

Abstract
Translesion synthesis (TLS) is a unique DNA damage tolerance mechanism involved in the replicative bypass of genetic lesions in favor of uninterrupted DNA replication. TLS is critical for the generation of mutations by many different chemical and physical agents, however, there is no information available regarding the role of TLS in carcinogenic metal-induced mutagenesis. Hexavalent chromium (Cr(VI))-containing compounds are highly complex genotoxins possessing both mutagenic and clastogenic activities. The focus of this work was to determine the impact that TLS has on Cr(VI)-induced mutagenesis in S. cerevisiae. Wild-type yeast and strains deficient in TLS polymerases (i.e. Polζ (rev3), Polη (rad30)) were exposed to Cr(VI) and monitored for cell survival and forward mutagenesis at the CAN1 locus. In general, TLS deficiency had little impact on Cr(VI)-induced clonogenic lethality or cell growth. rad30 yeast exhibited higher levels of basal and induced mutagenesis compared to Wt and rev3 yeast. In contrast, rev3 yeast displayed attenuated Cr(VI)-induced mutagenesis. Moreover, deletion of REV3 in rad30 yeast (rad30 rev3) resulted in a significant decrease in basal and Cr(VI) mutagenesis relative to Wt and rad30 single mutants indicating that mutagenesis primarily depended upon Polζ. Interestingly, rev3 yeast were similar to Wt yeast in susceptibility to Cr(VI)-induced frameshift mutations. Mutational analysis of the CAN1 gene revealed that Cr(VI)-induced base substitution mutations accounted for 83.9% and 100.0% of the total mutations in Wt and rev3 yeast, respectively. Insertions and deletions comprised 16.1% of the total mutations in Cr(VI) treated Wt yeast but were not observed rev3 yeast. This work provides novel information regarding the molecular mechanisms of Cr(VI)-induced mutagenesis and is the first report demonstrating a role for TLS in the fixation of mutations induced by a carcinogenic metal.
1. Introduction
The past several years have witnessed a tremendous increase in our knowledge of the cellular mechanisms involved in DNA damage tolerance. While this process is complex, translesion synthesis (TLS) is believed to play a central role in the tolerance of a wide variety of DNA base damage [1-3]. TLS involves the deployment of certain DNA polymerases that can accommodate a diverse array of damaged DNA bases [4-8] and mismatches [9] into their catalytic sites. In contrast to the major replicative DNA polymerases (i.e. Polδ), TLS polymerases are devoid of 3′-5′ proofreading activity and exhibit decreased processivity [10]. In S. cerevisiae, the TLS polymerases ζ, η and Rev1 are primarily involved in lesion bypass [3].
Rev1, Rev3 and Rev7 belong to the Rad6 epistasis group in yeast and the loss of any of these genes results in attenuated UV mutagenesis. Both Pols η and Rev1 belong to the Y-family of DNA polymerases [1]. Polη facilitates the bypass of several lesions including UV dimers [7-11], oxidized bases [12], cisplatin adducts [4] and thymine glycol [13]. In yeast, REV1, a deoxycytidyl transferase [14], displays bypass activity across UV DNA damage [15]. In addition, REV1, a phosphoprotein [16], might serve as a scaffold on which other TLS polymerases [17-18] and accessory proteins [19] interact at the site of replication arrest. Polζ, a B-family DNA polymerase, consists of a catalytic subunit, REV3, and a second subunit REV7 [18;20-22]. Polζ is generally considered to function in the extension phase of TLS [23]. Consistent with this, rev3 yeast are less susceptible to mutagenesis induced by a wide range of mutagens including UV [24], alkylating agents [25] and cisplatin [26]. The deployment of TLS polymerases at sites of replication arrest is believed to be controlled by the post-translational modification (i.e. ubiquitination) of proliferating cell nuclear antigen (PCNA) and 9-1-1 complex (reviewed in [1]) in yeast. It is postulated that Rev1 plays a central role in TLS [20] at the replication fork through its interactions with PCNA [19], Polη [27] and Polζ [9;28].
Some forms of hexavalent chromium [Cr(VI) ]-containing compounds are human respiratory carcinogens. The intracellular reduction of Cr(VI) to Cr(III) leads to the generation of oxidized DNA bases [29-31], Cr-DNA interstrand crosslinks (ICLs) [32;33] and DNA-protein crosslinks (DPCs) [34;35]. Moreover, Cr-DNA damage seems to involve the phosphodiester backbone [35] and has been reported to arrest DNA replication in vitro [33]. Reduced forms of Cr(VI) (i.e. Cr(V), Cr(III)), generate lesions that serve as a substrates for nucleotide excision repair (NER) [36-38] and base excision repair (BER) [39]. Cr(VI) treatment also induces the formation of DNA double-strand breaks in cells [40]. Accordingly, DSB repair via homologous recombination plays an important role in promoting cell survival after Cr(VI) insult in yeast [41]. NER has been shown to protect against Cr mutagenesis in mammalian cells using a shuttle vector system [42]. However, recent work has revealed that NER might play a role in Cr(VI) mutagenesis and clastogenesis in mammalian cells [43]. Consequently, the precise molecular mechanisms involved in Cr(VI) mutagenesis are still unclear.
Error-prone DNA damage tolerance mechanisms such as TLS play an important role in facilitating chemical mutagenesis. However, no information is available regarding the impact that TLS has on carcinogenic metal-induced mutagenesis. Given that several lesions generated by Cr(VI) reduction are DNA replication-arresting [44], we sought to determine whether the TLS-mediated bypass of this damage impacted Cr(VI) mutagenesis. Using a genetic approach in S. cerevisiae, we found that Cr(VI) mutagenesis was dependent on Polζ, but not Polη, -mediated TLS. Moreover, Cr(VI)-induced DNA insertions and deletions were not observed in rev3 (Polζ-deficient) yeast suggesting that these mutations are generated by TLS involving Polζ. To the best of our knowledge, this is the first report to demonstrate a role for TLS in carcinogenic metal-induced mutagenesis. The data also provide valuable insight into the molecular mechanisms of Cr(VI)-induced mutagenesis.
2. Materials and Methods
2.1 Yeast Strains and Methods
Yeast strains used in this study were either from the SJR0751 (MATα ade2-101oc his3Δ200 ura3ΔNco lys2ΔBgl leu2-R) or BY4741 (MATa his3 leu2 met15 ura3) (ATCC, Manassas, VA) haploid yeast backgrounds. The SJR strains were a kind gift of Dr. Sue Jinks-Robertson [45]. All strains were maintained on YEPD medium containing essential amino acids and nutrients.
