 2–5% of all malignant soft tissue tumors, with the majority of the pleomorphic subtype (5). Hence, due to the rarity of ARMS in the older adult and the relatively recent inclusion of molecular approaches in the diagnostic assessment of these neoplasms, very few cases of the head and neck region have been previously analyzed genetically (6–8).
In this study, the diagnosis of four ARMSs arising in the head and neck region of patients between 61 and 76 years of age was confirmed by the demonstration of PAX-FOXO1 fusion gene expression. Identification of the characteristic gene fusions in these tumors allowed for a rapid, definitive diagnosis and commencement of proper therapy. The clinicopathologic and genetic findings of these cases and those of previous isolated reports are reviewed (6–8).
Materials and Methods
Clinical Cases
A summary of the patient characteristics is presented in Table 1.
TABLE 1.
Adult Head and Neck ARMS Patient Characteristics
| Case No. | Age/Sex | Location | Size (cm) |
Metastases at Diagnosis | Treatment | Follow-up (mths) |
References |
|---|---|---|---|---|---|---|---|
| 1 | 76/F | Ethmoid sinus | 3 | Internal auditory canal | ChemoRT | 14 (DOD) |
Current study |
| 2 | 61/M | Nasopharynx | NA | Lymph nodes, bone, lung | ChemoRT | 10* (AWD) |
Current study |
| 3 | 61/F | Palate | 7.2 | Lymph node | ChemoRT | 13 (NED) |
Current study |
| 4 | 64/F | Maxillary sinus | 5 | None | ChemoRT | 12 (AWD) |
Current study |
| 5 | 68/F | Cervical paravertebra | 10 | Widespread: bone marrow, thoracic wall, stomach, pancreas | Chemo | 11 days (DOD) |
Stindl R, et al.8 |
| 6 | 57/F | Ethmoid sinus | NA | NA | NA | NA | Manucha V, et al.7 |
| 7 | 49/M | Nasal sinus | NA | None | Resection, chemo | 60 (AWD) |
Das KM, et al.6 |
ChemoRT, chemotherapy and radiation therapy; NA, not available; DOD, died of disease; AWD, alive with disease; NED, no evidence of disease
Patient lost to further follow-up after 10 months.
Case 1
A 76-year-old female presented with new onset of left ptosis, dysarthria, and headache. Radiographic studies revealed left ethmoid and sphenoid sinus opacification with contrast enhancement consistent with chronic sinus disease but concerning for neoplasm. Histopathologic evaluation of the surgically excised specimen revealed an infiltrative lesion composed of sheets of small round cells (Fig. 1A). Focally, fibrous tissue bands separated some cell aggregates (Fig. 1B). Examination of individual cells was limited by variable crush artifact and necrosis. The initial clinicohistopathologic impression was malignant undifferentiated tumor; a small cell carcinoma was favored. Immunohistochemistry was performed employing the following antibodies: epithelial membrane antigen (EMA): diverse keratin antibodies MAK6, AE1/AE3, CK7, CK20, CAM5.2: CD56: chromogranin: synaptophysin: glial fibrillary acidic protein (GFAP): S-100 protein: melan-A: CD45: Epstein-Barr viral LMP and myeloperoxidase. The tumor cells were positive solely for CD56 (Fig. 1C). Additional immunohistochemical studies demonstrated tumor immunoreactivity for vimentin, desmin, and myogenin (Fig. 1D). At the time of biopsy, a portion of the specimen was submitted for cytogenetic analysis. Fluorescence in situ hybridization (FISH) analysis was performed for assessment of the FOXO1 gene locus and reverse transcriptase-polymerase chain reaction (RT-PCR) studies for PAX-FOXO1 fusion gene expression following recognition of muscle marker immunoreactivity. These results provided a definitive ARMS diagnosis and the patient received multiagent chemotherapy and radiotherapy accordingly with significant shrinkage of the neoplasm. Fourteen months from initial consultation, however, the patient died of complications with disease.
Figure 1.
