

Abstract
Optical tweezers are ideally suited to perform force microscopy experiments that isolate a single biomolecule, which then provides multiple binding sites for ligands. The captured complex may be subjected to a spectrum of forces, inhibiting or facilitating ligand activity. In the following experiments, we utilize optical tweezers to characterize and quantify DNA binding of various ligands. High Mobility Group Type B (HMGB) proteins, which bind to double-stranded DNA, are shown to serve the dual purpose of stabilizing and enhancing the flexibility of double stranded DNA. Unusual intercalating ligands are observed to thread into and lengthen the double-stranded structure. Proteins binding to both double- and single-stranded DNA, such as the alpha polymerase subunit of E. coli Pol III, are characterized and the subdomains containing the distinct sites responsible for binding are isolated. Finally, DNA binding of bacteriophage T4 and T7 single-stranded DNA (ssDNA) binding proteins are measured for a range of salt concentrations, illustrating a binding model for proteins that slide along double-stranded DNA, ultimately binding tightly to ssDNA. These recently developed methods quantify both the binding activity of the ligand as well as the mode of binding.
Introduction
Once the structure of DNA was determined, the search for the specific mechanisms that copied and translated the genetic code began.1-3 Over time, it became understood that DNA was involved in many other processes as well. These interactions are not limited only to replication and transcription, but include DNA compaction, unwinding, chemical modification and sequence repair. The proteins that regulate and catalyze these reactions bind to DNA in ways that facilitate their function. Determining the specificity and affinity of binding ligands is a crucial step toward understanding their function. It is the goal of this work to summarize new developments that allow us to discern and quantify various types of ligand binding to both double- and single-stranded DNA.
Nucleic Acid Force Spectroscopy
Since their inception, optical trapping experiments have been utilized to isolate individual cells and their components.4-8 Evolving from the simplest single beam optical trap, an impressive variety of experiments have probed nucleic acid structure and ligand binding. The common thread to each experiment involves the isolation of a single biomolecule which is then subject to a specific tension or extension. Force microscopy experiments may attach a cantilever directly to a molecule, utilize fluid flow, or attach spherical beads that may then be manipulated by magnetic fields or focused lasers.9-15 The latter category of experiments have probed various DNA and RNA structures, and observed various ligands bound to those assemblies. Bound ligands may be observed to alter DNA structural or thermodynamic properties, and the kinetics and energetics of these interactions may be altered through the application of force. The aim of the work described here is to deduce distinct modes of binding to DNA and to quantify the affinity and thermodynamics of this process, to gain insight into ligand function.
After introducing methods for characterizing DNA stretching data and for using this data to quantify DNA binding, we will review three examples in which these methods have been used to elucidate DNA binding modes. We will first discuss the High Mobility Group Type B (HMGB) proteins, which bind to dsDNA and alter its flexibility and stability. We will then outline studies of DNA intercalators, which bind preferentially to dsDNA and significantly alter its overall structure. Finally, we will discuss experiments on the E. coli Pol III α polymerase subunit, which binds to both dsDNA and ssDNA, and we will show how these new methods are used to identify distinct regions of the protein that bind specifically to each form of DNA.
Optical tweezers apparatus
The apparatus utilized for these experiments is a dual counter-propagating beam instrument that is capable of high forces with relatively good force resolution (∼ 0.1 pN).16,17 Briefly, a pair of infrared laser beams is directed to a common, diffraction limited focus of ∼ 1 μm diameter. Though a pair of objectives provides this confocal arrangement with a numerical aperture of 1.0, only ∼ 0.6 of that aperture is used. Each beam provides 110 mW of power. After the beams exit the trap, polarization optics eject each beam from the counter-propagating path and onto a pair of lateral effect detectors. Far field optics ensure that small motions of the beam in the vicinity of the trap translate into easily detectable deflections at these micron resolution detectors.
Between the water-immersion objectives, a custom flow cell provides the sample volume. Varying solutions may be introduced through separate channels. Polystyrene beads of varying diameters (3 to 5 microns, typically) are trapped to serve as the anchor for the molecules to be studied. The following experiments study phage lambda DNA, which is attached to the trapped bead through a strepdavidin-biotin linkage. Another bead may be drawn onto a micropipette tip and the DNA is thus ‘caught’ between the fixed and moveable spheres. As the position of the stage is extended or relaxed, the rise in tension pulls the trapped bead, deflecting the trapping beams. The deflection may be calibrated with a viscous Stokes technique and a direct measurement of the trap stiffness using the bead immobilized upon the micropipette.18 Thus measurements of force versus extension are determined, as shown for several phage λ DNA molecules on Figure 1 (discussion of this data follow below). Alternatively, the extension may be measured for a given, fixed force and these experiments are also detailed below.
FIGURE 1.
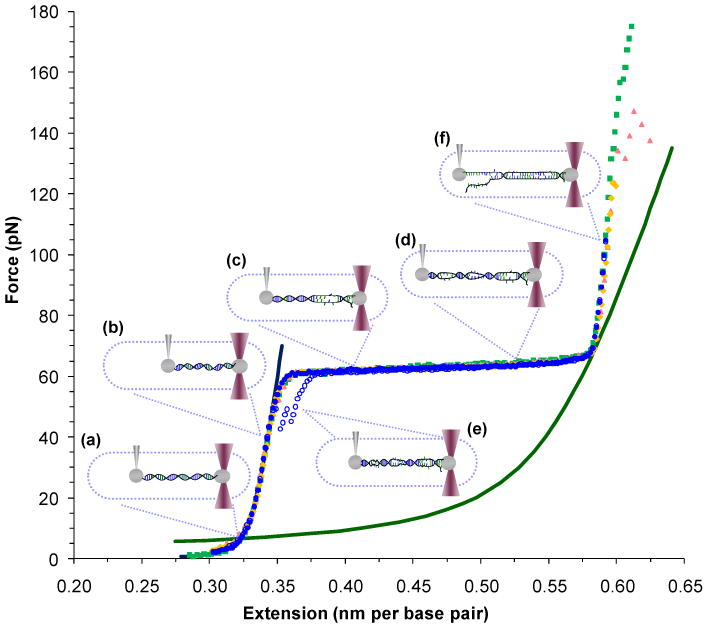
Force-extension experiments manipulate dsDNA as a function of constant force or extension. In this diagram of constant extension experiments, force-extension data are shown as solid symbols followed by relaxation data as open symbols. (a) DNA is tethered between two beads and (b) as the extension is increased, the tension rises (filled blue circles). (c) & (d) At a critical force, base pairing and stacking are disrupted as dsDNA is melted by force and converted into two parallel ssDNA strands. (e) If relaxed, these strands anneal and revert to dsDNA, though some hysteresis is evident (open blue circles). (f) Force-melted DNA may be stretched further until the oppositely labeled strands separate (green, yellow and red). Polymeric models that describe dsDNA (blue line) and ssDNA (green line) are shown and described in the text. All data is collected in 10 mM Hepes buffer, pH 7.5 with 100 mM Na+, unless noted.
Models of polymer elasticity and the properties of DNA
Several models have been proposed to characterize DNA force-extension curves. A successful model of DNA elasticity treats the polymer as an elastic continuum with a smooth distribution of bending angles. The Worm-Like Chain (WLC) model describes dsDNA in terms of its end to end distance, known as the contour length (Bds), and a measure of its flexibility, the persistence length (Pds).19-23 Though an exact solution has not been determined, approximations may be used that are appropriate for high or low force regimes. The observed length of dsDNA (bds) is determined by the tension (F) in the high force limit (kB is Boltzmann's constant and T is the temperature);
| (1) |
The parameters of this equation have been determined for a wide variety of conditions; values fit to the data in Figure 1 are Bds = 0.340 ± 0.001 nm/bp and Pds = 48 ± 2 nm. This model also features the addition of a phenomenological stretch modulus, Sds = 1200 ± 100 pN.11,19-24 Though successful at describing dsDNA, some discrepancies remain, particularly for short (n < 100 base pair) sequences, as the nature of DNA flexibility at this scale remains controversial.25-28 Furthermore, quantitative models that describe DNA-bound ligands remain elusive for all but the shortest sequences that can be crystallized. Finally, interpolations between the high and low force limits of the WLC model have enhanced the accuracy of the fits.24 This work will utilize long sequences (∼48 kbp) of DNA with multiple bound proteins that should be well described by changes to the parameters of the WLC model given by Equation 1.
Several experiments probe constructs that include some form of single-stranded DNA. The Freely-Jointed Chain (FJC) model describes a series of fixed segments that are free to independently rotate;9
| (2) |
Typical values for these parameters have also been determined, though less well when compared to the same numbers for dsDNA (Bss = 0.575 nm/bp, Pss = 0.75 nm and Sss = 800 pN).29 This model is plotted in Figure 1.
Force-induced melting of single DNA molecules
The earliest work acknowledged that stretching experiments probed first an entropic then an enthalpic stretching regime for dsDNA. Both are described by Equation 1 and shown on Figure 1a and 1b, respectively.19,21-23,30,31 While it was clear that DNA coupled to beads on opposite strands must separate (at ∼150 pN), the appearance of a constant force plateau raised questions. Some argued that the plateau represented a significant length change essentially from dsDNA to ssDNA (Figure 1). In this model, both base stacking and base pairing are disrupted for large domains, in a manner analogous to and consistent with the energies predicted from thermal melting experiments, and shown in Figure 1c and 1d.32-35 Experiments in varying salt concentrations, pH and temperature support the view that during overstretching, dsDNA is converted to ssDNA.19,34,35 At the end of this force-induced melting transition, only a few particularly stable (GC-rich) regions remain and the two strands are stretched in parallel until they separate (Figure 1f).14,34-37 If relaxed before separation, the double strands are posited to anneal, though some hysteresis will be evident (Figure 1e), and some peeling from the free ends of the molecule is also likely.38 Modeling studies39-41 and DNA ligand binding experiments42-48 have also supported this theory.