2.2 S. cerevisiae Survival
Cell survival was performed as we have previously described [41]. Briefly, overnight cultures (0.5 mL) were subcultured in 4.0 ml of fresh YEPD and cells were grown to mid-log phase (∼2×107 cells/ml) for ∼2 h. Cultures were then incubated for 2 h with the indicated concentration of Cr(VI) or MMS. After treatment, cells were collected by centrifugation and washed with 1× PBS. Approximately 400 cells were plated onto YEPC medium and allowed to grow for 3 days. Colonies were counted and data were expressed as percent survival of control (untreated) cultures.
2.3 Mutagenesis and Sequencing
Cultures were treated under the same conditions as the survival assays and washed in 1×PBS. Cultures were plated for clonogenic survival immediately after treatment and an aliquot (100 ul) was added to fresh YEPD broth and allowed to recover for ∼18 h to saturation. Cultures (1×107 cells) were then plated onto synthetic complete (SC)-media lacking arginine (SC-Arg) and containing L-canavanine (60 mg/ml) for up to 3-5 days at 30°C. Appropriate dilutions were also grown on YEPD plates for determining viability/plating efficiency. Spontaneous mutation frequencies were calculated using fluctuation analysis and the method of the median [46]. In general, the WT SJR strain (1.5×10-6) displayed a lower background mutation frequency than BY4741 (2.1×10-7) yeast. Colonies (L-canavanineR) were counted and the forward mutation frequency was calculated and expressed as the number of L-canR colonies/107 viable cells after correcting for spontaneous mutation frequency. DNA from L-canavanine-resistant clones was isolated using a commercial kit (MasterpureTM, Epicentre, Madison, WI). In yeast, the CAN1 gene (∼2 kb) was amplified by PCR using intron-based primers and samples were sequenced using the primers in Table I. Samples were cycle sequenced using Big Dye Terminator chemistry (Applied Biosystems, Foster City, CA) and resolved on an ABI Prism 310 Genetic Analyzer. For frameshift mutagenesis, yeast were processed exactly as described for CAN1 mutagenesis. After recovery, ∼1×107 cells were plated onto SC-Lys media and allowed to incubate for 5-7 days at 30°C. Reversion frequency at the lys2ΔBgl locus [45] was determined by the number of colonies observed on SC-Lys plates corrected for plating efficiency.
Table 1.
Primers used for PCR and sequencing
| Primer Set | Sequence (5′→3′) | Application |
|---|---|---|
| CAN1-5′ | CAGAGTAAACCGAATCAGGG | PCR* |
| CAN1-3′ | GCGTGGAAATGTGATCAAAGGT | PCR* |
| CanR1 | GTAACAGGGATGAATGTAGCC | Sequencing |
| CanR3 | GCCTGCAACACCAGTGAT | Sequencing |
| CanR2 | CCTTTGTACCAGAGTTCTC | Sequencing |
| CanR4 | CTCTCGTCGGGAAAGAGCGCAATG | Sequencing |
| CanF2 | GGCATATTCTGTCACGCAGT | Sequencing |
Intron-based primers
2.4 Statistical Analysis
A Student's t test (P<0.05) was used to compare differences of means between wild-type and mutant strains. Analysis of variance (ANOVA) was utilized for establishing the dose-effect within each strain (GraphPad Prism, GraphPad Software Inc., San Diego, CA). Mutational spectra were compared using publicly available software (http://www.ibiblio.org/dnam/mainpage.html) described in [47]. A Fisher's exact test was used for mutational analysis.
3. Results
3.1. TLS deficiency does not dramatically impact cell survival after Cr(VI) treatment
Previous work has suggested that TLS functions as a pro-survival pathway after genotoxic insult [48;49]. In order to determine whether TLS is important for cell survival after Cr(VI) treatment, we exposed Wt (BY4741 background), rev3 (Polζ-deficient), rad30 (Polη-deficient), and rev3 rad30 yeast to 1-10 mM Cr(VI) for 2 h and measured clonogenic survival. Rev3 is the catalytic subunit of Polζ and is known to interact with Rev1 [50]. Rad30, or Polη, is homologous to human XPV [51], also interacts with Rev1 [27] and is particularly efficient at bypassing UV-induced dimers [52]. As can be seen in Figure 1, all four strains generally displayed similar sensitivities towards Cr(VI) lethality. Although there was a slight, but not significant, increase in sensitivity of rev3 rad30 TLS mutants a 1 mM Cr(VI), this was not observed at higher concentrations (5-10 mM). Overall, clonogenic lethality did not exceed 30% at any of the concentrations examined and no differential impact of Cr(VI) on overall cell growth was observed (data not shown).
Figure 1.

TLS deficiency does not significantly impact cell survival after Cr(VI) exposure. Wt and TLS-deficient (rev3, rad30) yeast (BY4741) were exposed to the indicated amounts of Cr(VI) for 2 h at 30°C, washed and plated on nutrient-rich (YEPD) media. Plates were incubated at for 2-3 days at 30°C and colonies were counted. The data are presented as the mean ± SE of at least 3 independent experiments.
3.2 rev3, but not rad30, yeast exhibit decreased Cr(VI) mutagenicity
TLS is known to function in both an error-free and error-prone manner during the replicative bypass of DNA base damage generated by a variety of genotoxins. With regard to DNA damaging metals, Polη-dependent TLS has been implicated in the bypass of DNA adducts generated by the chemotherapeutic agent, cisplatin [53]. Beyond this, there is essentially no other information available on the role of TLS in bypassing metal-mediated DNA damage. In contrast to covalent platinum-DNA adducts, Cr(VI) is unique in that it has a propensity to form ternary DNA adducts involving the phosphodiester backbone [33;54]. To test whether TLS impacts Cr(VI) mutagenesis, we exposed Wt (BY4741 background) and TLS mutants (rev3, rad30, rev3 rad30) to Cr(VI) for 2 h and monitored forward mutagenesis at the CAN1 locus. Cr(VI) mutagenesis attained statistical significance at 5 mM in Wt cells (Figure 2). Interestingly, rev3 yeast were significantly attenuated in their sensitivity to Cr(VI) mutagenesis relative to Wt and rad30 strains. In contrast, rad30 yeast exhibited significantly higher basal and induced mutation frequencies compared to Wt and rev3 yeast. The loss of REV3 in rad30 yeast (rev3 rad30 double mutants) resulted in a significant decrease in Cr(VI)-induced mutagenesis relative to Wt and rad30 single mutants.