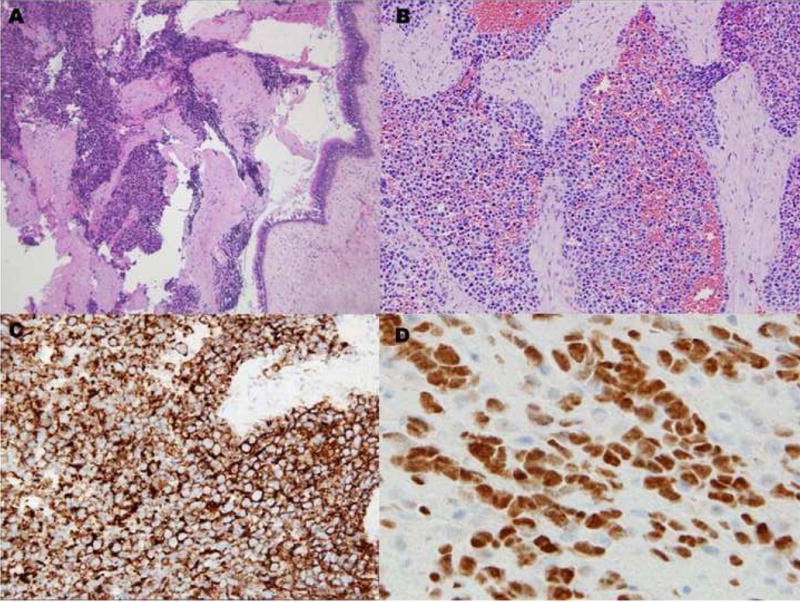
Case 1. A, Solid variant pattern of ARMS demonstrating focally crushed and fragmented tumor along with preserved sinonasal mucosa. B, Higher magnification illustrating a focus of fibrous septa separating nests of cells with relatively uniform round nuclei and scant cytoplasms. C, CD56 immunostaining. D, myogenin immunostaining.
Case 2
A 61-year-old male was found to have a large mass involving the paranasal sinuses with extension into the nasopharynx and adjacent orbit. The patient presented with metastastic disease involving level 1 and 2 cervical lymph nodes, the right ilium, the left lower lung lobe and multifocal spinal lesions resulting in a compression fracture at T11. A biopsy revealed sheets and nests of small undifferentiated cells admixed with slightly larger cells exhibiting relatively abundant clear cytoplasm (Fig. 2A). Immunohistochemically, the neoplastic cells were diffusely positive for MIB-1 and a cytokeratin cocktail (AE1/AE3, CAM5.2 and MAK6) (Fig. 2B), and focally positive for MAK6, CD56, synaptophysin (Fig. 2C), and S-100 protein. The neoplastic cells were negative for GFAP, neuron specific enolase (NSE), high molecular weight keratin (HMWCK), chromogranin, CD45, EMA, and thyroid transcription factor-1 (TTF-1). The initial diagnostic impression included small cell carcinoma and esthesioneuroblastoma. Following consultation, additional stains were pursued to exclude the following diagnoses: melanoma, Ewing’s sarcoma (pPNET), or an unusual myoepithelial tumor. The neoplastic cells demonstrated immunoreactivity for CD57 (Leu 7), cytokeratin cocktail, desmin, calponin, and myogenin. Smooth muscle actin (SMA), melan-A, CD45, and O13 (MIC2) immunohistochemical stains were negative. Based on these subsequent immunohistochemical findings, a diagnosis of RMS was favored and tissue was submitted for molecular (RT-PCR) analysis. Following genetic confirmation, the patient was treated with multiagent chemotherapy and radiotherapy to the primary tumor and right neck. Ten months after initial diagnosis the patient was alive with stable disease, but was then lost to subsequent follow-up.
Figure 2.
