

Summary
The string (stg) locus of Drosophila encodes a factor that is thought to trigger mitosis by activating the p34cdc2 protein kinase. stg is required for mitosis early in development and is transcribed in a dynamic pattern that anticipates the pattern of embryonic cell divisions. Here we show that differential cell cycle regulation during postblastoderm development (cell cycles 14–16) occurs in G2. We demonstrate that stg mRNA expressed from a heat shock promotor triggers mitosis, and an associated S phase, in G2 cells during these cycles. Hence, differential cell cycle timing at this developmental stage is controlled by stg. Finally, we use heat-induced stg expression to alter the normal pattern of embryonic mitoses. Surprisingly, the complex mitotic pattern evident during normal development is not essential for many features of pattern formation or for viability.
Introduction
Studies using different organisms and cell types have shown that the cell cycle can be regulated at several different points in its progression. In budding yeast and most mammalian cell lines, regulation occurs primarily at the G1/S boundary, whereas some cell lines, fission yeast, and the slime mold Physarum have cycles regulated at the G2/M boundary (for reviews see Pardee, 1989; Murray and Kirschner, 1989). During embryogenesis, the mode of cell cycle regulation switches several times in a stage-dependent and cell type–dependent manner. In Drosophila embryos, the subject of this paper, the first 13 cell cycles are driven by maternal products in an essentially unregulated fashion: they are rapid and synchronous, and lack G1 and G2 phases (Rabinowitz, 1941; Foe and Alberts, 1983). Following mitosis 13, the cell cycle switches to a more complex mode of regulation, as evidenced by the aquisition of G2 phases and the onset of differential, position-specific mitotic timing. Three rounds of patterned mitosis occur in the embryo after interphase 14 (mitoses 14, 15, and 16) over a period of about 4 hr. The highly invariant spatiotemporal pattern of mitosis 14 has been mapped in detail (Foe, 1989), and the patterns of mitoses 15 and 16 have been characterized in a somewhat more cursory fashion (Hartenstein and Campos-Ortega, 1985). Following mitosis 16, there is a second switch in the mode of cell cycle regulation, as many cells enter their first G1 phase (this paper). This is a terminal interphase for most cells, although many eventually undergo several rounds of polyploidization, especially during the larval period. Several cell lineages, such as those leading to the nervous system, do not enter a terminal interphase after mitosis 16, but continue to divide for some time according to an independent program (Hartenstein et al., 1987; Bodmer et al., 1989).
At present, little is known about the molecular basis for switches between modes of cell cycle regulation during development. Our studies of the string (stg) gene, however, offer a simple molecular explanation for the switch occurring in interphase 14 (Edgar and O’Farrell, 1989). stg encodes a protein belonging to a conserved family of mitotic regulators, the best studied of which is cdc25 of fission yeast (Fantes, 1981; Russell and Nurse, 1986; Russell et al., 1989; Sadhu et al., 1990; Ducommun et al., 1990). cdc25 is one of several factors, including cyclins, that are required to activate a highly conserved mitotic kinase, p34cdc2 (Moreno et al., 1989; Gould and Nurse, 1989). The active form of this kinase triggers mitotic events such as nuclear envelope breakdown, chromatin condensation, and spindle formation (for review see Murray and Kirschner, 1989). In yeast, removing cdc25+ function causes a first cycle arrest in the G2 phase. In Drosophila, removing zygotic stg function causes G2 arrest in interphase 14, just prior to the first mitosis that requires zygotic transcription (Edgar et al., 1986; Edgar and O’Farrell, 1989; O’Farrell et al., 1989). This arrest point is correlated with the abrupt degradation of maternal stg mRNA in early interphase 14. Zygotic stg expression normally begins later in interphase 14 and occurs in a spatial pattern that anticipates the spatial pattern of mitosis 14 (Edgar and O’Farrell, 1989). For the remainder of embryogenesis, stg mRNA is expressed in a dynamic series of patterns that are precisely correlated with mitotic patterns; in most cell cycles a brief pulse of stg transcription, giving rise to a very short-lived mRNA, occurs at the end of each G2 period (B. A. E., unpublished data). This information suggested that the switch from rapid, unregulated mitoses to slower, patterned mitoses in interphase 14 is actually a switch from cycles driven by ubiquitous maternal stg to cycles driven by tightly regulated zygotic stg. Most importantly, it led us to propose that regulated stg expression controls the spatiotemporal pattern of mitoses in the embryo.