Figure 2.

TLS deficiency impacts Cr(VI) mutagenesis in yeast. Wt and TLS-deficient (rev3, rad30) yeast (BY4741) were exposed to the indicated amounts of Cr(VI) and processed for CAN1 mutagenesis as described in Methods. * = significantly (P<0.05)) different from respective untreated control; # = significantly different from Wt cells; ‡ = significantly different than all other strains. The data are presented as the mean ± SE of at least 3 independent experiments.
3.3. rev3 yeast display attenuated Cr(VI) forward, but not frameshift, mutagenesis
Given that rev3 yeast, but not rad30, displayed less sensitivity towards Cr(VI) mutagenesis, we concluded that this process is dependent upon Polζ-mediated, but not Polη, TLS. To further confirm this finding and determine whether rev3 yeast are universally resistant to Cr(VI) mutagenesis, we investigated the sensitivity of WT and rev3 yeast to Cr(VI) forward and frameshift mutagenesis. In general, the SJR strains (Figure 3A) displayed similar sensitivity to Cr(VI) clonogenic lethality (1-10 mM) as BY4741 yeast (Figure 1). As shown in Figure 3A, rev3 yeast (BY4741 background) were also resistant to Cr(VI) CAN1 forward mutagenesis. We next sought to determine if Polζ was responsible for additional forms of Cr(VI)-induced mutations. The SJR751 strain harbors a reporter for monitoring frameshift (reversion) mutagenesis at the LYS allele [45]. As shown in Figure 3B, rev3 yeast displayed near Wt levels of sensitivity to Cr(VI)-induced frameshift mutations. The spontaneous frequency for lys2ΔBgl reversion rate in Wt and rev3 yeast was 4.3×10-8 and 8.0×10-8, respectively.
Figure 3.

Polζ deficiency (rev3) renders yeast resistant to Cr(VI) forward, but not frameshift, mutagenesis. Yeast (SJR751) were exposed to the indicated amounts of Cr(VI) and processed for (A) CAN1 mutagenesis and (B) frameshift mutagenesis as described in Methods. * = significantly (P<0.05)) different from respective untreated control; # = significantly different from Wt cells. The data are presented as the mean ± SE of at least 3 independent experiments.
3.4. Polζ-deficient yeast exhibit altered mutational spectra at the CAN1 locus
The specific lesions that contribute to Cr(VI) mutagenesis are not clear. To gain insight into the molecular mechanisms for Polζ-dependent, Cr mutagenesis we sequenced the complete can1 coding region from Wt (SJR751 background) and rev3 clones treated with a peak mutagenic concentration (10 mM, Figure 3A) of Cr(VI). In general, Cr(VI)-induced significantly different (p=0.013} mutational spectra in WT and rev3 yeast. As shown in Table 2, Cr(VI) treated Wt yeast harbored 61.3% and 22.6% transition and transversion mutations, respectively. In addition, deletions and insertions accounted for 16.1% of all mutations in Cr(VI)-treated Wt yeast. Much higher levels of transition mutations were found in rev3 yeast isolated from both untreated (90.0%) and Cr(VI) exposed (82.4%) cultures. Moreover, rev3 yeast exhibited fewer transversion mutations at GC base pairs in both spontaneous and Cr(VI)-induced mutants. Interestingly, no deletions/insertions were detected in Cr(VI) treated rev3 yeast which significantly differed from WT yeast (p<0.05). In both strains, Cr-induced mutations were most frequently observed within repetitive sequences, but exhibited no clear base-specificity (Figure 4). Figure 3B shows that there were no frameshift mutations detected in using a strain (SJR751) that is sensitive to these lesions [45]. Consistent with the increased sensitivity of Wt yeast towards Cr(VI) mutagenesis, the CAN1 gene was non-amplifiable in a greater number of Wt (∼28.1%) samples relative to clones isolated from rev3 (∼6.7%) cultures (data not shown).
Table 2.
Mutational analysis of the CAN1 coding region Wt and rev3 Strainsa
| Type of Mutationb | Wt Control |
Wt 10 mM Cr(VI) |
rev3 Control |
rev3 10 mM Cr(VI)‡ |
|---|---|---|---|---|
| Transitions | 75.9% (22) | 61.3% (19) | 90.0% (36) | 82.4% (27) |
| GC→AT | 27.6% (8) | 19.4% (6) | 32.5% (13) | 32.4% (11) |
| AT→GC | 48.3% (14) | 41.9% (13) | 57.5% (23) | 50.0% (17) |
| Transversions | 24.1% (7) | 22.6% (7) | 10.0% (4) | 17.6% (6) |
| GC→TA | 13.8% (3) | 6.5% (2) | 2.5% (1) | 5.9% (2) |
| GC→CG | 3.4 (1) | 6.5% (2) | 0 | 0 |
| AT→CG | 6.9% (2) | 0 | 0 | 8.8% (3) |
| AT→TA | 3.4% (1) | 9.7% (3) | 7.5% (3) | 2.9% (1) |
| Deletions | 0 | 12.9% (4) # | 0 | 0 |
| Insertions | 0 | 3.2 (1) | 0 | 0 |
Data from 10 independent mutant clones for each strain/condition
Mutational analysis of CAN1 coding region
Parenthesis indicate the number of mutants
Cr(VI)-induced deletions were significantly different from WT control (P=0.052), and rev3 10 mM (P=0.049) yeast.
The mutational spectra generated by Cr(VI) in WT and rev3 yeast were significantly different (P=0.013).
Figure 4.

CAN1 mutational spectra in Wt and rev3 yeast (SJR751) after Cr(VI) treatment. The complete coding region of the CAN1 gene was sequenced in Wt (red) and rev3 (blue) yeast exposed to 10 mM Cr(VI) for 2 hours as described in the Methods. Mutated bases are underlined in the non-transcribed strand. Mutations observed in Wt and rev3 yeast are shown above and below the sequence, respectively. Δ = denotes deletion. 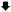 = denotes insertion. The data are described in Table 3. Mutational spectra were significantly different (P= 0.022).
= denotes insertion. The data are described in Table 3. Mutational spectra were significantly different (P= 0.022).