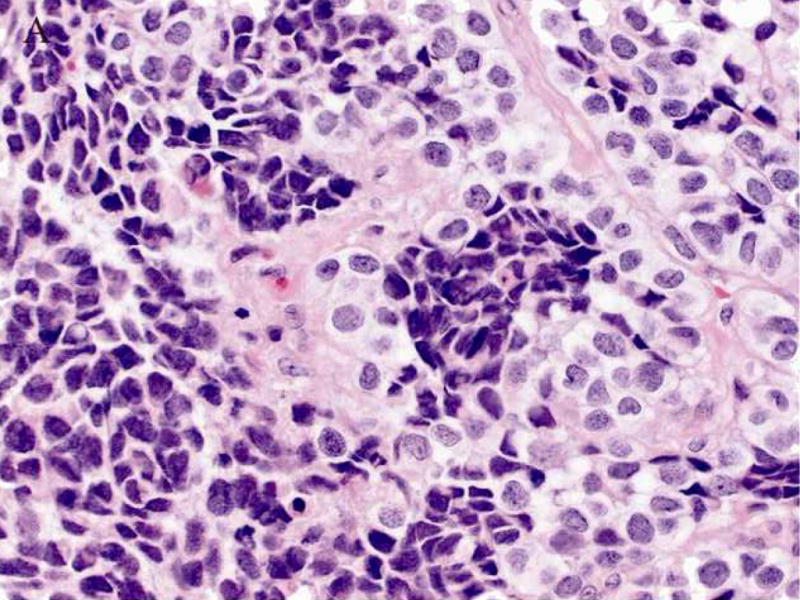
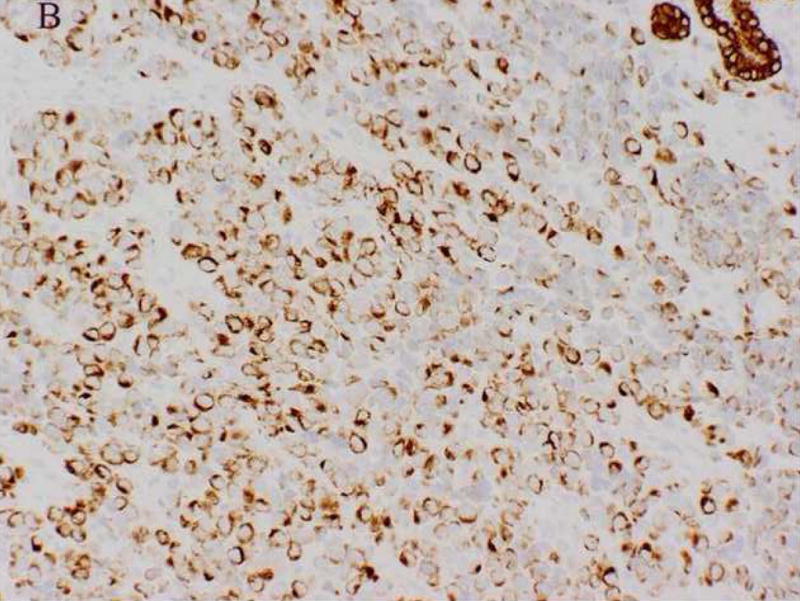
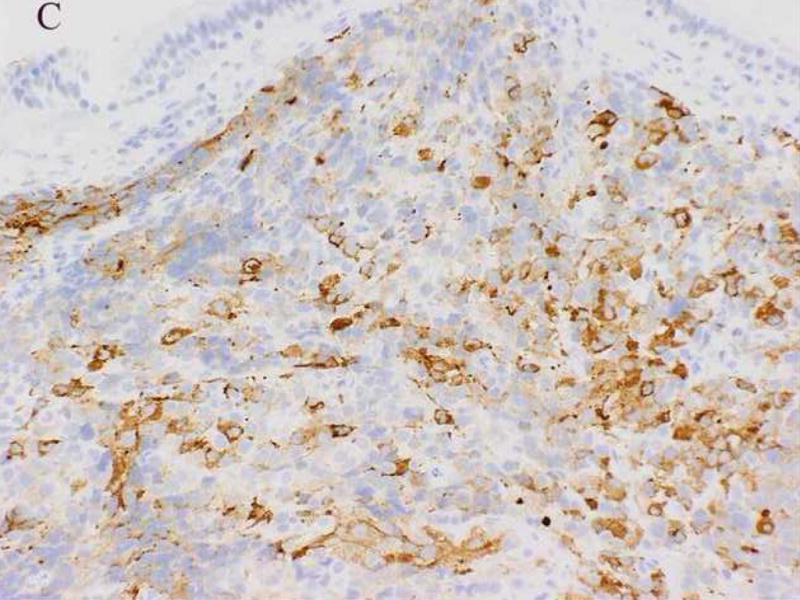
Case 2. A. High-power view illustrating cellular nests composed of round to polygonal cells with hyperchromatic nuclei and scant cytoplasm and slightly larger cells with abundant clear vacuolated cytoplasm. B, cytokeratin cocktail (AE1/AE3, CAM5.2 and MAK6) immunostaining. C, synaptophysin immunostaining.