An alternative model suggests that as separation occurs only at high forces, the plateau may be due to disruption of base stacking, but not base pairing.49-51 In this model, DNA lengthens during this transition, but assumes a new form, termed S-DNA.14,29,37,52 Early experiments on intercalating ligands at saturated DNA binding and molecular modeling supported this view.49-54 The authors of a recent study pulling short DNA duplexes at varying rates suggest that melting may occur at relatively slow rates, while faster stretching experiments (featuring loading rates > 106 pN/s, which are not accessible by the laser trapping instrument described above) may transiently stabilize a form that preserves base pairing.55 Others have hypothesized that overstretching may partially convert B-form DNA to S-DNA, dependent upon solution conditions.12,53,56-58
For the following work, it will be crucial to know the form assumed by the DNA molecule during the overstretching transition. A series of constant extension experiments were performed in the presence of glyoxal (C2H2O2). This molecule is known to bind to DNA by attaching only to exposed guanine bases, preventing base pairing.59-63 As the reaction is slow (> 30 minutes, but varying with solution conditions), the following experiments extended and held dsDNA to a fixed position. Figure 2a shows such an extension and the subsequent relaxation cycle in 100 mM Na+ solution. As the DNA is unable to re-anneal, it is clear that glyoxal has modified the exposed bases. Thus extension into the overstretching plateau must consist of substantial melting of dsDNA and subsequent exposure of individual bases.
FIGURE 2.

Glyoxal binds to force melted DNA. (a) An extension/relaxation cycle of DNA is shown as a solid/dotted line in black. After stretching DNA in the presence of 500 nM of glyoxal (blue solid line), the DNA is held at the extension marked by the blue arrow for 30 minutes. Upon relaxation (blue dotted line), the strands fail to anneal, as the exposed nucleotides have been modified by glyoxal. A subsequent extension/relaxation cycle (green lines) confirms that the modification is permanent. (b) Relaxation data for DNA stretched to the lengths denoted by the arrows and corresponding to the lengths of approximately ¼, ½ and ¾ of the overstretching transition (in blue, green and red, respectively). The data is fit to a superposition of the WLC and FJC models as described in the text, for data where bss > bds. The fraction of ssDNA stabilized by glyoxal is 0.22 ± 0.03, 0.33 ± 0.01 and 0.44 ± 0.05 respectively. Figures are adapted from Shokri, et al.65
A series of relaxation data that follows distinct fixed extensions is shown in Figure 2b. If these force-extension curves are due to a mixture of dsDNA and ssDNA, they should fit well to a linear combination of the WLC and FJC models;19,64-66
| (3) |
where b is the observed contour length, bds and bss are force-dependent parameters given by Eq. (1) and (2), and γss is the fraction of ssDNA stabilized by ligand binding. The data of Figure 2b appear to fit well to this model, though there is a change in Pss due to glyoxal modification. The fits show that for fractional melting of ¼, ½ and ¾ of the transition, the fraction of ssDNA stabilized by glyoxal is 0.22 ± 0.03, 0.33 ± 0.01 and 0.44 ± 0.05, respectively. The fact that the fraction stabilized appears to saturate at ∼ ½ may be due to the sequence specificity of glyoxal modification or possible formation of hairpin intermediates at the free ends. Experiments in low salt (5 mM Na+), where hairpins would be expected to be less stable, while the binding rate of glyoxal is unchanged, reveal more complete conversion to ssDNA (data not shown). Thus, force-induced melting represents the conversion of dsDNA into ssDNA, analogous to thermal melting.
Binding to the DNA Double Helix
High Mobility Group proteins are a numerous and diverse group of nuclear chromosomal proteins, divided into different subgroups according to unique binding motifs.67-70 The HMGB subgroup has been implicated in a striking variety of roles: disrupting chromatin,67-69,71-73 regulating transcription,74-83 enhancing DNA repair,84 and even as an extracellular signal facilitating immune response.85,86 The specific mechanisms of these functions are not clear. Yet it is known that the key to much of this activity is the protein's ability to bind in a manner independent of sequence to dsDNA through an 80 amino acid domain known as an HMG box, as shown in Figure 3a.87,88 Binding into the minor groove and slightly intercalating into dsDNA produces a strong deformation, evident as a pronounced bend in the backbone.16,87-93 The following experiments probe the flexibility and stability of DNA in the presence of the single box motif of HMGB2(box A) and the tandem double box of HMGB1(box A+B) to further understand the role these proteins play in the nucleus.
FIGURE 3.
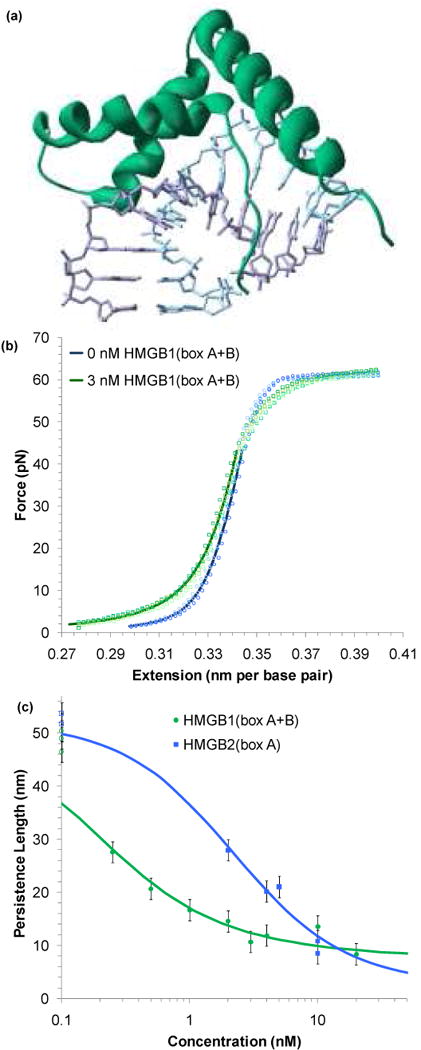
HMG proteins alter the flexibility of dsDNA. (a) Non-specific binding into the minor groove bends the backbone ∼ 111° in this crystal structure of HMG-D from D. melanogaster,89 which shows a box motif typical of the HMGB family (PDB code: 1QRV). (b) Four sets of extension data for dsDNA (blue circles) and dsDNA in the presence of 3 nM of HMGB1(box A+B) (green circles). The average of these data sets may be fit to the WLC model of Equation 1 (solid blue and green lines). The addition of protein reduces the persistence length from 46 ± 2 nm to 22 ± 2 nm. The contour length and bulk stiffness show less definite changes from bds = 0.339 ± 0.001 nm/bp and Kds = 1200 ± 50 pN to bds = 0.339 ± 0.003 nm/bp and Kds = 939 ± 100 pN for dsDNA with 3 nM HMGB1(box A+B). Fits apply only to data below 45 pN. (c) Fitted persistence lengths as a function of concentration for HMGB2(box A) (blue) and HMGB1(box A+B) (green). Uncertainties are determined from the fits to averaged data sets as shown above. The open circles along the left hand axis are the persistence lengths measured in the absence of any protein, shown as a reference. Fits to the concentration are described in the text and the data determine the equilibrium binding constant per ligand for HMGB1(box A+B) to be Kn = 5.9 ± 1.6 × 108 M-1 and Kn = 0.15 ± 0.06 × 108 M-1 for HMGB2(box A). Figures (b) and (c) adapted from McCauley, et al.91
HMGB proteins enhance DNA flexibility
As increasing concentrations of HMGB proteins are added to the solution surrounding a single DNA molecule, there is a clear change in the dsDNA force-extension data. Figure 3b shows this data for DNA in the absence and in the presence of the double box HMGB1(box A+B). To quantify the changes to the force-extension curves, the averages are fit to the WLC model of Equation 1. These fits show that the addition of protein decreases the persistence length. The persistence length is plotted as a function of protein concentration for both HMGB1(box A+B) and HMGB2(box A) on Figure 3c.91 A binding site in the absence of protein should have occupancy (γds) of zero and an observed persistence length (Pds) that matches the persistence length of bare dsDNA (PDNA). Upon protein binding, the persistence length changes (PPR), and averaged over multiple binding sites;94
| (4) |
The occupancy may be fit to the site exclusion binding isotherm of McGhee and von Hippel,95,96 which relates the equilibrium association constant per ligand (Kn) or per base pair or nucleotide (K) and the concentration (c);
| (5) |
Here the occupancy may refer to that for a general dsDNA binding site (γds), to an intercalation site (γint), or ssDNA binding site (γss), as necessary (see below). Fits to Equations 4 and 5 give an equilibrium association constant per ligand for HMGB1(box A+B) as Kn = 5.9 ± 1.6 × 108 M-1 and for HMGB2(box A), Kn = 0.15 ± 0.06 × 108 M-1.
Fits to the data in Figure 3c also yield the value of PPR, the persistence length when DNA is saturated by protein. From these numbers, we may deduce the average bending angle induced by protein binding;91,93,94,97
| (6) |
Equation 6 assumes bending is localized to independent sites on the DNA lattice. Thus protein concentrations are limited to values that show relatively little cooperative binding, which begins to dominate at higher concentrations. The value of the contour length at saturation, BPR, is determined from fits to Equation 1 (BPR = 0.380 ± 0.005 nm per base pair for both proteins, due to partial intercalation of several residues), while the binding site size is determined from previous estimates [n = 7 for HMGB2(box A) and n = 18 for HMGB1(box A+B)].88-90,92 From these values, average induced bending angles may be found for HMGB1(box A+B) as β = 77 ± 7°, while HMGB2(box A) induces β = 99 ± 9°. Thus HMGB appears to bend the DNA backbone and alter its flexibility, an effect found by other techniques as well.75,98-100
HMGB proteins stabilize the DNA double helix
In addition to altering the flexibility of dsDNA, changes to the force-induced melting plateau are evident as well. These changes are shown for both the single and double box motifs in Figure 4a and b. In both cases, protein binding stabilizes dsDNA, and higher forces are required to induce melting, as protein binding must be at least partially disrupted for melting to occur. Averaging the melting force about the midpoint of the transition shows an increase in force with protein concentration, as plotted in Figure 4c. This measured force (Fm) increases from the value observed in the absence of protein to a value when the DNA lattice appears to be saturated with protein . Assuming that the increase in the melting force is linearly proportional to the occupancy of the binding site (γds),101,102
FIGURE 4.