4. Discussion
TLS is known to play an integral role in both spontaneous and genotoxin-induced mutagenesis in yeast and mammalian cells. Specifically, TLS polymerases orchestrate the bypass of UV-, polycyclic aromatic hydrocarbon- and alkylating agent-induced DNA damage (reviewed in [55]). In addition, much progress has been made in our understanding of the molecular mechanisms of TLS using site-specifically damaged oligonucleotides and recombinant enzymes [56]. Given the importance of TLS to chemical mutagenesis, it is surprising that there is no information available on the role that TLS plays in carcinogenic metal-induced mutagenesis. Using a genetic approach in yeast, we investigated the relative contribution of two major TLS polymerases, Pols ζ and η, in Cr(VI) mutagenesis. Taken together, the data indicate that Polζ-dependent error-prone bypass of Cr-induced DNA damage is a primary factor in driving Cr(VI) mutagenesis in eukaryotic cells. In addition, these results also suggest that Polζ-mediated bypass of Cr-lesions may be responsible for Cr(VI)-induced deletions/insertions.
Cr(VI) is a complex DNA damaging agent generating a variety of genetic lesions including single and double-strand breaks, DNA interstrand crosslinks, DNA-protein crosslinks and oxidized bases (reviewed in [44]). Cr(VI) is ultimately reduced to Cr(III) within cells by reduced thiols (i.e. glutathione, cysteine) and ascorbic acid [57-60]. In contrast to many other carcinogens, Cr(III) does not form covalent bonds with DNA bases. Instead, Cr(III) participates in coordinate covalent interactions with various cellular macromolecules (reviewed in [33;35]). In addition, Cr(III), a trivalent cation, displays an affinity for the phosphodiester backbone which is believed to be a primary mode of initial attachment to DNA by Cr [33;35;54;61]. A number of lesions generated by Cr can inhibit replication fork progression [32;33;62;63] and our work is consistent with Polζ-dependent TLS playing an important, albeit mutagenic, role in bypassing Cr-induced replication arresting lesions. Given the complexity of Cr(VI) genotoxicity, it is likely that several lesions contribute to Cr mutagenesis through a Polζ-dependent mechanism. However, based on our knowledge of the substrate specificity of Polζ, certain Cr-induced lesions are potential candidates. For instance, Polζ is thought to participate in the bypass of ICLs [64] and abasic sites [65;66] which are both generated by Cr [64;67-69]. Future work employing site-specific Cr-lesions will help to elucidate the precise mechanism of Polζ -medicated TLS after Cr(VI) exposure in cells.
Polη-deficient strain displayed a higher level of spontaneous and Cr(VI)-induced mutagenesis relative to WT and rev3 yeast. This suggests that Polη might mediate the error-free replication past Cr damage, which would be consistent with the role that this enzyme is known to play in the error-free bypass of UV dimers [7] and ionizing radiation-induced intrastrand crosslinks [70]. The substrate specificity of Polη seems to be broad in that this enzyme has also been implicated, with NER, in the processing of mitomycin C-induced ICLs [71]. In addition, recent work indicates that Polη might facilitate the error-free bypass of 8-oxo-dG in mammalian cells [72]. Therefore, there are several potential lesions generated by Cr on DNA that might serves as substrates for Polη including Cr(III) ternary adducts with the DNA phosphodiester backbone including ICLs [32;73] as well as Cr(VI)-mediated oxidation products of DNA such as 8-oxo-dG. Consequently, Polη likely participates in the accurate bypass of numerous lesions formed by Cr on DNA and functions as an important mechanism for preventing Cr(VI)-induced mutagenesis in eukaryotic cells.
There were two noteworthy differences in the CAN1 mutational spectra between Wt and rev3 yeast. First, rev3 yeast harbored a lower relative percentage of transversion mutations, but a much higher level of transition mutations (G:C base pairs) relative to Wt yeast. This preponderance of transition mutations in the absence of Polζ is consistent with similar work using UVC and simulated sunlight (SSL) in S. cerevisiae [74]. Secondly, insertions/deletions (1-10 bp) accounted for 16.1% of the total mutations generated by Cr(VI) in Wt yeast, but these mutations were not observed in rev3 yeast. This is in line with previous work which found that benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide-induced fewer insertions/deletions (1-3 bp) in rev3 yeast relative to Wt controls [75]. At the molecular level, it is plausible that Cr-induced insertions and deletions are related to the generation of ICLs, or DPCs, by Cr(VI). It is well known that crosslinking agents, including Cr(VI) [76], induce deletions in cells [77] and Polζ has been postulated to promote TLS through ICLs [78]. Furthermore, ICLs can lead to the formation of DNA DSBs which are resolved by homology-directed repair (HDR) leading to both insertions and deletions in cells [79].
Because Polζ seems to be required for ICL-induced HDR in mammalian cells [79;80], it is possible that Polζ-mediated bypass of these helix-distorting crosslinks results in the loss of one, or several, nucleotides flanking the lesion. Polζ primarily functions in the extension step of TLS [81], consequently, our results suggest that Polζ is likely involved in the extension of misinsertions by other TLS polymerases across Cr-DNA damage. Rev1 is indispensable for Polζ-mediated TLS [82], however, this does not require its DNA synthetic activity [23;83]. Instead, Rev1 seems to play a role in Polζ TLS involving the its direct binding to Rev3 thereby targeting the enzyme to the replication fork [9]. In a recent review, Polζ-dependent TLS was proposed to involve the interaction between Rev3 with ubiquitinated PCNA, via Rev1, at the site of replication arrest. This then leads to nucleotide insertion across the damaged/non-coding base (by Polζ, Rev1 or other polymerase) and extension of the primer terminus by Polζ [20].
Recent studies have demonstrated that a model high-valent Cr(V) complex (i.e. Cr(VI)) further oxidizes 8-oxo-G to guanidinohydantoin (GH) and spiroiminodihydantoin (Sp) [30;31]. These lesions lead to both G→C transversions and G→T transversions which are commonly observed in cells after Cr(VI) treatment [84-86]. One explanation for the G→T mutations could be the so-called “A rule” wherein DNA polymerases preferentially insert adenine nucleotides across from damaged guanines [87]. In addition, it is plausible that Polζ-mediated TLS through GH and/or Sp may lead to the generation of G:C transversions as has been reported for these lesions [88-90]. Consistent with this, we found that rev3 yeast exhibited a slight decrease in transversions at G:C base-pairs (Wt = 13.0%, rev3 = 5.9%), and a concomitant shift in the mutational spectrum towards GC→AT transitions after Cr(VI) treatment in rev3 yeast. We postulate that this shift could be related to the bypass of Cr-DNA damage by Polη, which is thought to generate GC→AT mutations, in Polζ-deficient cells after DNA damage [91]. Alternatively, G:C transition mutations have also been linked to the deamination of cytosine which has a propensity to generate GC→AT mutations [92]. However, no evidence is yet available indicating that Cr is capable of facilitating the deamination of cytosine.