Case 3
A 61-year-old woman noted irritability of her right upper denture palate for approximately one month. Her symptoms progressed to severe pain and swelling of the right side of her face. Clinical examination revealed an obvious subcutaneous mass involving the right cheek with infiltration of the right upper alveolar ridge. A CT scan demonstrated a soft tissue mass involving the subcutaneous fat overlying the bones of the right maxilla with invasion of the hard palate and alveolar ridge in the pterygomaxillary space. Multiple enlarged lymph nodes suspicious for metastatic disease could also be appreciated. A biopsy of the lesion revealed cellular groups of small round cells with scant cytoplasm and the initial clinicohistopathologic impression was high grade neuroendocrine carcinoma. Immunohistochemically, the neoplastic cells were diffusely positive for CD56 and vimentin. The neoplastic cells were negative for NSE, synaptophysin, and chromogranin. Additional immunohistochemical studies revealed that the tumor cells expressed muscle-specific actin and myogenin, but failed to express either desmin or myoglobin. Based on the histological features and the immunostains a diagnosis of RMS was favored. Following molecular (RT-PCR) confirmation, the patient received multiagent chemotherapy and radiotherapy resulting in significant shrinkage of the neoplasm. She is alive with no evidence of disease 13 months from initial consultation.
Case 4
A 64-year-old female complained of sinus pain. Radiographic studies revealed complete opacification of the right maxillary sinus. A right maxillary sinus biopsy revealed a dense population of neoplastic cells with small round, regular nuclei and little cytoplasm infiltrating nasal mucous membrane. Focal tumor necrosis and numerous mitoses could be appreciated. Initial immunohistochemical stains demonstrated that the neoplastic cells were positive for CD56 but negative for CD2, CD3, CD5, CD8, CD10, CD20, LCA, pancytokeratin, chromogranin, synaptophysin and granzyme B. Due to the virtual absence of all lymphoid markers with the exception of CD56 and lack of apparent epithelial cell groups, stains for muscle markers were performed and revealed immunoreactivity for desmin and myogenin consistent with a diagnosis of RMS. Tissue was subsequently submitted for molecular studies. Following molecular confirmation, the patient received multiagent chemotherapy and radiotherapy and is alive with disease 12 months from initial consultation.
Cytogenetic Analysis
Standard culture and harvesting procedures were performed on a representative portion of the neoplasm from Case 1, as described previously (9). Metaphase cells were banded with Giemsa Wright stain, and the karyotypes were expressed according to the International System for Human Cytogenetic Nomenclature 2005 (10).
Fluorescence in Situ Hybridization (FISH)
FISH was performed on an unstained, paraffin-embedded tissue section of Case 1 using the Vysis LSI ® FKHR (13q14) Dual Color, Break Apart Rearrangement Probe (Abbott Molecular Inc., Des Plaines, IL) according to the manufacturer’s instructions. Hybridization signals were assessed in 200 interphase nuclei with strong, well-delineated signals and distinct nuclear borders by two different individuals as previously described (9).
RNA Isolation and Reverse Transcriptase – Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from representative, formalin-fixed, paraffin-embedded tissue of each case with Trizol reagent (Gibco BRL, Gaithersburg, MD, USA). For one-step RT-PCR analysis, 0.5 μg of total RNA (2.5μL of 0.2 μg/μL solution) was reverse transcribed and amplified using 5 units of Superscript II RT/TAQ (Invitrogen, Carlsbad, CA) and the following primers: 5′CCG ACA GCA GCT CTG CCT AC3′ and 5′ATG AAC TTG CTG TGT AGG GAC AG3’′. After reverse transcription at 50°C for 30 min, reactions were denatured at 94°C for 2 min. Subsequently, 35 thermal cycles were used at the following temperatures: denaturation at 94°C for 30 sec; annealing at 65°C for 1 min; and extension at 72°C for 1 min, with a final extension at 72°C for 10 min. The amplified fragments were identified by gel electrophoresis and ethidium bromide staining. The integrity of the mRNA was assessed by an independent RT-PCR reaction using primers to the ubiquitously expressed hypoxanthine phosphoribosyltransferase gene. The positive control was a known PAX3-FOXO1 fusion transcript positive ARMS cell line. The negative control was devoid of template. Confirmation of the presence of a PAX3-FOXO1 versus a PAX7-FOXO1 fusion transcript was performed by subsequent restriction enzyme digestion (Sau96I and HhaI respectively).
Results
Cytogenetic Analysis
Case 1 exhibited the following abnormal chromosomal complement: 82–104, XXXX, t(2;13)(q35;q14)×2,+der(2)t(2;13),+3,+7,+7,+12,+12,+4–9mar[cp2], Fig. 3.
Figure 3.

Partial karyotypes of two GW-banded metaphase cells from Case 1 illustrating multiple copies of the t(2;13)(q35;q14) characteristic of ARMS.
FISH
FISH analysis of Case 1 revealed a rearrangement of the FOXO1 gene locus (13q14) in 90% of the interphase cells analyzed. Specifically, 2–5 copies of a rearranged FOXO1 gene locus (split probe signals) with one fused probe signal set (non-rearranged FOXO1 gene locus) were identified, Fig. 4.