HMG proteins stabilize DNA melting. (a) Increasing concentrations of HMGB1(box A+B) raise the force necessary to melt DNA. Extension/relaxation data are shown as solid/dotted lines. To emphasize this change, the graph is split; data to the right of the dotted line is expanded on the force axis shown to the right. (b) Increasing concentrations of HMGB2(box A) reveal little change in the melting force until much higher concentrations are added to solution. (c) The averaged midpoint of the melting force for a range of protein concentrations for HMGB1(box A+B) (green) and HMGB2(box A) (blue). Error bars are determined from standard deviations of at least four measured melting curves. Fits to the data yield an equilibrium association constant for HMGB1(box A+B) (Kn = 7.2 ± 1.7 × 108 M-1) and for HMGB2(box A) (Kn = 0.28 ± 0.10 × 108 M-1). Figures adapted from McCauley, et al.91
| (7) |
Fits to Equation 5 and 7 assume the same binding site sizes as above and determine an equilibrium association binding constant of Kn = 7.2 ± 1.7 × 108 M-1 for HMGB1(box A+B) and a much weaker Kn = 0.28 ± 0.10 × 108 M-1 for HMGB2(box A).
HMGB protein single and double box motifs
HMGB binding to DNA has been implicated as a precursor to transcription, by disrupting the structure of chromatin.67-69,71-73 Thus it seems fitting that these proteins induce bends and increase the lateral flexibility of dsDNA. It is initially surprising that the double box shows a weaker bending angle compared to the single box, as one may simply expect that more boxes should give more bending. That is clearly not the case here. Then what is the benefit of having the additional binding motif in the double box protein? The most likely explanation is that the binding affinity of a protein containing two equivalent binding sites should scale quadratically with the number of binding sites.103-105 Thus, even if the total bending angle induced by the double box protein is lower, its overall DNA binding affinity may be higher, as shown above. On the other hand, the increase in binding with an additional box is not as much as one would expect for two identical binding sites. Therefore, we cannot think of the double box protein as simply having two identical binding sites. In addition to the likely case that both binding motifs are distinct, we must take into account the flexible tether of the double box motif, which can reduce binding relative to that of the individual boxes together. Also, it appears that at least one of the binding sites must be disrupted to melt the intervening DNA, while DNA can be melted in the presence of the single box without completely dissociating the protein at low protein concentrations. Thus, for the single box motif to stabilize melting must require box to box interactions that may occur only at high protein concentrations and that mimic the presence of the flexible tether in the double box case. Indeed, still higher concentrations of HMGB2(box A) have been observed to show fully cooperative binding that forms a filament around dsDNA, a function seen in other proteins as well.106,107 Thus while it appears that both proteins stabilize dsDNA and enhance its local flexibility, HMGB2(box A) appears to induce stronger bending, while HMGB1(box A+B) seems to be much more effective at stabilizing dsDNA and also exhibits much stronger binding.
Intercalation into the DNA Double Helix
DNA intercalation describes a process that inserts a molecule between adjacent bases.108 Though base pairing is preserved, overall DNA structure is altered by the disruption of adjacent base stacking, lengthening of the DNA, and stabilization of the double helix. Intercalation therefore diminishes the capacity for DNA replication and gene expression.109 Thus intercalation has emerged as a potential tool for targeting cancer. The effectiveness of these treatments is determined in part by the affinity of the ligand for DNA, and the rate of dissociation from the DNA.110 Ruthenium complexes consist of a ruthenium atom surrounded by aromatic rings, which serve as attachment points for varying intercalating motifs.111-114 To test the effectiveness of these motifs as intercalators, the following experiments stretch DNA in the presence of various complexes. Below we describe experiments on two types of intercalators. In the presence of mononuclear intercalators, application of force lowers the free energy of the final intercalated state, resulting in strongly enhanced equilibrium intercalation at high forces. In the presence of binuclear intercalators consisting of a pair of ruthenium coordinated rings, which require fluctuational DNA melting to precede intercalation, applying a force lowers the barrier to intercalation, resulting in enhanced interacalation rates.
Studies of mononuclear intercalators
Averages of multiple force-extension curves in the presence of [Ru(phen)2dppz]2+ are shown in Figure 5a, with the structure of this known intercalator shown in the inset.112,115 These data show a clear change to the contour length of dsDNA. As [Ru(phen)2dppz]2+ intercalates, the distance between adjacent bases is widened to accommodate the planar extension of this molecule (which contains the dppz moiety). Ruthenium complexes without the dppz extension were previously thought to be unable to intercalate, but it was recently shown that the intercalation is just strongly weakened.115-118 As the concentration of the ligand increases, the melting force also increases, until a critical concentration is reached (approximately 100 nM in this case).119 Beyond this critical point no force-induced melting takes place because it is the length change from dsDNA to ssDNA that drives the transition, and at the critical point dsDNA and ssDNA are equal in length.
FIGURE 5.
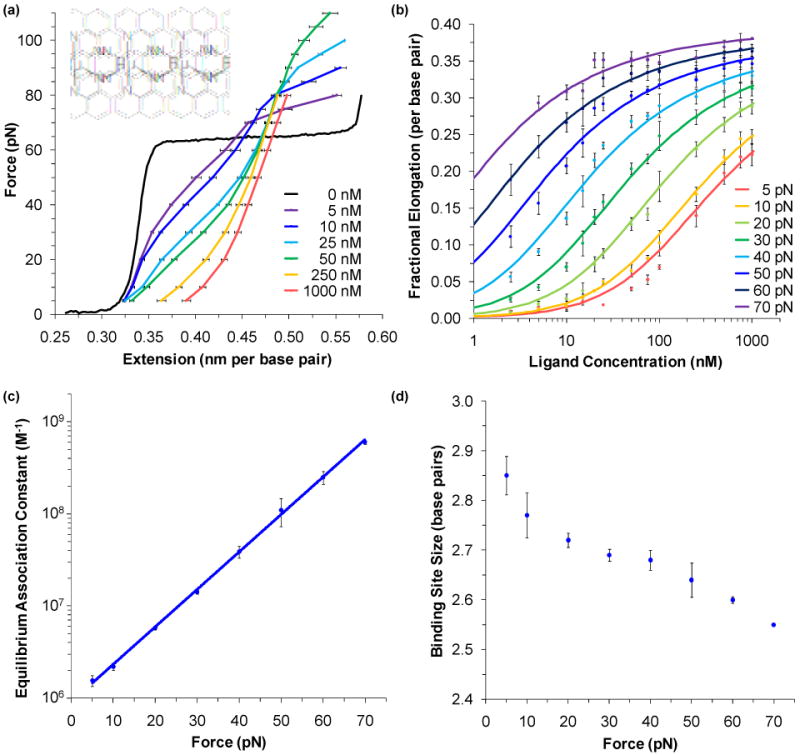
Intercalating ligands characterized by the change in the length of DNA. (a) Averages of the force-extension data for DNA in the presence of varying amounts of the intercalating ligand [Ru(phen)2dppz]2+. Each curve is the average of at least three different extension cycles. DNA lengthens as ligand is added. As the applied force increases the length change increases for a given concentration, indicating that higher forces enable greater binding. (b) Data points are determined from the fractional elongation of DNA as a function of ligand concentration shown in panel a. Solid lines are fits to the data as described in the text. Parameters obtained from these fits are plotted versus force, including the (c) equilibrium association binding constant and (d) binding site size. Higher forces favor binding and reduce the binding site size, which decreases from 2.9 ± 0.1 to 2.6 ± 0.1 base pairs. Fitting the equilibrium association constant to exponential force dependence gives a binding constant in the absence of force of 9.0 ± 1.0 × 105 M-1. Figures from Mihailovic, et al112 and Vladescu, et al.115
The application of force not only stabilizes ssDNA, but also lowers the energy of the fully intercalated state, resulting in further intercalation at higher forces. Furthermore, higher forces allow intercalation into sites that may be sterically hindered at low forces. Figure 5b plots the observed fractional increase in the contour length versus ligand concentration for increasing forces. As the change in the observed length with concentration bds(c) should be proportional to the occupancy (γint);112
| (8) |
Each constant force isotherm may be fitted to Equations 5 and 8, and the results produce equilibrium association binding constants and binding site sizes, which are graphed, in turn, in Figure 5c and d. These graphs confirm that the application of force does increase binding activity and decrease the binding site size. Furthermore, a linear fit to the logarithm of the equilibrium binding constant allows us to extrapolate to the rate in the absence of force, 9.0 ± 1.0 × 105 M-1. Thus the application of force allows intercalation to be studied, even in cases of relatively weak binding, and the results compare well with bulk studies.118,120,121
Studies of binuclear intercalators
Covalently linking two [Ru(phen)2dppz]2+ complexes creates a dumb-bell shaped molecule, Δ,Δ-[μ-bidppz(phen)4Ru2]4+ [bidppz=11,11′ – bi(dipyrido[3,2-a: 2′3′-c]phenazinyl)] (which we will refer to as ΔΔ-P).122 Though this molecule seems an unlikely intercalator, and early experiments suggested binding only into the major groove, intercalation is the dominant binding mode, as suggested by the diagram in Figure 6a.123 It is believed that intercalation takes place only slowly, as ΔΔ-P must wait for thermal fluctuations to transiently melt local regions of dsDNA.123-125 This large molecule then threads between adjacent bases before they anneal. Dissociation is also necessarily slow, as this process also requires fluctuational opening of the DNA base pairs.125,126
FIGURE 6.