There were no clear “hotspots” observed for Cr-induced mutations in the current investigation. This is consistent with other studies that evaluated the mutational spectra of Cr(VI)-containing compounds within the HPRT locus in CHO cells. Specifically, Yang et al found that 90% of Cr(VI)-induced mutations occurred at A/T base-pairs [93], whereas Chen and Thilly observed that Cr(VI) mutagenesis occurred at both A/T and G/C base-pairs [84;85]. These conflicting results probably reflect the fact that Cr displays very little base-specific DNA binding (reviewed in [44]). Instead Cr(III) interacts with the phosphodiester backbone indicating that the contribution of Cr base-specific binding, via coordinate covalent interactions, to Cr genotoxicity and mutagenesis is relatively minor [33;35]. In addition to its DNA binding activity, Cr, possibly through the generation of Cr(V), is capable of oxidizing DNA bases. Cr(VI) in the presence of hydrogen peroxide, has been reported to oxidize both G as well as T in DNA [94;95]. Moreover, Cr(V)-mediated H abstraction on deoxyribose occurs to a greater extent on T and C leading to preferential release of these bases from dsDNA [29]. Collectively, the results from these investigations suggest that mutational base-specificity is unlikely to be observed after Cr-induced DNA damage.
The molecular factors involved in carcinogenic metal-induced mutagenesis and carcinogenesis are complex. Cr(VI)-induced DNA damage is collectively repaired by several DNA repair pathways due to the wide variety of lesions generated by this metal [44]. While NER and BER are undoubtedly involved in the removal of Cr lesions from DNA, the work presented here suggests that error-prone TLS plays a central role in Cr mutagenesis. Interestingly, NER protects against Cr mutagenesis in shuttle vector studies [96]. However, Cr(VI) clastogenesis and mutagenesis (i.e. HPRT gene) are less inducible in NER deficient mammalian cells relative to WT controls, implicating NER in Cr mutagenesis [43]. The molecular mechanisms of Cr(VI) mutagenesis are highly complex and likely involve several lesions/mechanisms. This work highlights the central role that TLS plays in Cr(VI) mutagenesis and suggests that Polζ is likely an important factor contributing to metal-induced carcinogenesis.
Acknowledgments
The authors would like to thank Rachelle Oliver for technical help and Gina Chun for critical comments. This work was supported by ES-05304 to SRP from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andersen PL, Xu F, Xiao W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008;18:162–173. doi: 10.1038/cr.2007.114. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, Green CM. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair (Amst) 2007;6:891–899. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 4.Alt A, Lammens K, Chiocchini C, Lammens A, Pieck JC, Kuch D, Hopfner KP, Carell T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science. 2007;318:967–970. doi: 10.1126/science.1148242. [DOI] [PubMed] [Google Scholar]
- 5.Chiapperino D, Kroth H, Kramarczuk IH, Sayer JM, Masutani C, Hanaoka F, Jerina DM, Cheh AM. Preferential misincorporation of purine nucleotides by human DNA polymerase eta opposite benzo[a]pyrene 7,8-diol 9,10-epoxide deoxyguanosine adducts. J Biol Chem. 2002;277:11765–11771. doi: 10.1074/jbc.M112139200. [DOI] [PubMed] [Google Scholar]
- 6.Chiapperino D, Cai M, Sayer JM, Yagi H, Kroth H, Masutani C, Hanaoka F, Jerina DM, Cheh AM. Error-prone translesion synthesis by human DNA polymerase eta on DNA-containing deoxyadenosine adducts of 7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. J Biol Chem. 2005;280:39684–39692. doi: 10.1074/jbc.M508008200. [DOI] [PubMed] [Google Scholar]
- 7.Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Pol eta. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- 8.Rechkoblit O, Zhang Y, Guo D, Wang Z, Amin S, Krzeminsky J, Louneva N, Geacintov NE. trans-Lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J Biol Chem. 2002;277:30488–30494. doi: 10.1074/jbc.M201167200. [DOI] [PubMed] [Google Scholar]
- 9.Acharya N, Johnson RE, Prakash S, Prakash L. Complex formation with Rev1 enhances the proficiency of Saccharomyces cerevisiae DNA polymerase zeta for mismatch extension and for extension opposite from DNA lesions. Mol Cell Biol. 2006;26:9555–9563. doi: 10.1128/MCB.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci USA. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu SL, Johnson RE, Prakash S, Prakash L. Requirement of DNA polymerase eta for error-free bypass of UV-induced CC and TC photoproducts. Mol Cell Biol. 2001;21:185–8. doi: 10.1128/MCB.21.1.185-188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kino K, Ito N, Sugasawa K, Sugiyama H, Hanaoka F. Translesion synthesis by human DNA polymerase eta across oxidative products of guanine. Nucleic Acids Symp Ser (Oxf) 2004:171–172. doi: 10.1093/nass/48.1.171. [DOI] [PubMed] [Google Scholar]
- 13.Kusumoto R, Masutani C, Iwai S, Hanaoka F. Translesion synthesis by human DNA polymerase eta across thymine glycol lesions. Biochemistry. 2002;41:6090–6099. doi: 10.1021/bi025549k. [DOI] [PubMed] [Google Scholar]
- 14.Nelson JR, Lawrence CW, Hinkle DC. Deoxycytidyl transferase activity of yeast REV1 protein. Nature. 1996;382:729–731. doi: 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]
- 15.Vaisman A, Frank EG, Iwai S, Ohashi E, Ohmori H, Hanaoka F, Woodgate R. Sequence context-dependent replication of DNA templates containing UV-induced lesions by human DNA polymerase iota. DNA Repair (Amst) 2003;2:991–1006. doi: 10.1016/s1568-7864(03)00094-6. [DOI] [PubMed] [Google Scholar]