Figure 4.

FISH analysis of Case 1 with the Vysis LSI ® FKHR (13q14) Dual Color, Break Apart Rearrangement Probe revealed multiple split red and green signals indicative of disruption of the FOXO1 gene locus. A single fused red and green signal was also identified in the majority of aberrant interphase cells examined consistent with the presence of a nonrearranged FOXO1 gene locus.
RT-PCR
Three cases (Cases 1, 3, and 4) were positive for a PAX3-FOXO1 fusion transcript and one case (Case 2) was positive for a PAX7-FOXO1 fusion transcript (Fig. 5). A summary of the molecular data is presented in Table 2.
Figure 5.

Ethidium-stained gels of the RT-PCR results (a separate gel was run for each patient at the time of diagnostic assay). Total RNA was extracted from formalin-fixed, paraffin-embedded tumor tissue and subjected to RT-PCR using a consensus PAX3/PAX7 primer and FOXO1 primer. M, DNA molecular weight marker (100 bp ladder); Lane 1 in each case represents the respective patient sample (PAX3-FOXO1 fusion product is 172 bp and PAX7-FOXO1 fusion product is 160 bp); Lane 2, PAX3-FOXO1 positive control; and Lane 3, no template negative control.
TABLE 2.
Pathologic and Molecular Data
| Case | Tissue Analyzed |
Initial DDx | Pattern | Immunohistochemical Findings | Karyotype | FISH | RT-PCR | |
|---|---|---|---|---|---|---|---|---|
| CD56 | Synaptophysin |
FOXO1 locus rearrangement |
||||||
| 1 | P | High grade neuroendocrine tumor | Solid | + | − | 82–104, XXXX, t(2;13) (q35;q14)x2,+der(2) t(2;13)(q35;q14),+3,+7 +7,+12,+12,+4–9mar | + | PAX3-FOXO1 |
| 2 | P | Small cell carcinoma, esthesioneuroblastoma | Solid | + | + | NP | NP | PAX7-FOXO1 |
| 3 | P | High grade neuroendocrine tumor | Solid | + | − | NP | NP | PAX3-FOXO1 |
| 4 | P | Lymphoma | Solid | + | − | NP | NP | PAX3-FOXO1 |
| 5 | M | Acute hematologic malignancy | NA | NP | NP | 80–92, XXXX, i(1)(q10), t(2;13)(q35;q14)x2, inc | +* | NP |
| 6 | P | Undifferentiated small round cell tumor | Solid | + | − | 45, XX, −5, −13,+der(16) t(1;16)(q21;q13) | + | NP |
| 7 | P, M† | Small cell carcinoma, lymphoma, epithelial neoplasm, rhabdomyosarcoma | NA | NP | − | t(2;13)(q35;q14) | + | NP |
DDx, differential diagnosis; NP, not performed; M, metastasis; NA, not available.
Metaphase FISH with paint probes confirmed the presence of a t(2;13) in this case.
Conventional cytogenetic studies were performed on the primary tumor of this case and FISH studies on a neck metastasis 5 years later.
Discussion
RMSs in older adults are distinctly unusual, and typically are pleomorphic with high-grade cytologic features and differing biological characteristics (2,11–13). Because of their extreme rarity, inclusion of the ARMS subtype in the differential diagnosis of small round cell tumors of the head and neck region in patients over the age of 45 years is often neglected. Similarly, Nakhleh et al (14) concluded in their study of younger adult patients with RMS (between 18 and 36 years of age) that a round cell neoplasm in a nasal, orbital or lymph node biopsy specimen is unlikely to suggest the immediate possibility of a RMS. Other malignancies such as small cell undifferentiated or neuroendocrine carcinoma, lymphoma, olfactory neuroblastoma, and melanoma are more likely to be considered in this location and older age group. In the pediatric population, most sinonasal ARMSs are of the solid form (lack a recognizable alveolar pattern) and are poorly differentiated, thereby complicating diagnostic efforts (15). Cytogenetic and molecular genetic studies may aid in the diagnosis of head and neck ARMS, however, with the exception of isolated case descriptions, these studies are lacking in the older adult (6–8).