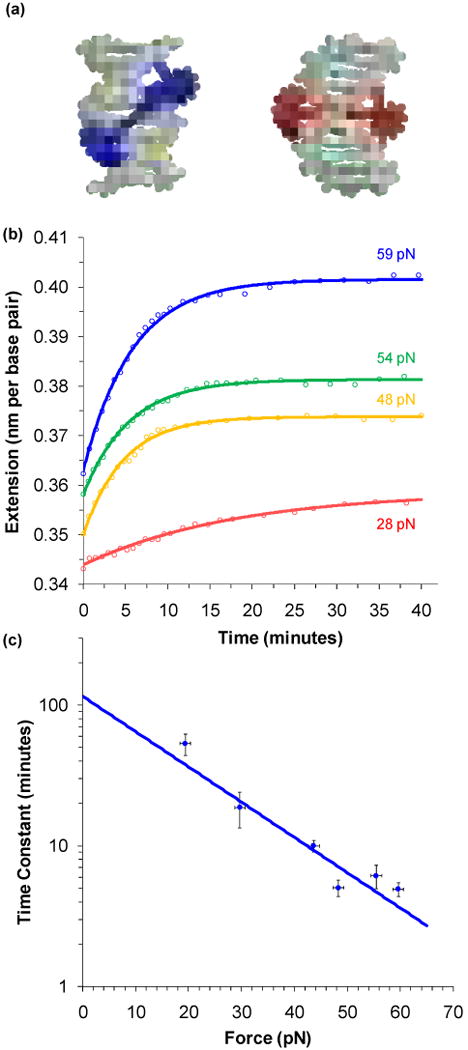
Constant force experiments measure the intercalation rate of a threading intercalator. (a) A pair of covalently linked [Ru(phen)2dppz]2+ molecules yield Δ,Δ-[μ-bidppz(phen)zRu2]4+ (shortened to ΔΔ-P). These complexes initially bind into the major groove, as shown by a predicted model in blue. Over several hours, the complex threads and intercalates into the double helix, resulting in a structure modeled in red. (b) A series of constant force experiments reveal an increase in extension over time as intercalation occurs. The fits shown are to a single exponential dependence on time. (c) Fitted time constants as a function of pulling force. An exponential fit to this data gives a length change due to threading of 0.239 ± 0.025 nm, or the number of base pairs that must melt for each intercalation event must be 1.08 ± 0.11. Extrapolating to zero force gives a reaction time constant of 2.0 ± 0.5 hours. These figures adapted from Paramanathan et al.127
The rate of DNA binding by ΔΔ-P should be both slow and strongly dependent upon force. Using feedback to keep the tension constant allows the extension to be monitored as a function of time. Figure 6b shows the change in the extension as a function of time for a selection of given forces.127 Fits are to simple exponentials and yield single time constants for each constant force. The time constant determined from the data of Figure 6b are shown for each force on Figure 6b. Intercalation of ΔΔ-P into DNA becomes faster with the application of greater force. Here the rate limiting step for intercalation is the requirement for transient melting of a small segment of dsDNA, as indicated in Figure 6a. Force lowers the barrier to melting DNA, decreasing the free energy barrier by the factor FnΔx,38 where n is the number of melted base pairs required for intercalation and Δx is the change in length from dsDNA to ssDNA (and is 0.22 nm).16,43 If this is the case, then applied tension should reduce the observed time constant (τ) exponentially:
| (9) |
This fit is shown on Figure 6c and is used to determine the value of nΔx = 0.239 ± 0.025 nm, which in turn means that n = 1.08 ± 0.11. Thus, for threaded intercalation to occur, only a single base pair needs to melt. The fit also determines the reaction rate in the absence of force to be τ0 = 2.0 ± 0.5 hours.
Distinct Domains Bind to Double and Single Stranded DNA
The Escherichia coli replicative DNA polymerase is a complex holoenzyme composed of ten subunits, which are collectively responsible for both leading and lagging strand synthesis.128-131 The polymerase subunit, α, consists of 1160 residues that adopt the classical polymerase fold, resembling a right hand, as shown in Figure 7a and 7b.132 However, this subunit bears surprisingly little sequence similarity to other DNA polymerases of known structure.132-134 Furthermore, while the basic mechanism of polymerase activity is well understood, many of the interactions of specific polymerases that occur during replication and repair processes are not known. In the case of the E. coli Pol III α subunit, DNA binding has been hypothesized to occur at specific locations on the enzyme, but this binding activity has not been directly demonstrated or quantified. Modeling has suggested that two distinct regions, the Helix-hairpin-Helix (HhH)2 motif135,136 and a predicted oligosaccharide-oligonucleotide domain (OB-domain)132-134,137-140 will each bind DNA, as shown in Figure 7c and 7d. The following experiments determine binding of α and of isolated fragments of α to DNA, to isolate domains of the protein that may bind specifically to either double- or single-stranded DNA.
FIGURE 7.
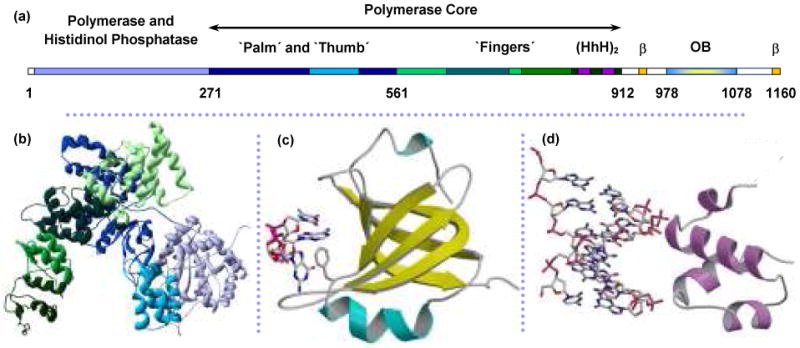
Structure of Pol III catalytic α polymerase subunit from E. coli. (a) Linear structure of α with structural domains indicated from (b) crystal structure of α amino acids 1-917.132 The polymerase core includes the palm, thumb and fingers domains that assume the classical right-hand fold. Residue numbers marking the boundaries of these regions are from the crystal structure (PDB code: 2HNH). (c) Homology model of the C-terminus predicts an OB-fold domain (not present in the crystal structure) for residues 978-1078. ssDNA is shown interacting with the residue F1031.152,162,163 (d) dsDNA is predicted to bind nonspecifically with the residues 833-889, which contains the dual helix-hairpin-helix motif, noted as (HhH)2 within the sequence map.135,136 Figures (c) and (d) are from McCauley, et al.64
Binding to ssDNA created by force-induced melting
The cycle of DNA stretching and relaxation show two clear deviations in the presence of the α subunit relative to that observed for DNA alone, as shown in Figure 8a. In the experiment, DNA is initially double-stranded and this form is at first stabilized by protein binding, as will be discussed below. However, after ssDNA is created by force-induced melting, as shown in Figure 8a, the same protein binds preferentially to ssDNA and stabilizes this form. This process is demonstrated in Figure 8a. In the absence of protein, the melted strands are able to anneal upon relaxation after force-induced melting. However, when the α subunit is introduced into solution, complete reannealing does not take place, and the relaxation curve appears to be a linear combination of dsDNA that has reannealed and ssDNA that has been stabilized by binding to the α subunit. Though some additional reannealing of melted DNA does take place between cycles, subsequent extensions show that the much of the protein remains bound, Finally, α does not actively melt the DNA, as it does not lower the melting force, and the protein will only bind to ssDNA that has been previously melted by force.
FIGURE 8.

Pol III α polymerase domains bind to single- and double-stranded DNA. (a) A sequence of extension and relaxation cycles (solid and dotted lines) for DNA in 100 nM full length α. The initial extension (blue) shows little change from bare DNA (black), though relaxation shows that the force-melted DNA has failed to reanneal. Subsequent cycles (green, yellow and red) show some protein unbinding and subsequent DNA reannealing, but binding is complete at the final cycle. An increase in the melting force indicates stabilization of dsDNA. The figure is split about the dotted line; data to the right are expanded onto the scale shown. (b) Extension and relaxation cycles (blue, green and red) of N-terminal construct α1-917 show complete annealing upon relaxation and no protein binding to ssDNA. Stabilization of dsDNA is evident, indicating that a dsDNA binding site is contained within the construct. (c) Three data cycles (blue, green, and red) for the N-terminal construct α1-835 show no detectable binding to either ssDNA or dsDNA. Comparing the data from panel (b) and (c) indicates a dsDNA binding site within the residues 836-917. Thus the (HhH)2 motif is predominantly responsible for dsDNA binding. These figures are adapted from McCauley, et al.64
Binding to dsDNA increases the DNA melting force
Another effect visible in Figure 8a is the stabilization of dsDNA, evident as an increase in the DNA melting force. As shown with HMGB (above), this rise in the average melting force in the presence of the protein is due to additional force required to disrupt protein binding before DNA may be melted. This result has been observed in the presence of protein at salt concentrations ranging from 10 mM to 250 mM Na+, and the average melting force appears to be constant within uncertainty, while in the absence of protein the force varies strongly with salt concentration32,94 (data not shown).64 Primer extension assays have confirmed that the polymerase is active over these Na+ ranges as well.64
Isolating distinct binding sites
To determine the location of the DNA binding sites within α, fragments of the α subunit were constructed and purified. Force-induced melting experiments were performed in the presence of these constructs. Figure 8b shows the result of a series of extension/relaxation cycles for DNA stretched in the presence of the N-terminal construct α1-917. In contrast to data from the full length protein, this N-terminal construct shows no binding to ssDNA, within the uncertainty of the experiment. However, the stabilization of dsDNA is still evident. Therefore, the protein domains that bind to dsDNA and ssDNA must be distinct, and dsDNA binding is primarily found in this N-terminal region. The N-terminal construct α1-835 exhibits no detectable binding to either dsDNA or ssDNA (Figure 8c). Thus dsDNA binding is limited primarily to the residues 836-917, a region that contains the (HhH)2 motif.132 Similar experiments with additional constructs limit the ssDNA binding to a short region containing the OB-fold domain.
To further characterize DNA binding, full length α, the N-terminal constructs α1-917 and α1-835, and the C-terminal construct α917-1160 were titrated in DNA stretching experiments. The results for these binding experiments are shown for binding to dsDNA in Figure 9a, while binding to ssDNA is shown in Figure 9b. As mentioned above, binding to dsDNA is quantified by Equations 5 and 7. Binding to ssDNA is quantified by fits to the relaxation data with the linear combination model of Equation 3 (fits not shown). The contour length obtained from these fits (b) are proportional to the fraction of force-melted DNA that has been stabilized by the binding of protein (γss),64
FIGURE 9.