- 16.Sabbioneda S, Bortolomai I, Giannattasio M, Plevani P, Muzi-Falconi M. Yeast Rev1 is cell cycle regulated, phosphorylated in response to DNA damage and its binding to chromosomes is dependent upon MEC1. DNA Repair (Amst) 2007;6:121–127. doi: 10.1016/j.dnarep.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, Kisker C, Friedberg EC. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murakumo Y, Ogura Y, Ishii H, Numata S, Ichihara M, Croce CM, Fishel R, Takahashi M. Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J Biol Chem. 2001;276:35644–35651. doi: 10.1074/jbc.M102051200. [DOI] [PubMed] [Google Scholar]
- 19.Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, Ulrich HD, Friedberg EC. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell. 2006;23:265–271. doi: 10.1016/j.molcel.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 20.Gan GN, Wittschieben JP, Wittschieben BO, Wood RD. DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res. 2008;18:174–183. doi: 10.1038/cr.2007.117. [DOI] [PubMed] [Google Scholar]
- 21.Gibbs PE, McGregor WG, Maher VM, Nisson P, Lawrence CW. A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase zeta. Proc Natl Acad Sci USA. 1998;95:6876–80. doi: 10.1073/pnas.95.12.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murakumo Y, Roth T, Ishii H, Rasio D, Numata S, Croce CM, Fishel R. A human REV7 homolog that interacts with the polymerase zeta catalytic subunit hREV3 and the spindle assembly checkpoint protein hMAD2. J Biol Chem. 2000;275:4391–4397. doi: 10.1074/jbc.275.6.4391. [DOI] [PubMed] [Google Scholar]
- 23.Haracska L, Unk I, Johnson RE, Johansson E, Burgers PM, Prakash S, Prakash L. Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 2001;15:945–54. doi: 10.1101/gad.882301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison A, Christensen RB, Alley J, Beck AK, Bernstine EG, Lemontt JF, Lawrence CW. REV3, a Saccharomyces cerevisiae gene whose function is required for induced mutagenesis, is predicted to encode a nonessential DNA polymerase. J Bacteriol. 1989;171:5659–67. doi: 10.1128/jb.171.10.5659-5667.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson RE, Yu SL, Prakash S, Prakash L. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol Cell Biol. 2007;27:7198–7205. doi: 10.1128/MCB.01079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu F, Lin X, Okuda T, Howell SB. DNA polymerase zeta regulates cisplatin cytotoxicity, mutagenicity and the rate of development of cisplatin resistance. Cancer Res. 2004;64:8029–8035. doi: 10.1158/0008-5472.CAN-03-3942. [DOI] [PubMed] [Google Scholar]
- 27.Acharya N, Haracska L, Prakash S, Prakash L. Complex formation of yeast Rev1 with DNA polymerase eta. Mol Cell Biol. 2007;27:8401–8408. doi: 10.1128/MCB.01478-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garg P, Burgers PM. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc Natl Acad Sci USA. 2005;102:18361–18366. doi: 10.1073/pnas.0505949102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugden KD, Wetterhahn KE. Direct and hydrogen peroxide-induced chromium(V) oxidation of deoxyribose in single-stranded and double-stranded calf thymus DNA. Chem Res Toxicol. 1997;10:1397–406. doi: 10.1021/tx970135r. [DOI] [PubMed] [Google Scholar]
- 30.Sugden KD, Campo CK, Martin BD. Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: a possible mechanism for chromate genotoxicity. Chem Res Toxicol. 2001;14:1315–22. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- 31.Sugden KD, Martin BD. Guanine and 7,8-dihydro-8-oxo-guanine-specific oxidation in DNA by chromium(V) Environ Health Perspect. 2002;110 5:725–8. doi: 10.1289/ehp.02110s5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bridgewater LC, Manning FC, Patierno SR. Base-specific arrest of in vitro DNA replication by carcinogenic chromium: relationship to DNA interstrand crosslinking. Carcinogenesis. 1994;15:2421–7. doi: 10.1093/carcin/15.11.2421. [DOI] [PubMed] [Google Scholar]
- 33.O'Brien T, Mandel HG, Pritchard DE, Patierno SR. Critical Role of Chromium (Cr)-DNA Interactions in the Formation of Cr- Induced Polymerase Arresting Lesions. Biochemistry. 2002;41:12529–37. doi: 10.1021/bi020452j. [DOI] [PubMed] [Google Scholar]
- 34.Voitkun V, Zhitkovich A, Costa M. Complexing of amino acids to DNA by chromate in intact cells. Environ Health Perspect. 1994;102 3:251–5. doi: 10.1289/ehp.94102s3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhitkovich A, Voitkun V, Costa M. Formation of the amino acid-DNA complexes by hexavalent and trivalent chromium in vitro: importance of trivalent chromium and the phosphate group. Biochemistry. 1996;35:7275–82. doi: 10.1021/bi960147w. [DOI] [PubMed] [Google Scholar]
- 36.O'Brien TJ, Brooks BR, Patierno SR. Nucleotide excision repair functions in the removal of chromium-induced DNA damage in mammalian cells. Mol Cell Biochem. 2005;279:85–95. doi: 10.1007/s11010-005-8225-0. [DOI] [PubMed] [Google Scholar]
- 37.O'Brien TJ, Jiang G, Chun G, Mandel HG, Westphal CS, Kahen K, Montaser A, States JC, Patierno SR. Incision of trivalent chromium [Cr(III) ]-induced DNA damage by Bacillus caldotenax UvrABC endonuclease. Mutat Res. 2006;610:85–92. doi: 10.1016/j.mrgentox.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 38.Reynolds M, Peterson E, Quievryn G, Zhitkovich A. Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J Biol Chem. 2004;279:30419–30424. doi: 10.1074/jbc.M402486200. [DOI] [PubMed] [Google Scholar]
- 39.Hailer MK, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair (Amst) 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 40.Ha L, Ceryak S, Patierno SR. Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis. 2004;25:2265–2274. doi: 10.1093/carcin/bgh242. [DOI] [PubMed] [Google Scholar]
- 41.O'Brien TJ, Fornsaglio JL, Ceryak S, Patierno SR. Effects of hexavalent chromium on the survival and cell cycle distribution of DNA repair-deficient S. cerevisiae. DNA Repair (Amst) 2002;1:617–27. doi: 10.1016/s1568-7864(02)00078-2. [DOI] [PubMed] [Google Scholar]
- 42.Reynolds M, Peterson E, Quievryn G, Zhitkovich A. Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J Biol Chem. 2004;279:30419–30424. doi: 10.1074/jbc.M402486200. [DOI] [PubMed] [Google Scholar]