In the current study, the initial diagnostic work-up of four ARMSs arising in the head and neck region of patients between 61 and 76 years of age did not include myogenic markers. Review of the clinicohistopathologic findings of these four cases and three additional genetically characterized head and neck ARMSs from older adults revealed that all cases showed a solid pattern of small, relatively uniform, round-to-ovoid cells with scant cytoplasm. A subset of the neoplastic cells from one ARMS (Case 2) also exhibited pale-staining or clear cytoplasm, a rare histologic feature that may contribute to diagnostic confusion. In contrast to most ERMSs and spindle cell RMSs (the latter also have an affinity for the head and neck region in adults) (16), rhabdomyoblasts were conspicuously absent as were multinucleated giant cells. Neoplastic cells expressed CD56 in 5/5 cases, synaptophysin in 1/6, and cytokeratin in 2/6. As a consequence of nonimmunoreactivity for other examined epithelial, lymphoma, or malignant melanoma markers, the differential diagnosis was expanded to include RMS and additional corresponding myogenic immunostains and molecular studies were pursued.
CD56 expression has an established role in the diagnosis of non-Hodgkin lymphoma (NHL)-natural killer cell type and other hematologic malignancies. The sinonasal region is notably a common location for natural killer/T-cell lymphomas, particularly in certain regions of the world (17). Interestingly, diagnostic delays have been reported in occasional patients whose initial ARMS clinicohistopathologic presentation simulated a leukemia or lymphoma (18–22). CD56 immunoreactivity is of limited value in the differential diagnosis of sarcomas because it is widely expressed in these neoplasms. It may be useful however, in conjunction with assessment of CD99 expression in distinguishing Ewing’s sarcoma from RMS. Only 10–25% of Ewing’s sarcomas are CD56 positive in contrast to 100% of primitive embryonal and alveolar RMSs (2,23,24).
CD56 is also commonly expressed in neuroendocrine tumors. A combination of CD56 and/or synaptophysin immunoreactivity is suggestive of a neoplasm of neuronal or neuroendocrine differentiation which led to the initial diagnostic impression of small cell carcinoma or esthesioneuroblastoma for several of the ARMSs in the current study. Reports of synaptophysin expression in ARMS are few (25,26). Conversely, rhabdomyoblastic differentiation may occur in olfactory neuroblastoma (27).
Poorly differentiated round cell neoplasms constitute a common problem in differential diagnosis and for some cases, morphologic evaluation alone is insufficient, and further investigations are required. Distinguishing ERMS and ARMS is especially problematic but is essential for prognostic consideration and provision of optimal therapy. Thus, a valuable diagnostic adjunct in ARMS is the identification of translocations t(2;13)(q35;q14) and t(1;13)(p36;q14), and the associated PAX3-FOXO1 and PAX7-FOXO1 fusion transcripts, respectively (9,28). Recognition of these specific translocations is also prognostically important as PAX3-FOXO1 expression is an independent indicator of adverse outcome for patients with ARMS, while PAX7-FOXO1 gene fusion may predict a more favorable outcome (20,29,30). Molecular confirmation of the presence of a PAX-FOXO1 fusion transcript was fundamental to classifying each neoplasm of the current study. Unexpectedly, one case (Case 2), exhibited a PAX7-FOXO1 fusion gene, a finding corresponding with the younger cohort of one ARMS study (31).
In summary, RMSs of the head and neck region of older adults are extremely rare and the five-year survival is dismal (8 percent or less) (32). For each of the patients in the current study, the diagnosis was histologically challenging and more common tumors in these anatomic sites of older adults such as lymphoma, small cell carcinoma and/or esthesioneuroblastoma were initially considered. The utility of molecular detection of PAX-FOXO1 gene fusions in ARMS of childhood and young adults is well documented. The current cases illustrate the important adjunctive role molecular characterization also serves in the classification of RMS in patients over the age of 45 years. The value of these ancillary molecular studies are further appreciated in light of the predominantly solid histologic pattern each of these ARMSs exhibited. The prognostic value of the fusion gene status in this series, however, is less clear as the limited study size and short follow-up period preclude any definitive conclusions.
Acknowledgments
The authors gratefully acknowledge the assistance of Drs. Thomas Seemayer, Sonny Johansson, and Neil Abrahams at the University of Nebraska Medical Center for their helpful suggestions and contributions. They also thank Dr. Stewart Cramer at Viahealth Pathology and Laboratory Medicine, Rochester General Hospital and Dr. Deepak Sahasrabudhe at Strong Health, Strong Memorial Hospital, Department of Medicine, Division of Hematology/Oncology for contributing case materials.
This work was supported in part by Eppley Cancer Center Pediatric Pilot Award, State of Nebraska LB595, and NIH/NCI P30 CA 36727. T.Y. was supported by the Gladys Pearson Fellowship Award.
