

Abstract
We have used isothermal titration calorimetry (ITC) to study the thermodynamics of binding of twelve bisphosphonates to human bone. The ITC results show that there are two binding sites. Site A is the weak, highly populated site seen by NMR and is characterized by an average ΔG of binding of −5.2 kcal. Site B is a strong binding site characterized by a ΔG of binding of −8.5 kcal. Binding to both sites is overwhelmingly entropy driven. Using a thermodynamic group approach and a linear regression method we predict the ΔG of binding of all twelve compounds with an R2=0.95 (a 0.19 kcal error variance estimate, about 3% of the total ΔG range), opening up the way to designing novel chemotherapy, immunotherapy and anti-infectious disease drugs having weak bone binding affinity.
Bisphosphonates are the major drugs used to treat boneresorption diseases1. They act by preventing osteoclastic bone resorption, inhibiting the enzyme farnesyl diphosphate synthase (FPPS). Bisphosphonates also kill tumor cells2 and many parasitic protozoa3 and can activate γδ T cells of the immune system4 to kill tumor cells5 and bacteria6. There is thus interest in their use for immuno-chemotherapy of cancer7, and in the treatment of parasitic protozoan diseases8, where less avid bone binding might be advantageous. In earlier work9, we used NMR to probe how different bisphosphonates bind to bone. We found that the 31P magic-angle sample-spinning NMR spectra of bound bisphosphonates exhibited a single broad peak, and that there was ~0.8 phosphate (Pi) released per bisphosphonate bound. These and other NMR results led to a model9 in which a bisphosphonate –PO32− group displaced Pi, while the cationic side-chains interacted electrostatically with anionic surface groups. However, a puzzling observation was that the free energy for binding was low (~−4.3 kcal, for pamidronate). Here, we investigate this topic further, by using isothermal titration calorimetry (ITC), which might yield information on any additional, tight binding site(s) that – if at low occupancy, would be difficult to detect via NMR.
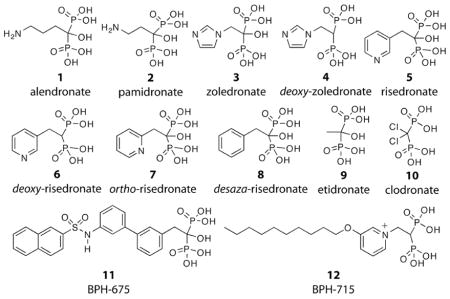
We investigated by ITC the interaction of the twelve bisphosphonates (1–12) shown above with human bone mineral, which enabled us to study the effects of having a 1-OH group removed (4,6), changing the position of the ring nitrogen in risedronate (7), removing the ring nitrogen in risedronate (8), and truncating the risedronate side-chain (9), in addition to studying several other bisphosphonates of interest (10–12)11. Representative ITC results for three compounds(1,2,4), together with their corresponding fitting curves, are shown in Figure 1A–B (all twelve fitting curves arse in Figure S1 in the Supporting Information), and the ΔG, ΔH and ΔS values so derived10 are given in Table 1.
Figure 1.

(A) ITC data for bisphosphonates 1,3,4 binding to human bone. (B) representative fitting curves.
Table 1.
Thermodynamic parameters for ligand bindinga
| Site Ab | Site Bc | |||||||
|---|---|---|---|---|---|---|---|---|
| ΔG | ΔH | ΔS | TΔS | ΔG | ΔH | ΔS | TΔS | |
| 1 | −6.5 | −1.7 | 15 | 4.7 | −10.4 | 0.60 | 35 | 11 |
| 2 | −5.7 | −1.4 | 14 | 4.3 | −9.3 | 0.41 | 31 | 9.8 |
| 3 | −5.4 | −1.0 | 14 | 4.4 | −8.4 | 0.54 | 29 | 8.9 |
| 4 | −5.2 | 0.49 | 18 | 5.6 | − | - | - | - |
| 5 | −4.7 | −0.69 | 13 | 4.0 | −7.3 | 0.89 | 27 | 8.2 |
| 6 | −4.9 | 0.56 | 18 | 5.4 | - | - | - | - |
| 7 | −5.0 | −1.3 | 12 | 3.7 | −8.0 | 0.68 | 28 | 8.7 |
| 8 | −4.8 | 0.86 | 18 | 5.6 | - | - | - | - |
| 9 | −5.6 | −1.4 | 14 | 4.2 | −7.7 | 0.74 | 27 | 8.4 |
| 10 | −5.1 | −2.4 | 8.7 | 2.7 | - | - | - | - |
| 11 | −4.7 | −2.1 | 8.4 | 2.6 | - | - | - | - |
| 12 | −5.1 | −1.6 | 11 | 3.5 | - | - | - | - |
| Avg | −5.2 | −0.96 | 14 | 4.2 | −8.5 | 0.64 | 30 | 9.2 |
Units of ΔG, ΔH, and TΔS are kcal mol−1, for ΔS, cal deg−1 mol−1.