Equilibrium binding to DNA for various constructs of α. (a) Binding to dsDNA is characterized by the change in the observed melting force. Data was collected for full length α (blue), the N-terminal constructs α1-917 (green) and α1-835 (yellow), and the C-terminal construct α917-1160 (red). Error bars are the standard error of four extension cycles. Fits to the binding isotherm described in the text give the equilibrium association constant for full length α as Kds = 28 ± 7 × 106 M-1, while the N-terminal constructs α1-917 and α1-835 measured Kds = 3.5 ± 0.9 × 106 M-1 and Kds = 0.4 ± 0.1 × 106 M-1, respectively. The C-terminal fragment α917-1160 showed little binding and an upper limit was set of Kds < 0.03 × 106 M-1. (b) ssDNA binding was quantified by fits to the WLC and FJC models as described in the text. Data was collected for full length α (blue), the N-terminal constructs α1-917 (green) and α1-835 (yellow) and the C-terminal construct α917-1160 (red). Error bars are determined from uncertainties of the fits. Equilibrium association binding constants for full length α showed Kss = 22 ± 6 × 106 M-1 and α917-1160, which contains the putative OB-fold region, gave Kss = 10 ± 3 × 106 M-1. N-terminal constructs showed little binding to ssDNA. α1-917 and α1-835 were limited to Kss < 0.1 × 106 M-1 and Kss < 0.01 × 106 M-1, respectively. Figures are from McCauley, et al.64
| (10) |
Equilibrium association binding constants for full length α showed Kss = 22 ± 6 × 106 M-1 and Kds = 28 ± 7 × 106 M-1. The N-terminal construct α1-917 measured Kss < 0.1 × 106 M-1 and Kds = 3.5 ± 0.9 × 106 M-1, while α1-835 gave Kss < 0.01 × 106 M-1and Kds = 0.4 ± 0.1 × 106 M-1. The C-terminal fragment α917-1160 bound strongly to ssDNA, as Kss = 10 ± 3 × 106 M-1, though binding to dsDNA was weak, with an upper limit was set of Kds < 0.03 × 106 M-1.
Combined with the data described in the previous sections, the two distinct binding regions predicted by the models in Figure 7c and 7d have been confirmed. Binding to ssDNA is confined to the C-terminal region containing the OB-fold domain. The (HhH)2 motif is responsible for much of the binding activity to dsDNA. A recent co-crystal structure of the Thermus aquaticus α subunit with DNA supports this result and suggests that the OB-fold binds template DNA.141 Thus the binding to ssDNA might be expected to be passive, responding to ssDNA created during replication, in contrast to other ssDNA binding proteins, which can actively destabilize dsDNA, as shown in the final section.
Domains that Bind to Both Double and Single Stranded DNA
DNA replication studies have utilized two archetypal bacteriophage systems, T4 and T7, to identify and elucidate the roles of various subunits.142 Specific proteins, the T4 gene 32 protein (gp32) and T7 gene 2.5 protein (gp 2.5), were found to bind preferentially to ssDNA, serving to protect the single strand, prevent formation of secondary structures and coordinate with other replicative subunits and are also involved in recombination and DNA repair.129,143-150 Sharing a common domain structure, both proteins include an OB-fold consisting of aromatic residues which project from a basic core.151,152 Truncated structures of these proteins lack the acidic C-terminus and are known to bind more strongly to DNA; gp32 (*I) is shown in Figure 10a while gp2.5 - Δ26C is shown in Figure 10b. Details of the interaction of these proteins with DNA are unclear.153-155 The following experiments seek to quantify the salt-dependent ssDNA and dsDNA binding for the full length and truncated proteins, to determine the mechanics of binding.
FIGURE 10.
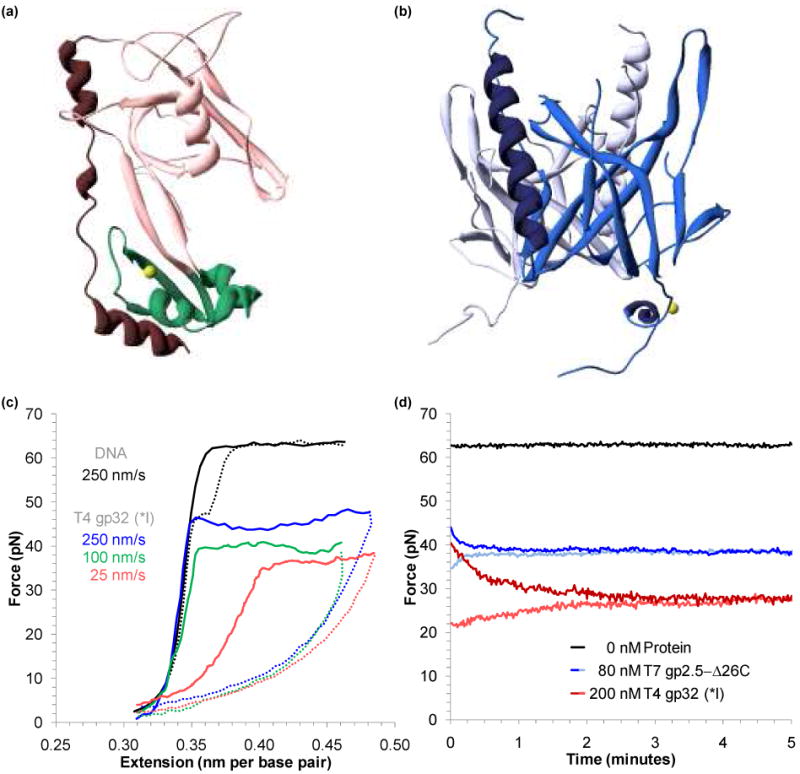
Finding the equilibrium melting force for proteins that bind to ssDNA and dsDNA. (a) Structure of the fragment of T4 gene 32 protein (gp32) known as *III, which excludes the N-terminal residues 1-20 (present in the fragment *I) and C-terminal residues 254-301 present in the full structure.152 The major lobe, containing an OB-fold, is shown in pink, while the minor, zinc – coordinating lobe is in green. A C-terminal binding flap is shown in red. DNA binds into the region between the two lobes (PDB code: 1GPC). (b) Structure of the fragment of the T7 gene 2.5 protein (gp2.5), which lacks 26 residues at the C-terminus that are believed to participate in the formation of the dimer (shown in blue and violet).151 Each 206 residue protein consists of a β-barrel (OB-fold, in cyan) and an α-helix (dark blue), while the long tail coordinates a calcium atom (yellow). DNA binds into the structure above center, interacting with structurally conserved aromatic residues that surround the basic core of the OB-fold (PDB code: 1JE5). (c) Extension (solid lines) and relaxation (dotted lines) for DNA (black) and DNA in the presence of 200 nM of T4gp32 (*I). The non-equilibrium melting force varies strongly with the pulling rate (250 nm/s in blue, 100 nm/s in green, and 25 nm/s in red). (d) The equilibrium melting force for DNA (black), T4gp32 (*I) in 100 mM Na+ (red) and T7gp2.5-Δ26C in 25 mM Na+ (blue) are found by holding a constant extension at the midpoint of the melting transition. The lightly shaded curves were collected by pausing during relaxation. Figures (c) and (d) adapted from Pant, et al.45 and Shokri, et al.48
Binding to ssDNA lowers the DNA melting force
While the data of Figure 10c clearly indicates binding to ssDNA by T4 gp32 (*I), this data reveals significant contrasts from the passive ssDNA binding data of the α subunit.43,45 Unlike the data shown in the previous sections, here the observed melting force significantly decreases and clearly varies with the velocity of DNA extension (the pulling rate). Thus *I actively destabilizes dsDNA. Furthermore, the stretching curves and the observed melting force are not in equilibrium. To find the equilibrium melting force, the DNA molecule is extended to the midpoint of the transition, and held fixed. Alternatively, the DNA molecule may be extended fully through the melting transition, and then relaxed to the midpoint. The observed force decreases (or increases) exponentially to reveal the equilibrium melting force for the truncated proteins shown in Figure 10d.
The equilibrium melting force (Fm) may be plotted for various protein concentrations and salt conditions, as for the truncated proteins in Figure 11a. A model based on thermal melting analysis may be fit to the data to determine Kssω, the cooperative equilibrium association binding constant to ssDNA (neglecting dsDNA binding);47,156,157
FIGURE 11.

Determining equilibrium binding to ssDNA and dsDNA for various salt concentrations. (a) Measured equilibrium melting force as a function of protein concentration in 100 mM, 75 mM and 50 mM Na+ (data points in green, yellow and red, respectively for T4 gp32 (*I) and violet, blue and cyan for T7 gp 2.5 - Δ26C). Fits shown are to Equation 11. (b) Fitted values of the equilibrium association binding constant (including cooperativity) for T7 gp2.5 (blue) and T4 gp32 (red) to ssDNA. Uncertainties are on the order of the symbols shown. (c) The melting force dependence upon the pulling rate. Selected data shows 50 nM (red), 100 nM (yellow) and 200 nM (green) of gp32 (*I) in 100 mM Na+, and 230 nM (cyan), 300 nM (blue) and 460 nM (violet) of gp2.5 – Δ26C in 50 mM Na+. DNA in the absence of protein is shown in black. Dotted lines represent fits to Equation 12. (d) Equilibrium association binding constant to dsDNA for T4 gp32 (red) and T7 gp2.5 (blue). Uncertainties are on the order of the symbols shown. Linear fits are shown as solid lines for wild type proteins and dotted lines for truncated fragments. Figures adapted from Pant, et al.44 and Shokri, et al.48
| (11) |
where c is the protein concentration, Δx is the length increase upon melting, and is the melting force in the absence of protein. The binding site size is generally assumed from previous studies to be nss = 7 base pairs, for each of the proteins studied. Fits to typical data are shown in Figure 11a, and the full spectrum of equilibrium association binding constants for the full length proteins and the truncated constructs for various salt concentrations are shown in Figure 11b.44,48
Varying the pulling rate reveals weak dsDNA binding
The rate dependence of the melting force reveals the kinetics of protein binding. In this regime, the pulling rate (υ) is faster than the rate at which fluctuations may be stabilized by ssDNA binding protein. Since this rate dependent force (Fk(υ)) is lower than the equilibrium force in the absence of protein , the binding that does occur takes place primarily at the ends of the DNA molecule. If the rate of a protein finding a binding site is ka, then the rate dependence of the force should be;43,44,48
| (12) |
Representative data for the truncated proteins and their fits are shown in Figure 11c, yielding values of ka in the range of 102 -104 s-1. Comparison of these rates with the 3D diffusion limit (109 M-1s-1) suggests that these proteins find the ssDNA binding site by first binding to dsDNA and diffusing along molecule to the ssDNA binding site.43,44,48 Assuming a sliding rate of ks ∼ 107 s-1, the binding site size (nds) and the occupancy (γ) will give the binding rate;
| (13) |
Equation 13, combined with the isotherm of Equation 5, may be fit to the data of the binding rate versus protein concentration (not shown, though all four of the proteins exhibit this binding effect), to determine the equilibrium association binding constant to dsDNA. Kds is shown in Figure 11d for each protein in various Na+ concentrations.44,48
Equilibrium binding to ssDNA and dsDNA
The clearest result in Figures 11b and 11d is the disparity between the equilibrium association binding constants to ssDNA and dsDNA. All four proteins studied exhibit binding to the ssDNA that is ∼ 10,000× stronger than dsDNA binding. Furthermore, the truncated fragments exhibit stronger binding than the wild type proteins, suggesting that the acidic C-terminus must be displaced before binding to either dsDNA or ssDNA. Yet gp32 is known to bind with high cooperativity (∼103), while gp2.5 shows almost none.158-161 Furthermore, gp2.5 is a dimer in solution, while gp32 is not. Thus direct binding with ssDNA is weaker for gp32, and unbinding from ssDNA is slower versus gp2.5. Finally, *I exhibits the strongest salt dependence indicating higher cationic charge of the core binding site for gp32 versus gp2.5.44
Conclusions
Stretching DNA and melting it by force in the presence of a binding ligand produces data with a signature unique to the mode of binding. Thus, nonspecific binding to dsDNA, DNA intercalation (a specific dsDNA binding mode), and ssDNA binding may be resolved when a particular ligand exhibits a combination of these binding modes, even if these modes are confined within the same binding site. The strength of the binding affinity for each mode can be quantified and the kinetics and thermodynamics of binding may be measured in some cases. Furthermore, by altering the structure of a binding ligand, either chemically or by expressing fragments of a full protein, it is possible to isolate the local DNA binding site. Finally, the changes to the force-extension and relaxation data provide quantitative insight into the function of DNA binding proteins.