Footnotes
This work was presented in part at the 97th annual meeting of the United States and Canadian Academy of Pathology in Denver, CO, March 1 to 7, 2008.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Russell WO, Cohen HJ, Enzinger FM, et al. A clinical and pathological staging system for soft tissue sarcomas. Cancer. 1997;40:1562–1570. doi: 10.1002/1097-0142(197710)40:4<1562::aid-cncr2820400428>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Weiss SW, Goldblum J. Rhabdomyosarcoma. In: Weiss SW, Goldblum JR, editors. Enzinger and Weiss’s Soft Tissue Tumors. Philadelphia, PA: Mosby Elsevier Inc; 2008. pp. 595–631. [Google Scholar]
- 3.Crist W, Gehan EA, Ragab AH, et al. The third intergroup rhabdomyosarcoma study. J Clin Oncol. 1995;13:610–630. doi: 10.1200/JCO.1995.13.3.610. [DOI] [PubMed] [Google Scholar]
- 4.Gurney JG, Young JL, Roffers SD, et al. Soft Tissues Sarcomas. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995. In: Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, editors. National Cancer Institute, SEER Program. Bethesda, MD: 1999. NIH Pub. No. 99–4649. [Google Scholar]
- 5.Hawkins WG, Hoos A, Antonescu CR, et al. Clinicopathologic analysis of patients with adult rhabdomyosarcoma. Cancer. 2001;91:794–803. [PubMed] [Google Scholar]
- 6.Das KM, Mirani N, Hameed M, Plinerr L, Aisner SC. Fine-needle aspiration cytology of alveolar rhabdomyosarcoma utilizing ThinPrep liquid-based sample and cytospin preparations: a case confirmed by FKHR break apart rearrangement by FISH Probe. Diagn Cytopathol. 2006;34:704–706. doi: 10.1002/dc.20545. [DOI] [PubMed] [Google Scholar]
- 7.Manucha V, Castellani R, Sun CC. Alveolar rhabdomyosarcoma of the paranasal sinuses in a 57-year-old woman with 1;16 translocation. Int J Surg Pathol. 2006;14:238–242. doi: 10.1177/1066896906290560. [DOI] [PubMed] [Google Scholar]
- 8.Stindl R, Fiegl M, Regele H, et al. Alveolar rhabdomyosarcoma in a 68-year-old patient identified by cytogenetic analysis of bone marrow. Cancer Genet Cytogenet. 1998;107:43–47. doi: 10.1016/s0165-4608(98)00061-2. [DOI] [PubMed] [Google Scholar]
- 9.Nishio J, Althof PA, Bailey JM, et al. Use of a novel FISH assay on paraffin-embedded tissues as an adjunct to diagnosis of alveolar rhabdomyosarcoma. Lab Invest. 2006;86:547–556. doi: 10.1038/labinvest.3700416. [DOI] [PubMed] [Google Scholar]
- 10.Shaffer LG, Tommerup N, editors. An International System for Human Cytogenetics Nomenclature. Basel: Karger; 2005. ISCN 2005. [Google Scholar]
- 11.Nayar RC, Prudhomme F, Parise O, Gandia D, Luboinski B, Schwaab G. Rhabdomyosarcoma of the head and neck in adults: a study of 26 patients. Laryngoscope. 1993;103:1362–1366. doi: 10.1288/00005537-199312000-00008. [DOI] [PubMed] [Google Scholar]
- 12.El-Naggar AK, Batsakis JG, Ordonez NG, Luna MA, Goepfert H. Rhabdomyosarcoma of the adult head and neck: a clinicopathologic and DNA ploidy study. J Laryngol Otol. 1993;107:716–720. doi: 10.1017/s0022215100124223. [DOI] [PubMed] [Google Scholar]
- 13.Parham DM, Ellison DA. Rhabdomyosarcoma in adults and children: an update. Arch Pathol Lab Med. 2006;130:1454–1465. doi: 10.5858/2006-130-1454-RIAACA. [DOI] [PubMed] [Google Scholar]
- 14.Nakhleh RE, Swanson PE, Dehner LP. Juvenile (embryonal and alveolar) rhabdomyosarcoma of the head and neck in adults. A clinical, pathologic, and immunohistochemical study of 12 cases. Cancer. 1991;67:1019–1024. doi: 10.1002/1097-0142(19910215)67:4<1019::aid-cncr2820670426>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 15.Ahmed AA, Tsokos M. Sinonasal rhabdomyosarcoma in children and young adults. Int J Surg Pathol. 2007;15:160–165. doi: 10.1177/1066896906299122. [DOI] [PubMed] [Google Scholar]