Site A is the weak binding, highly populated site;
Site B is the strong binding site
There are several observations. First, there are only two types of ITC curve seen. Binding of half of the compounds (1–3, 5, 7 and 9) is characterized by both weak (Site A, Table 1) and strong (Site B, Table 1) interactions (two independent sites), while the other six compounds (4, 6, 8, 10–12) bind to only the weak Site A (e.g. 4, in Figure 1B). Second, in most cases, binding is overwhelmingly entropy driven, that is, ΔG ~ −TΔS. Third, there is a rather small range in ΔS in both sites. In the weak binding Site A, ΔGavg is ~−5.2 kcal and ΔSavg is 14 cal K−1 mole−1, while in the strong binding Site B ΔGavg is ~−8.5 kcal, and ΔSavg is 30 cal K−1 mole−1, almost twice that seen in the weak binding site.
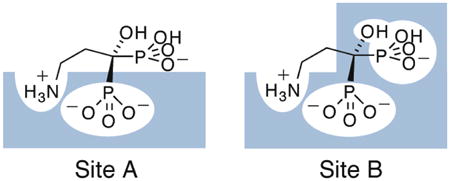
Based on these results, and those described previously9, we propose the bisphosphonate binding model shown (for pamidronate) in Figure 2. The weak binding Site A originates via displacement of ~1 Pi per bisphosphonate bound. It is the one that is most highly populated (Supporting Information Table S1), and is that which is observed by NMR. One phosphonate group binds into the bone mineral matrix and most of the binding free energy arises due to release of Pi and corresponds to the ΔS of ~14 cal K−1 mole−1 (−TΔSavg = −4.2 kcal mole−1; ΔGavg=−5.2 kcal mole−1).
Figure 2.

A, Schematic of the weak (Site A, left) and strong (Site B, right) pamidronate binding sites on human bone; B, ΔG and C, ΔH, - TΔS experimental versus calculated results for 1–12 binding to bone.
The observation that binding to the strong binding Site B is again overwhelmingly entropy driven (ΔS ~30 cal K−1 mole−1, −TΔS=−9.3 kcal mole−1) and that this ΔS value is about twice that seen in the weak binding site, and that only the small 1- OH containing species bind to this site, strongly suggests the binding mode shown in Figure 2A (Site B). Here, both phosphonates (and OH) bury into the bone mineral, resulting in release of ~2Pi (or 1 Pi + 1 CO32−) and thus, a ~2x increase in ΔS (from ~14 to ~30 cal K−1 mole−1). A 1-OH group is thus critical for bone mineral binding (as seen in vivo) although, interestingly, is not required for FPPS inhibition.
The results shown in Table 1 also indicate that the ΔG for binding of alendronate (1) to Site B is very large (−10.4 kcal mole−1), followed by that for pamidronate (2; −9.3 kcal mole−1) while that for zoledronate (3) is less strong (−8.4 kcal mole−1) and that for risedronate (5, −7.3 kcal mole−1) is relatively weak. This binding strength behavior is what might be anticipated based on the pKa values of the side-chains. That is, the strongly basic species (pKa ~11) are fully protonated at pH = 7, so make a strong electrostatic contribution to binding; zoledronate (imidazole pKa ~6.7) is ~33% protonated, while risedronate is only weakly protonated (pKa ~5.5). In the case of the weak binding site, the ΔG values for the OH-containing species (3,5) are only ~0.2 kcal mole−1 different from those observed with the corresponding deoxy analogs (4,6), consistent with a weak OH interaction with the surface. Of course, based on this model, it might be expected that the phenyl analog (8) of risedronate (5) should also bind to the strong binding site. This appears not to be the case, however, presumably because the bulky phenyl group is involved in a net repulsive (anion-π) interaction with the bone surface. Removal of this bulky phenyl group results in etidronate (9), which binds to both sites, consistent with this idea.