- 43.Brooks B, O'Brien TJ, Ceryak S, Wise JP, Sr, Wise SS, Wise JP, Jr, DeFabo E, Patierno SR. Excision repair is required for genotoxin-induced mutagenesis in mammalian cells. Carcinogenesis. 2008;29:1064–1069. doi: 10.1093/carcin/bgn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Brien TJ, Ceryak S, Patierno SR. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res. 2003;533:3–36. doi: 10.1016/j.mrfmmm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Swanson RL, Morey NJ, Doetsch PW, Jinks-Robertson S. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:2929–35. doi: 10.1128/mcb.19.4.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. J Genet. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 47.Cariello NF, Piegorsch WW, Adams WT, Skopek TR. Computer program for the analysis of mutational spectra: application to p53 mutations. Carcinogenesis. 1994;15:2281–2285. doi: 10.1093/carcin/15.10.2281. [DOI] [PubMed] [Google Scholar]
- 48.Grossmann KF, Ward AM, Moses RE. Saccharomyces cerevisiae lacking Snm1, Rev3 or Rad51 have a normal S- phase but arrest permanently in G2 after cisplatin treatment. Mutat Res. 2000;461:1–13. doi: 10.1016/s0921-8777(00)00035-5. [DOI] [PubMed] [Google Scholar]
- 49.Grossmann KF, Ward AM, Matkovic ME, Folias AE, Moses RE. S. cerevisiae has three pathways for DNA interstrand crosslink repair. Mutat Res. 2001;487:73–83. doi: 10.1016/s0921-8777(01)00106-9. [DOI] [PubMed] [Google Scholar]
- 50.Acharya N, Haracska L, Johnson RE, Unk I, Prakash S, Prakash L. Complex formation of yeast Rev1 and Rev7 proteins: a novel role for the polymerase-associated domain. Mol Cell Biol. 2005;25:9734–9740. doi: 10.1128/MCB.25.21.9734-9740.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–4. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 52.Washington MT, Johnson RE, Prakash S, Prakash L. Accuracy of thymine-thymine dimer bypass by Saccharomyces cerevisiae DNA polymerase eta. Proc Natl Acad Sci USA. 2000;97:3094–3099. doi: 10.1073/pnas.050491997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alt A, Lammens K, Chiocchini C, Lammens A, Pieck JC, Kuch D, Hopfner KP, Carell T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science. 2007;318:967–970. doi: 10.1126/science.1148242. [DOI] [PubMed] [Google Scholar]
- 54.Voitkun V, Zhitkovich A, Costa M. Cr(III)-mediated crosslinks of glutathione or amino acids to the DNA phosphate backbone are mutagenic in human cells. Nucleic Acids Res. 1998;26:2024–30. doi: 10.1093/nar/26.8.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andersen PL, Xu F, Xiao W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008;18:162–173. doi: 10.1038/cr.2007.114. [DOI] [PubMed] [Google Scholar]
- 56.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 57.O'Brien T, Xu J, Patierno SR. Effects of glutathione on chromium-induced DNA crosslinking and DNA polymerase arrest. Mol Cell Biochem. 2001;222:173–82. [PubMed] [Google Scholar]
- 58.Standeven AM, Wetterhahn KE. Ascorbate is the principal reductant of chromium (VI) in rat liver and kidney ultrafiltrates. Carcinogenesis. 1991;12:1733–7. doi: 10.1093/carcin/12.9.1733. [DOI] [PubMed] [Google Scholar]
- 59.Standeven AM, Wetterhahn KE. Ascorbate is the principal reductant of chromium(VI) in rat lung ultrafiltrates and cytosols, and mediates chromium-DNA binding in vitro. Carcinogenesis. 1992;13:1319–24. doi: 10.1093/carcin/13.8.1319. [DOI] [PubMed] [Google Scholar]
- 60.Zhitkovich A, Shrager S, Messer J. Reductive metabolism of Cr(VI) by cysteine leads to the formation of binary and ternary Cr--DNA adducts in the absence of oxidative DNA damage. Chem Res Toxicol. 2000;13:1114–24. doi: 10.1021/tx0001169. [DOI] [PubMed] [Google Scholar]
- 61.Sugden KD, Wetterhahn KE. EPR Evidence for Chromium(V) Binding to Phosphate and Pyrophosphate: Implications for Chromium(V)-DNA Interactions. Inorg Chem. 1996;35:3727–3728. doi: 10.1021/ic960199+. [DOI] [PubMed] [Google Scholar]
- 62.Bridgewater LC, Manning FC, Woo ES, Patierno SR. DNA polymerase arrest by adducted trivalent chromium. Mol Carcinog. 1994;9:122–33. doi: 10.1002/mc.2940090304. [DOI] [PubMed] [Google Scholar]
- 63.O'Brien T, Xu J, Patierno SR. Effects of glutathione on chromium-induced DNA crosslinking and DNA polymerase arrest. Mol Cell Biochem. 2001;222:173–182. [PubMed] [Google Scholar]
- 64.McHugh PJ, Sarkar S. DNA interstrand cross-link repair in the cell cycle: a critical role for polymerase zeta in G1 phase. Cell Cycle. 2006;5:1044–1047. doi: 10.4161/cc.5.10.2763. [DOI] [PubMed] [Google Scholar]
- 65.Garg P, Stith CM, Majka J, Burgers PM. Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase zeta. J Biol Chem. 2005;280:23446–23450. doi: 10.1074/jbc.C500173200. [DOI] [PubMed] [Google Scholar]
- 66.Haracska L, Unk I, Johnson RE, Johansson E, Burgers PM, Prakash S, Prakash L. Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 2001;15:945–954. doi: 10.1101/gad.882301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chaney SG, Vaisman A. Specificity of platinum-DNA adduct repair. J Inorg Biochem. 1999;77:71–81. doi: 10.1016/s0162-0134(99)00149-x. [DOI] [PubMed] [Google Scholar]
- 68.Garg P, Stith CM, Majka J, Burgers PM. Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase zeta. J Biol Chem. 2005;280:23446–23450. doi: 10.1074/jbc.C500173200. [DOI] [PubMed] [Google Scholar]
- 69.Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 2006;25:1285–1294. doi: 10.1038/sj.emboj.7600993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Colis LC, Raychaudhury P, Basu AK. Mutational specificity of gamma-radiation-induced guanine-thymine and thymine-guanine intrastrand cross-links in mammalian cells and translesion synthesis past the guanine-thymine lesion by human DNA polymerase eta. Biochemistry. 2008;47:8070–8079. doi: 10.1021/bi800529f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L. Nucleotide excision repair- and polymerase eta-mediated error-prone removal of mitomycin C interstrand cross-links. Mol Cell Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee DH, Pfeifer GP. Translesion synthesis of 7,8-dihydro-8-oxo-2′-deoxyguanosine by DNA polymerase eta in vivo. Mutat Res. 2008;641:19–26. doi: 10.1016/j.mrfmmm.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bridgewater LC, Manning FC, Patierno SR. Arrest of replication by mammalian DNA polymerases alpha and beta caused by chromium-DNA lesions. Mol Carcinog. 1998;23:201–6. [PubMed] [Google Scholar]