Acknowledgments
The authors would like to thank P. R. Hardwidge and L. J. Maher, 3rd for providing HMGB1(box A+B) and HMGB2(box A). Furthermore, I. D. Vladescu and T. Paramanathan collected the data on [Ru(phen)2dppz]2+ and ΔΔ-P binding to DNA, respectively. We would also like to extend our gratitude to Megan E. Nuñez for providing [Ru(phen)2dppz]2+ samples and to Fredrik Westerlund and Per Lincoln for ΔΔ-P samples and data analysis. We are also grateful to P. J. Beuning, who provided full length α protein subunits, assisted with the interpretation of α-DNA binding experiments, and critically read this manuscript. We would also like to thank Kiran Pant, who collected the data on T4 gp 32 and T4 gp32 (*I). Additionally, we appreciate the work of L. Shokri who assisted with α protein purification, collected the data on glyoxal binding to DNA, as well as the data on T7 gp2.5 and T7gp2.5 – Δ26C. Finally, we gratefully thank I. Rouzina for analysis on DNA intercalation, ssDNA binding proteins, force melting and glyoxal modification as well as helpful discussions on all aspects of this work. This work was funded by NIH (GM75965) and NSF (MCB-0744456).
References
- 1.Franklin R, Gosling RG. Nature. 1953;171:740–741. doi: 10.1038/171740a0. [DOI] [PubMed] [Google Scholar]
- 2.Wilkins MHF, Stokes AR, Wilson HR. Nature. 1953;171:738–740. doi: 10.1038/171738a0. [DOI] [PubMed] [Google Scholar]
- 3.Watson JD, Crick FHC. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 4.Ashkin A, Dziedzic JM. Science. 1987;235:1517–1520. doi: 10.1126/science.3547653. [DOI] [PubMed] [Google Scholar]
- 5.Ashkin A, Dziedzic JM. Proc Natl Acad Sci U S A. 1989;86:7914–7918. doi: 10.1073/pnas.86.20.7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashkin A, Dziedzic JM, Bjorkholm JE, Chu S. Opt Lett. 1986;11:288–290. doi: 10.1364/ol.11.000288. [DOI] [PubMed] [Google Scholar]
- 7.Ashkin A, Dziedzic JM, Yamane T. Nature. 1987;330:769–771. doi: 10.1038/330769a0. [DOI] [PubMed] [Google Scholar]
- 8.Ashkin A, Schutze K, Dziedzic JM, Euteneuer U, Schliwa M. Nature. 1990;348:346–348. doi: 10.1038/348346a0. [DOI] [PubMed] [Google Scholar]
- 9.Smith SB, Finzi L, Bustamante C. Science. 1992;258:1122–1126. doi: 10.1126/science.1439819. [DOI] [PubMed] [Google Scholar]
- 10.Neuman KC, Block SM. Rev Sci Instrum. 2004;75:2787–2809. doi: 10.1063/1.1785844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang MD, Yin H, Landick R, Gelles J, Block SM. Biophys J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bustamante C, Bryant Z, Smith SB. Nature. 2003;421:423–427. doi: 10.1038/nature01405. [DOI] [PubMed] [Google Scholar]
- 13.van Oijen AM. Biopolymers. 2007;85:144–153. doi: 10.1002/bip.20624. [DOI] [PubMed] [Google Scholar]
- 14.Clausen-Schaumann H, Rief M, Tolksdorf C, Gaub HE. Biophys J. 2000;78:1997–2007. doi: 10.1016/S0006-3495(00)76747-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Block SM, Goldstein LS, Schnapp BJ. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- 16.McCauley MJ, Williams MC. Biopolymers. 2007;85:154–168. doi: 10.1002/bip.20622. [DOI] [PubMed] [Google Scholar]
- 17.Smith SB, Cui Y, Bustamante C. Methods Enzymol. 2003;361:134–162. doi: 10.1016/s0076-6879(03)61009-8. [DOI] [PubMed] [Google Scholar]
- 18.Williams MC. In: Biophysics Textbook Online. Schwille P, editor. Biophysical Society; Bethesda, MD: 2002. [Google Scholar]
- 19.Wenner JR, Williams MC, Rouzina I, Bloomfield VA. Biophys J. 2002;82:3160–3169. doi: 10.1016/S0006-3495(02)75658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumann CG, Smith SB, Bloomfield VA, Bustamante C. Proc Natl Acad Sci U S A. 1997;94:6185–6190. doi: 10.1073/pnas.94.12.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Odijk T. Macromolecules. 1995;28:7016–7018. [Google Scholar]
- 22.Podgornik R, Hansen PL, Parsegian PA. J Chem Phys. 2000;113:9343–9350. [Google Scholar]
- 23.Marko JF, Siggia ED. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 24.Bouchiat C, Wang MD, Allemand JF, Strick TR, Block SM, Croquette V. Biophys J. 1999;76:409–413. doi: 10.1016/s0006-3495(99)77207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cloutier TE, Widom J. Mol Cell. 2004;14:355–362. doi: 10.1016/s1097-2765(04)00210-2. [DOI] [PubMed] [Google Scholar]
- 26.Du Q, Smith C, Shiffeldrim N, Vologodskaia M, Vologodskii A. Proc Natl Acad Sci U S A. 2005;102:5397–5402. doi: 10.1073/pnas.0500983102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiggins PA, van der Heijden T, Moreno-Herrero F, Spakowitz A, Phillips R, Widom J, Dekker C, Nelson PC. Nat Nanotechnol. 2006;1:137–141. doi: 10.1038/nnano.2006.63. [DOI] [PubMed] [Google Scholar]
- 28.Podgornik R, Hansen PL, Parsegian PA. J Chem Phys. 2000;113:9343–9350. [Google Scholar]
- 29.Smith SB, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 30.Smith SB, Cui YJ, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 31.Bustamante C, Marko JF, Siggia ED, Smith S. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 32.Rouzina I, Bloomfield VA. Biophys J. 2001;80:894–900. doi: 10.1016/S0006-3495(01)76068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rouzina I, Bloomfield VA. Biophys J. 2001;80:882–893. doi: 10.1016/S0006-3495(01)76067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams MC, Wenner JR, Rouzina I, Bloomfield VA. Biophys J. 2001;80:1932–1939. doi: 10.1016/S0006-3495(01)76163-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams MC, Wenner JR, Rouzina I, Bloomfield VA. Biophys J. 2001;80:874–881. doi: 10.1016/S0006-3495(01)76066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hegner M, Smith SB, Bustamante C. Proc Natl Acad Sci U S A. 1999;96:10109–10114. doi: 10.1073/pnas.96.18.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 38.Williams MC, Rouzina I, Bloomfield VA. Acc Chem Res. 2002;35:159–166. doi: 10.1021/ar010045k. [DOI] [PubMed] [Google Scholar]
- 39.Harris SA, Sands ZA, Laughton CA. Biophys J. 2005;88:1684–1691. doi: 10.1529/biophysj.104.046912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heng JB, Aksimentiev A, Ho C, Marks P, Grinkova YV, Sligar S, Schulten K, Timp G. Biophys J. 2006;90:1098–1106. doi: 10.1529/biophysj.105.070672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piana S. Nucleic Acids Res. 2005;33:7029–7038. doi: 10.1093/nar/gki1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krautbauer R, Fischerländer S, Allen S, Gaub HE. Single Mol. 2002;3:97–103. [Google Scholar]
- 43.Pant K, Karpel RL, Rouzina I, Williams MC. J Mol Biol. 2004;336:851–870. doi: 10.1016/j.jmb.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 44.Pant K, Karpel RL, Rouzina I, Williams MC. J Mol Biol. 2005;349:317–330. doi: 10.1016/j.jmb.2005.03.065. [DOI] [PubMed] [Google Scholar]
- 45.Pant K, Karpel RL, Williams MC. J Mol Biol. 2003;327:571–578. doi: 10.1016/s0022-2836(03)00153-0. [DOI] [PubMed] [Google Scholar]
- 46.Rouzina I, Pant K, Karpel RL, Williams MC. Biophys J. 2005;89:1941–1956. doi: 10.1529/biophysj.105.063776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shokri L, Marintcheva B, Richardson CC, Rouzina I, Williams MC. J Biol Chem. 2006;281:38689–38696. doi: 10.1074/jbc.M608460200. [DOI] [PubMed] [Google Scholar]
- 48.Shokri L, Marintcheva B, Eldib M, Hanke A, Rouzina I, Williams MC. Nucleic Acids Res. 2008;17:5668–5677. doi: 10.1093/nar/gkn551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konrad MW, Bolonick JI. J Am Chem Soc. 1996;118:10989–10994. [Google Scholar]
- 50.Kosikov KM, Gorin AA, Zhurkin VB, Olson WK. J Mol Biol. 1999;289:1301–1326. doi: 10.1006/jmbi.1999.2798. [DOI] [PubMed] [Google Scholar]