The question then arises: can we construct a quantitative model to predict the ΔG, ΔH and ΔS for all 12 compounds binding into Site A and/or Site B? To do this, we used a thermodynamic group approach combined with a linear regression method, assuming that there are transferable or additive group ΔG, ΔH and ΔS values for the cationic head-group, each phosphonate, the 1-OH group, together with an additional term to describe the presence of a hydrophobic (1-H or phenyl) interaction. The matrix we constructed is shown in Supporting Table S2 and simply represents the presence (1) or absence (0) of an interaction, with the cation term (ammonium, etc.) scaled to account for pKa differences. Four coefficients are derived from a partial least squares analysis and are given in Supporting Table S3. Figures 2b,c show experimental vs. predicted ΔG (Fig. 2b) and ΔH, −TΔS (Figure 2c) results and there is good accord between experiment and prediction. For example, for ΔG, we find the R2 value is 0.95, F-value=88, p<0.00001 and the error is 0.19 kcal, about 3% of the overall range, using a −1.6 kcal ΔG for binding for the - NH3+ groups in alendronate and pamidronate, −4.8 kcal for the ~−2 charged Site A phosphonate, −3.1 kcal for the Site B phosphonate plus the OH group, and −0.05 kcal for the hydrophobic group. Good results are also obtained for ΔH, − TΔS, Figure 2c.
These results are of broad general interest since they provide the first detailed thermodynamic description of the binding of a widely used class of drug molecules, bisphosphonates, to human bone. We find the presence of two different binding sites, with binding to each being readily explained based on the structural features present in each molecule, opening the way to the design of novel, potent enzyme and cell growth inhibitors that have weak bone-binding affinity, of interest in the context of chemotherapy11, immunotherapy7, and anti-infective drug development8.
Supplementary Material
Acknowledgments
This work was supported by the U.S. Public Health Service (NIH grant GM65307). S.M. was an American Heart Association, Midwest Affiliate, Pre-doctoral Fellow (grant number 0615564Z).
Footnotes
Supporting Information Available: ITC methods, data, and fitting curves for 1–12 binding to human bone, and computational results, and complete ref 7. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Russell RG. Ann N Y Acad Sci. 2006;1068:367–401. doi: 10.1196/annals.1346.041. [DOI] [PubMed] [Google Scholar]
- 2.Green JR. Oncologist. 2004;9 Suppl 4:3–13. doi: 10.1634/theoncologist.9-90004-3. [DOI] [PubMed] [Google Scholar]
- 3.Martin MB, Grimley JS, Lewis JC, Heath HT, 3rd, Bailey BN, Kendrick H, Yardley V, Caldera A, Lira R, Urbina JA, Moreno SN, Docampo R, Croft SL, Oldfield E. J Med Chem. 2001;44:909–16. doi: 10.1021/jm0002578. [DOI] [PubMed] [Google Scholar]
- 4.Kunzmann V, Bauer E, Wilhelm M. N Engl J Med. 1999;340(9):737–8. doi: 10.1056/NEJM199903043400914. [DOI] [PubMed] [Google Scholar]
- 5.Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. Blood. 2000;96:384–92. [PubMed] [Google Scholar]
- 6.Wang L, Kamath A, Das H, Li L, Bukowski JF. J Clin Invest. 2001;108:1349–57. doi: 10.1172/JCI13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gnant M, et al. N Engl J Med. 2009;360:679–91. doi: 10.1056/NEJMoa0806285. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez N, Bailey BN, Martin MB, Oldfield E, Urbina JA, Docampo R. J Infect Dis. 2002;186:138–40. doi: 10.1086/341074. [DOI] [PubMed] [Google Scholar]
- 9.Mukherjee S, Song Y, Oldfield E. J Am Chem Soc. 2008;130:1264–73. doi: 10.1021/ja0759949. [DOI] [PubMed] [Google Scholar]
- 10.Yin F, Cao R, Goddard A, Zhang Y, Oldfield E. J Am Chem Soc. 2006;128:3524–5. doi: 10.1021/ja0601639. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, et al. J Am Chem Soc. 2009;131:5153–62. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.