- 74.Kozmin SG, Pavlov YI, Kunkel TA, Sage E. Roles of Saccharomyces cerevisiae DNA polymerases Poleta and Polzeta in response to irradiation by simulated sunlight. Nucleic Acids Res. 2003;31:4541–4552. doi: 10.1093/nar/gkg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie Z, Braithwaite E, Guo D, Zhao B, Geacintov NE, Wang Z. Mutagenesis of benzo[a]pyrene diol epoxide in yeast: requirement for DNA polymerase zeta and involvement of DNA polymerase eta. Biochemistry. 2003;42:11253–11262. doi: 10.1021/bi0346704. [DOI] [PubMed] [Google Scholar]
- 76.Liu S, Dixon K. Induction of mutagenic DNA damage by chromium (VI) and glutathione. Environ Mol Mutagen. 1996;28:71–9. doi: 10.1002/(SICI)1098-2280(1996)28:2<71::AID-EM2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 77.Bredberg A, Sandor Z, Brant M. Mutational response of Fanconi anaemia cells to shuttle vector site-specific psoralen cross-links. Carcinogenesis. 1995;16:555–561. doi: 10.1093/carcin/16.3.555. [DOI] [PubMed] [Google Scholar]
- 78.McHugh PJ, Sarkar S. DNA interstrand cross-link repair in the cell cycle: a critical role for polymerase zeta in G1 phase. Cell Cycle. 2006;5:1044–1047. doi: 10.4161/cc.5.10.2763. [DOI] [PubMed] [Google Scholar]
- 79.Jonnalagadda VS, Matsuguchi T, Engelward BP. Interstrand crosslink-induced homologous recombination carries an increased risk of deletions and insertions. DNA Repair (Amst) 2005;4:594–605. doi: 10.1016/j.dnarep.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 80.Zhang N, Liu X, Li L, Legerski R. Double-strand breaks induce homologous recombinational repair of interstrand cross-links via cooperation of MSH2, ERCC1-XPF, REV3, and the Fanconi anemia pathway. DNA Repair (Amst) 2007;6:1670–1678. doi: 10.1016/j.dnarep.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haracska L, Unk I, Johnson RE, Johansson E, Burgers PM, Prakash S, Prakash L. Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 2001;15:945–954. doi: 10.1101/gad.882301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lawrence CW, Christensen RB. Ultraviolet-induced reversion of cyc1 alleles in radiation-sensitive strains of yeast. I. rev1 Mutant strains. J Mol Biol. 1978;122:1–21. doi: 10.1016/0022-2836(78)90104-3. [DOI] [PubMed] [Google Scholar]
- 83.Baynton K, Bresson-Roy A, Fuchs RP. Distinct roles for Rev1p and Rev7p during translesion synthesis in Saccharomyces cerevisiae. Mol Microbiol. 1999;34:124–33. doi: 10.1046/j.1365-2958.1999.01583.x. [DOI] [PubMed] [Google Scholar]
- 84.Chen J, Thilly WG. Mutational spectrum of chromium(VI) in human cells. Mutat Res. 1994;323:21–7. doi: 10.1016/0165-7992(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 85.Chen J, Thilly WG. Use of denaturing-gradient gel electrophoresis to study chromium-induced point mutations in human cells. Environ Health Perspect. 1994;102 3:227–9. doi: 10.1289/ehp.94102s3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu S, Medvedovic M, Dixon K. Mutational specificity in a shuttle vector replicating in chromium(VI)- treated mammalian cells. Environ Mol Mutagen. 1999;33:313–9. doi: 10.1002/(sici)1098-2280(1999)33:4<313::aid-em8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 87.Taylor JS. New structural and mechanistic insight into the A-rule and the instructional and non-instructional behavior of DNA photoproducts and other lesions. Mutat Res. 2002;510:55–70. doi: 10.1016/s0027-5107(02)00252-x. [DOI] [PubMed] [Google Scholar]
- 88.Henderson PT, Delaney JC, Gu F, Tannenbaum SR, Essigmann JM. Oxidation of 7,8-dihydro-8-oxoguanine affords lesions that are potent sources of replication errors in vivo. Biochemistry. 2002;41:914–921. doi: 10.1021/bi0156355. [DOI] [PubMed] [Google Scholar]
- 89.Leipold MD, Muller JG, Burrows CJ, David SS. Removal of hydantoin products of 8-oxoguanine oxidation by the Escherichia coli DNA repair enzyme FPG. Biochemistry. 2000;39:14984–14992. doi: 10.1021/bi0017982. [DOI] [PubMed] [Google Scholar]
- 90.Muller JG, Duarte V, Hickerson RP, Burrows CJ. Gel electrophoretic detection of 7,8-dihydro-8-oxoguanine and 7, 8-dihydro-8-oxoadenine via oxidation by Ir (IV) Nucleic Acids Res. 1998;26:2247–2249. doi: 10.1093/nar/26.9.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kozmin SG, Pavlov YI, Kunkel TA, Sage E. Roles of Saccharomyces cerevisiae DNA polymerases Poleta and Polzeta in response to irradiation by simulated sunlight. Nucleic Acids Res. 2003;31:4541–4552. doi: 10.1093/nar/gkg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kreutzer DA, Essigmann JM. Oxidized, deaminated cytosines are a source of C --> T transitions in vivo. Proc Natl Acad Sci USA. 1998;95:3578–3582. doi: 10.1073/pnas.95.7.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang JL, Hsieh YC, Wu CW, Lee TC. Mutational specificity of chromium(VI) compounds in the hprt locus of Chinese hamster ovary-K1 cells. Carcinogenesis. 1992;13:2053–7. doi: 10.1093/carcin/13.11.2053. [DOI] [PubMed] [Google Scholar]
- 94.Rodriguez H, Holmquist GP, D'Agostino R, Jr, Keller J, Akman SA. Metal ion-dependent hydrogen peroxide-induced DNA damage is more sequence specific than metal specific. Cancer Res. 1997;57:2394–403. [PubMed] [Google Scholar]
- 95.Rodriguez H, Holmquist GP, D'Agostino R, Jr, Keller J, Akman SA. Metal ion-dependent hydrogen peroxide-induced DNA damage is more sequence specific than metal specific. Cancer Res. 1997;57:2394–2403. [PubMed] [Google Scholar]
- 96.Reynolds M, Peterson E, Quievryn G, Zhitkovich A. Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J Biol Chem. 2004;279:30419–30424. doi: 10.1074/jbc.M402486200. [DOI] [PubMed] [Google Scholar]