- 51.Lebrun A, Lavery R. Nucleic Acids Res. 1996;24:2260–2267. doi: 10.1093/nar/24.12.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cluzel P, Lebrun A, Heller C, Lavery R, Viovy JL, Chatenay D, Caron F. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 53.Storm C, Nelson PC. Phys Rev E. 2003;67:051906. doi: 10.1103/PhysRevE.67.051906. [DOI] [PubMed] [Google Scholar]
- 54.Cizeau P, Viovy JL. Biopolymers. 1997;42:383–385. [Google Scholar]
- 55.Albrecht CH, Neuert G, Lugmaier RA, Gaub HE. Biophys J. 2008;94:4766–4774. doi: 10.1529/biophysj.107.125427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cocco S, Yan J, Leger JF, Chatenay D, Marko JF. Phys Rev E. 2004;70:011910. doi: 10.1103/PhysRevE.70.011910. [DOI] [PubMed] [Google Scholar]
- 57.Strick TR, Dessinges MN, Charvin G, Dekker NH, Allemand JF, Bensimon D, Croquette V. Rep Prog Phys. 2003;66:1–45. [Google Scholar]
- 58.Whitelam S, Pronk S, Geissler PL. Biophys J. 2008;94:2452–2469. doi: 10.1529/biophysj.107.117036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hutton JR, Wetmur JG. Biochemistry. 1973;12:558–563. doi: 10.1021/bi00727a032. [DOI] [PubMed] [Google Scholar]
- 60.Borovik AS, Kalambet YA, Lyubchenko YL, Shitov VT, Golovanov EI. Nucleic Acids Res. 1980;8:4165–4184. doi: 10.1093/nar/8.18.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Borovik AS, Liubchenko Iu L, Naroditskii BS, Tikhonenko TI. Mol Biol (Mosk) 1984;18:1634–1638. [PubMed] [Google Scholar]
- 62.Broude NE, Budowsky EI. Biochim Biophys Acta. 1971;254:380–388. doi: 10.1016/0005-2787(71)90868-9. [DOI] [PubMed] [Google Scholar]
- 63.Broude NE, Budowsky EI. Biochim Biophys Acta. 1973;294:378–384. doi: 10.1016/0005-2787(73)90092-0. [DOI] [PubMed] [Google Scholar]
- 64.McCauley MJ, Shokri L, Sefcikova J, Venclovas C, Beuning PJ, Williams MC. ACS Chem Biol. 2008;3:577–587. doi: 10.1021/cb8001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shokri L, McCauley MJ, Rouzina I, Williams MC. Biophys J. 2008;95:1248–1255. doi: 10.1529/biophysj.108.132688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Williams MC, Rouzina I. Curr Opin Struct Biol. 2002;12:330–336. doi: 10.1016/s0959-440x(02)00340-8. [DOI] [PubMed] [Google Scholar]
- 67.Bustin M. Mol Cell Biol. 1999;19:5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bustin M. Trends Biochem Sci. 2001;26:152–153. doi: 10.1016/s0968-0004(00)01777-1. [DOI] [PubMed] [Google Scholar]
- 69.Bustin M, Reeves R. Prog Nucleic Acid Res Mol Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 70.Johns E, editor. HMG Chromosomal Proteins. Academic Press; New York: 1982. [Google Scholar]
- 71.Catez F, Yang H, Tracey KJ, Reeves R, Misteli T, Bustin M. Mol Cell Biol. 2004;24:4321–4328. doi: 10.1128/MCB.24.10.4321-4328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ragab A, Travers A. Nucleic Acids Res. 2003;31:7083–7089. doi: 10.1093/nar/gkg923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bianchi ME, Beltrame M. EMBO Reports. 2000;1:109–114. doi: 10.1093/embo-reports/kvd030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.LeRoy G, Orphanides G, Lane WS, Reinberg D. Science. 1998;282:1900–1904. doi: 10.1126/science.282.5395.1900. [DOI] [PubMed] [Google Scholar]
- 75.Ross ED, Hardwidge PR, Maher LJ., 3rd Mol Cell Biol. 2001;21:6598–6605. doi: 10.1128/MCB.21.19.6598-6605.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Agresti A, Bianchi ME. Curr Opin Genet Dev. 2003;13:170–178. doi: 10.1016/s0959-437x(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 77.Bianchi ME, Beltrame M. Am J Hum Genet. 1998;63:1573–1577. doi: 10.1086/302170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jayaraman L, Moorthy NC, Murthy KG, Manley JL, Bustin M, Prives C. Genes Dev. 1998;12:462–472. doi: 10.1101/gad.12.4.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kruppa M, Moir RD, Kolodrubetz D, Willis IM. Mol Cell. 2001;7:309–318. doi: 10.1016/s1097-2765(01)00179-4. [DOI] [PubMed] [Google Scholar]
- 80.Laser H, Bongards C, Schuller J, Heck S, Johnsson N, Lehming N. Proc Natl Acad Sci U S A. 2000;97:13732–13737. doi: 10.1073/pnas.250400997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mitsouras K, Wong B, Arayata C, Johnson RC, Carey M. Mol Cell Biol. 2002;22:4390–4401. doi: 10.1128/MCB.22.12.4390-4401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paull TT, Carey M, Johnson RC. Genes Dev. 1996;10:2769–2781. doi: 10.1101/gad.10.21.2769. [DOI] [PubMed] [Google Scholar]
- 83.Vijayanathan V, Thomas T, Shirahata A, Thomas TJ. Biochemistry. 2001;40:13644–13651. doi: 10.1021/bi010993t. [DOI] [PubMed] [Google Scholar]
- 84.Lange SS, Mitchell DL, Vasquez KM. Proc Natl Acad Sci U S A. 2008;105:10320–10325. doi: 10.1073/pnas.0803181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lotze MT, Tracey KJ. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 86.Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. Trends Immunol. 2005;26:381–387. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 87.Murphy FVI, Sweet RM, Churchill ME. EMBO Journal. 1999;18:6610–6618. doi: 10.1093/emboj/18.23.6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stott K, Tang GSF, Lee KB, Thomas JO. J Mol Bio. 2006;360:90–104. doi: 10.1016/j.jmb.2006.04.059. [DOI] [PubMed] [Google Scholar]
- 89.Murphy FVI, Sweet RM, Churchill ME. EMBO J. 1999;18:6610–6618. doi: 10.1093/emboj/18.23.6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klass J, Murphy FI, Fouts S, Serenil M, Changela A, Siple J, Churchill ME. Nucleic Acids Res. 2003;31:2852–2864. doi: 10.1093/nar/gkg389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McCauley MJ, Zimmerman J, Maher LJ, 3rd, Williams MC. J Mol Biol. 2007;374:993–1004. doi: 10.1016/j.jmb.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Churchill ME, Changela A, Dow LK, Krieg AJ. Methods Enzymol. 1999;304:99–103. doi: 10.1016/s0076-6879(99)04009-4. [DOI] [PubMed] [Google Scholar]
- 93.McCauley M, Hardwidge PR, Maher LJ, 3rd, Williams MC. Biophys J. 2005;89:353–364. doi: 10.1529/biophysj.104.052068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rouzina I, Bloomfield VA. Biophys J. 1998;74:3152–3164. doi: 10.1016/S0006-3495(98)78021-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McGhee JD, von Hippel PH. J Mol Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 96.McGhee JD. Biopolymers. 1976;15:1345–1375. doi: 10.1002/bip.1976.360150710. [DOI] [PubMed] [Google Scholar]
- 97.Schellman JA. Biopolymers. 1974;13:217–226. doi: 10.1002/bip.1974.360130115. [DOI] [PubMed] [Google Scholar]
- 98.Hardwidge PR, Kahn JD, Maher LJ, 3rd, Ross ED. Biochemistry. 2002;41:8277–8288. doi: 10.1021/bi020185h. [DOI] [PubMed] [Google Scholar]
- 99.Hardwidge PR, Parkhurst KM, Parkhurst LJ, Maher LJ., 3rd Biopolymers. 2003;69:110–117. doi: 10.1002/bip.10321. [DOI] [PubMed] [Google Scholar]
- 100.Dragan AI, Read CM, Makeyeva EN, Milgotina EI, Churchill ME, Crane-Robinson C, Privalov PL. J Mol Biol. 2004;343:371–393. doi: 10.1016/j.jmb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 101.Cruceanu M, Gorelick RJ, Musier-Forsyth K, Rouzina I, Williams MC. J Mol Biol. 2006;363:867–877. doi: 10.1016/j.jmb.2006.08.070. [DOI] [PubMed] [Google Scholar]
- 102.Cruceanu M, Urbaneja MA, Hixson CV, Johnson DG, Datta SA, Fivash MJ, Stephen AG, Fisher RJ, Gorelick RJ, Casas-Finet JR, Rein A, Rouzina I, Williams MC. Nucleic Acids Res. 2006;34:593–605. doi: 10.1093/nar/gkj458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Crothers DM, Metzger H. Immunochemistry. 1972;9:341–357. doi: 10.1016/0019-2791(72)90097-3. [DOI] [PubMed] [Google Scholar]
- 104.Cui T, Wei S, Brew K, Leng F. J Mol Biol. 2005;325:629–645. doi: 10.1016/j.jmb.2005.07.048. [DOI] [PubMed] [Google Scholar]
- 105.Grasser KD, Teo SH, Lee KB, Broadhurst RW, Rees C, Hardman CH, Thomas JO. Eur J Biochem. 1998;253:787–795. doi: 10.1046/j.1432-1327.1998.2530787.x. [DOI] [PubMed] [Google Scholar]