- 16.Nascimento A, Fletcher CDM. Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol. 2005;29:1106–1113. [PubMed] [Google Scholar]
- 17.Gaal K, Sun NCJ, Hernandez AM, Arber DA. Sinonasal NK/T-cell lymphomas in the United States. Am J Surg Pathol. 2000;24:1511–1517. doi: 10.1097/00000478-200011000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Shah Ho, Lin JH. Alveolar rhabdomyosarcoma with concurrent metastases to bone marrow and lymph nodes simulating acute hematologic malignancy. J Pediatr Hematol Oncol. 2004;26:696–697. doi: 10.1097/01.mph.0000140654.50344.92. [DOI] [PubMed] [Google Scholar]
- 19.Pinto A, Tallini G, Novak RW, Bowen T, Parham DM. Undifferentiated rhabdomyosarcoma with lymphoid phenotype expression. Med Pediatr Oncol. 1997;28:165–170. doi: 10.1002/(sici)1096-911x(199703)28:3<165::aid-mpo1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 20.Sorensen PHB, Lynch JC, Qualman SJ, et al. PAX3- FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Onco. 2002;20:2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- 21.Tsai SC, Reale LD, Flomenberg N, Schwarting R, Enck RE. Alveolar rhabdomyosarcoma mimicking a lymphoma at presentation. J Clin Oncol. 2006;24:4031–4032. doi: 10.1200/JCO.2006.05.8917. [DOI] [PubMed] [Google Scholar]
- 22.Yamaguchi K, Koga Y, Suminoe A, et al. Alveolar rhabdomyosarcoma of unknown origin mimicking acute leukemia at the initial presentation. Rinsho Ketsueki. 2007;48:315–320. [PubMed] [Google Scholar]
- 23.Garin-Chesa P, Fellinger EJ, Huvos AG, et al. Immunohistochemical analysis of neural cell adhesion moleculars: differential expression in small round cell tumors of the childhood and adolescence. Am J Pathol. 1991;139:275–286. [PMC free article] [PubMed] [Google Scholar]
- 24.Miettinen M, Coup W. Neural cell adhesion molecule distribution in soft tissue tumors. Hum Pathol. 1993;24:62–66. doi: 10.1016/0046-8177(93)90064-n. [DOI] [PubMed] [Google Scholar]
- 25.Leroy X, Petit M-L, Fayoux P, Aubert S, Escanda F. Aberrant diffuse expression of synaptophysin in a sinonasal alveolar rhabdomyosarcoma. Pathology. 2007;39:275–276. doi: 10.1080/00313020701230781. [DOI] [PubMed] [Google Scholar]
- 26.Bahrami A, Gown AM, Baird GS, Hicks MJ, Folpe AL. Aberrant expression of epithelial and neuroendocrine markers in alveolar rhabdomyosarcoma: a potentially serious diagnostic pitfall. Mod Pathol. 2008;21:795–806. doi: 10.1038/modpathol.2008.86. [DOI] [PubMed] [Google Scholar]
- 27.Slootweg PJ, Lubsen H. Rhabdomyoblasts in olfactory neuroblastoma. Histopathology. 1991;19:182–184. doi: 10.1111/j.1365-2559.1991.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 28.Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20:5736–5746. doi: 10.1038/sj.onc.1204599. [DOI] [PubMed] [Google Scholar]
- 29.Davicioni E, Finckenstein FG, Shahbazian V, et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006;66:6936–6946. doi: 10.1158/0008-5472.CAN-05-4578. [DOI] [PubMed] [Google Scholar]
- 30.Kazanowska B, Reich A, Stegmaier S, et al. PAX3-FKHR and PAX7-FKHR fusion genes impact outcome of alveolar rhabdomyosarcoma in children. Fetal Pediatr Pathol. 2007;26:17–31. doi: 10.1080/15513810701394702. [DOI] [PubMed] [Google Scholar]
- 31.Kelly KM, Womer RB, Sorensen PH, Xiong QB, Barr FG. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol. 1997;15:1831–1836. doi: 10.1200/JCO.1997.15.5.1831. [DOI] [PubMed] [Google Scholar]
- 32.El-Ghazali AMS, McLaren KM. Embryonal rhabdomyosarcoma of adult nasopharynx. J Laryngol Otol. 2005;119:639–642. doi: 10.1258/0022215054516142. [DOI] [PubMed] [Google Scholar]