- 106.Paull TT, Haykinson MJ, Johnson RC. Genes Dev. 1993;7:1521–1534. doi: 10.1101/gad.7.8.1521. [DOI] [PubMed] [Google Scholar]
- 107.Van Noort J, Verbrugge S, Goosen N, Dekker C, Dame RT. Proc Natl Acad Sci U S A. 2004;101:6969–6974. doi: 10.1073/pnas.0308230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lerman LS. J Mol Biol. 1961;3:18–30. doi: 10.1016/s0022-2836(61)80004-1. [DOI] [PubMed] [Google Scholar]
- 109.Waring MJ. Annu Rev Biochem. 1981;50:159–192. doi: 10.1146/annurev.bi.50.070181.001111. [DOI] [PubMed] [Google Scholar]
- 110.Muller W, Crothers DM. J Mol Biol. 1968;35:251–290. doi: 10.1016/s0022-2836(68)80024-5. [DOI] [PubMed] [Google Scholar]
- 111.Friedman AE, Chambron JC, Sauvage JP, Turro NJ, Barton JK. J Am Chem Soc. 1990;112:4960–4962. [Google Scholar]
- 112.Mihailovic A, Vladescu I, McCauley M, Ly E, Williams MC, Spain EM, Nunez ME. Langmuir. 2006;22:4699–4709. doi: 10.1021/la053242r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dupureur CM, Barton JK. Inorg Chem. 1997;36:33–43. [Google Scholar]
- 114.Haq I, Lincoln P, Suh D, Nordén B, Chowdhry B, Chaires JB. J Am Chem Soc. 1995;117:4788–4796. [Google Scholar]
- 115.Vladescu I, McCauley M, Nunez ME, Rouzina I, Williams MC. Nat Meth. 2007;4:517–522. doi: 10.1038/nmeth1044. [DOI] [PubMed] [Google Scholar]
- 116.Lincoln P, Nordén B. J Phys Chem B. 1998;102:9583–9594. [Google Scholar]
- 117.Pyle AM, Rehmann JP, Meshoyrer R, Kumar CV, Turro NJ, Barton JK. J Am Chem Soc. 1989;111:3051–3058. [Google Scholar]
- 118.Satyanarayana S, Dabrowaik JC, Chaires JB. Biochemistry. 1992;31:9319–9324. doi: 10.1021/bi00154a001. [DOI] [PubMed] [Google Scholar]
- 119.Vladescu ID, McCauley MJ, Rouzina I, Williams MC. Phys Rev Lett. 2005;95:158102. doi: 10.1103/PhysRevLett.95.158102. [DOI] [PubMed] [Google Scholar]
- 120.Berman HM, Young PR. Ann Rev Biophysics Bioeng. 1981;10:87–114. doi: 10.1146/annurev.bb.10.060181.000511. [DOI] [PubMed] [Google Scholar]
- 121.Mahadevan S, Palaniandavar M. Bioconjug Chem. 1996;7:138–143. doi: 10.1021/bc950090a. [DOI] [PubMed] [Google Scholar]
- 122.Lincoln P, Norden B. Chem Commun. 1996:2145–2146. [Google Scholar]
- 123.Wilhelmsson LM, Westerlund F, Lincoln P, Norden B. J Am Chem Soc. 2002;124:12092–12093. doi: 10.1021/ja027252f. [DOI] [PubMed] [Google Scholar]
- 124.Nordell P, Westerlund F, Wilhelmsson LM, Norden B, Lincoln P. Angewandte Chemie International ed. 2007;46:2203–2206. doi: 10.1002/anie.200604294. [DOI] [PubMed] [Google Scholar]
- 125.Westerlund F, Nordell P, Norden B, Lincoln P. J Phys Chem B. 2007;111:9132–9137. doi: 10.1021/jp072126p. [DOI] [PubMed] [Google Scholar]
- 126.Westerlund F, Wilhelmsson LM, Norden B, Lincoln P. J Am Chem Soc. 2003;125:3773–3779. doi: 10.1021/ja029243c. [DOI] [PubMed] [Google Scholar]
- 127.Paramanathan T, Westerlund F, McCauley MJ, Rouzina I, Lincoln P, Williams MC. J Am Chem Soc. 2008;130:3752–3753. doi: 10.1021/ja711303p. [DOI] [PubMed] [Google Scholar]
- 128.Johnson A, O'Donnell M. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 129.Kornberg A, Baker TA. DNA Replication. W.H. Freeman & Company; New York: 1992. [Google Scholar]
- 130.Kelman Z, O'Donnell M. Annu Rev Biochem. 1995;64:171–200. doi: 10.1146/annurev.bi.64.070195.001131. [DOI] [PubMed] [Google Scholar]
- 131.Rothwell PJ, Waksman G. Adv Protein Chem. 2005;71:401–440. doi: 10.1016/S0065-3233(04)71011-6. [DOI] [PubMed] [Google Scholar]
- 132.Lamers MH, Georgescu RE, Lee SG, O'Donnell M, Kuriyan J. Cell. 2006;126:881–892. doi: 10.1016/j.cell.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 133.Bailey S, Wing RA, Steitz TA. Cell. 2006;126:893–904. doi: 10.1016/j.cell.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 134.Leipe DD, Aravind L, Koonin EV. Nucleic Acids Res. 1999;27:3389–3401. doi: 10.1093/nar/27.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Doherty AJ, Serpell LC, Ponting CP. Nucleic Acids Res. 1996;24:2488–2497. doi: 10.1093/nar/24.13.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shao X, Grishin NV. Nucleic Acids Res. 2000;28:2643–2650. doi: 10.1093/nar/28.14.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Arcus V. Curr Opin Struct Biol. 2002;12:794–801. doi: 10.1016/s0959-440x(02)00392-5. [DOI] [PubMed] [Google Scholar]
- 138.Murzin AG. EMBO J. 1993;12:861–867. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Theobald DL, Mitton-Fry RM, Wuttke DS. Annu Rev Biophys Biomol Struct. 2003;32:115–133. doi: 10.1146/annurev.biophys.32.110601.142506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhao XQ, Hu JF, Yu J. Genomics Proteomics and Bioinf. 2006;4:203–211. doi: 10.1016/S1672-0229(07)60001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wing RA, Bailey S, Steitz TA. J Mol Biol. 2008;382:859–869. doi: 10.1016/j.jmb.2008.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Benkovic SJ, Valentine AM, Salinas F. Annu Rev Biochem. 2001;70:181–208. doi: 10.1146/annurev.biochem.70.1.181. [DOI] [PubMed] [Google Scholar]
- 143.Alberts BM, Frey L. Nature. 1970;227:1313–1318. doi: 10.1038/2271313a0. [DOI] [PubMed] [Google Scholar]
- 144.Karpel RL. T4 Bacteriophage Gene 32 Protein. CRC Press; Boca Raton: 1990. [Google Scholar]
- 145.Karpel RL, Henderson LE, Oroszlan S. J Biol Chem. 1987;262:4961–4967. [PubMed] [Google Scholar]
- 146.Kim YT, Richardson CC. J Biol Chem. 1994;269:5270–5278. [PubMed] [Google Scholar]
- 147.Kim YT, Tabor S, Churchich JE, Richardson CC. J Biol Chem. 1992;267:15032–15040. [PubMed] [Google Scholar]
- 148.Kong D, Richardson CC. EMBO J. 1996;15:2010–2019. [PMC free article] [PubMed] [Google Scholar]
- 149.Notarnicola SM, Mulcahy HL, Lee J, Richardson CC. J Biol Chem. 1997;272:18425–18433. doi: 10.1074/jbc.272.29.18425. [DOI] [PubMed] [Google Scholar]
- 150.Rezende LF, Willcox S, Griffith JD, Richardson CC. J Biol Chem. 2003;278:29098–29105. doi: 10.1074/jbc.M303374200. [DOI] [PubMed] [Google Scholar]
- 151.Hollis T, Stattel JM, Walther DS, Richardson CC, Ellenberger T. Proc Natl Acad Sci U S A. 2001;98:9557–9562. doi: 10.1073/pnas.171317698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shamoo Y, Friedman AM, Parsons MR, Konigsberg WH, Steitz TA. Nature. 1995;376:362–366. doi: 10.1038/376362a0. [DOI] [PubMed] [Google Scholar]
- 153.Jensen DE, Kelly RC, von Hippel PH. J Biol Chem. 1976;251:7215–7228. [PubMed] [Google Scholar]
- 154.von Hippel PH, Delagoutte E. Cell. 2001;104:177–190. doi: 10.1016/s0092-8674(01)00203-3. [DOI] [PubMed] [Google Scholar]
- 155.Waidner LA, Flynn EK, Wu M, Li X, Karpel RL. J Biol Chem. 2001;276:2509–2516. doi: 10.1074/jbc.M007778200. [DOI] [PubMed] [Google Scholar]
- 156.Frank-Kamenetskii MD, Anshelevich VV, Lukashin AV. Sov Phys. 1987;151:595–618. [Google Scholar]
- 157.Jensen DE, von Hippel PH. J Biol Chem. 1976;251:7198–7214. [PubMed] [Google Scholar]
- 158.Kim YT, Tabor S, Bortner C, Griffith JD, Richardson CC. J Biol Chem. 1992;267:15022–15031. [PubMed] [Google Scholar]
- 159.Kowalczykowski SC, Lonberg N, Newport JW, Paul LS, von Hippel PH. Biophys J. 1980;32:403–418. doi: 10.1016/S0006-3495(80)84964-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kowalczykowski SC, Lonberg N, Newport JW, von Hippel PH. J Mol Biol. 1981;145:75–104. doi: 10.1016/0022-2836(81)90335-1. [DOI] [PubMed] [Google Scholar]
- 161.Lonberg N, Kowalczykowski SC, Paul LS, von Hippel PH. J Mol Biol. 1981;145:123–138. doi: 10.1016/0022-2836(81)90337-5. [DOI] [PubMed] [Google Scholar]
- 162.Bochkareva E, Belegu V, Korolev S, Bochkarev A. EMBO J. 2001;20:612–618. doi: 10.1093/emboj/20.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Singleton MR, Scaife S, Wigley DB. Cell. 2001;107:79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]