Given this proposal, the regulation of stg expression patterns becomes an interesting problem. Although the regulation of stg has not yet been studied in detail, several observations suggest that stg expression is controlled at the transcriptional level by combinations of segmentation and homeotic gene products (Edgar and O’Farrell, 1989; O’Farrell et al., 1989; B. A. E., unpublished data). A second major question concerns the generality of stg as a cell cycle regulator during development. Does stg regulate cell cycle patterns throughout development, or is it only truly rate limiting at one particular stage, such as cycle 14? In this report, we present a combination of descriptive and experimental data concerning the regulation of mitotic cycles 14, 15, and 16. We demonstrate that G2/M is the only differentially regulated transition in cycle progression during these cycles, and that stg activity is rate limiting for this transition. We also document the switch to G1 regulation that occurs during cycle 16. Finally, we present experiments that address the significance of mitotic patterning as a developmental process.
Results
Cell Cycle Progression Is Regulated in G2 during Embryonic Cell Cycles 14, 15, and 16
The first 13 cell cycles in Drosophila development do not have discrete G1 or G2 periods and are synchronous. The cycles following these, however, are differentially timed. To determine whether differences in cell cycle timing arise from variable G1, S, or G2 phases, we compared the spatiotemporal pattern of S phases 14 through 17 with the spatiotemporal patterns of the corresponding mitoses (13 through 16).
Earlier studies of S phase 14 indicated that DNA replication begins immediately after mitosis 13, and that the bulk of replication occurs within the first 40 min of interphase, which lasts 70–170 min in different cells (Blumenthal et al., 1974; McKnight and Miller, 1977; Edgar and Schubiger, 1986). These studies, however, used methods that could not detect spatial differences in replication patterns, and thus would not have uncovered any differential regulation of S phases if it existed. To visualize the spatial patterns of DNA synthesis in situ, we pulse labeled living embryos with 5-bromo-2′-deoxyuridine (BrdU) for 20–30 min. Following a pulse labeling, we fixed the embryos and visualized the BrdU-labeled DNA by immunofluorescence. Pulse labelings beginning in mitosis 13 (130 min after egg deposition [AED]) showed that all of the blastoderm nuclei enter S phase 14 synchronously, early in interphase (Figure 1A). Labelings beginning in late S phase (150–160 min AED) detected only a single tiny speck of late replicating, centromeric heterochromatin in each interphase chromosome, and showed that S phase ends at virtually the same time in all of the blastoderm nuclei (Figure 1B). From assessing the labeling patterns in carefully staged embryos, we determined that the end of S phase 14 occurs 35–45 min after mitosis 13 (165–175 min AED). Following S phase 14, there is a true G2 period, during which no labeling is observed, that lasts at least 30 min (Figure 1C). Since cells enter mitosis 14 over a period of greater than 100 min, this means that the G2 period of cycle 14 is variable, lasting between 30 and 130 min in different cells.
Figure 1. S Phase 14 Begins and Ends Synchronously without a G1 Period.

We show three embryos pulse labeled with BrdU for 20 min at progressively later stages of interphase 14. The BrdU is detected by immunoflourescence, and labeled nuclei appear white.
(A) An embryo labeled for the first 20 min of interphase 14 (130–150 min after egg deposition [AED]) shows all blastoderm nuclei heavily and uniformly labeled.
(B) An embryo labeled at about 160–180 min AED shows spots of late replicating centromeric heterochromatin in each blastoderm nucleus. Note that nuclei in all regions of the blastoderm are similarly labeled, indicating that S phase completion is not spatially patterned. The yolk nuclei and pole cell nuclei are heavily labeled in this embryo, indicating that they replicate their DNA later than the blastoderm nuclei.
(C) An embryo labeled at about 180–200 min AED shows most of the blastoderm nuclei unlabeled, indicating a true G2 period. Patches of labeled nuclei in the anterior are those that have already progressed through mitosis 14 and entered S phase 15. This photo is overexposed to show cytoplasmic background staining.
Twenty minute pulse labelings ending at a series of times (210, 240, and 270 min AED) in S phase 15 resulted in labeling patterns that corresponded exactly to the pattern of mitosis 14 (Figures 2D, 2E, and 2F), as described by Foe (1989). These labeling patterns were also exactly inverse to the staining pattern of cyclin A (Figures 2A, 2B, and 2C), an antigen that accumulates uniformly during interphase 14 and is quantitatively degraded at the following metaphase (Lehner and O’Farrell, 1989). Since the spatial pattern of S phase 15 is identical to that of mitosis 14, the time interval between mitosis and S phase initiation must be constant in all cells. By comparing the distribution of mitotic figures and BrdU-labeled nuclei in single embryos with the temporal map of mitosis generated by Foe (1989), we determined that the interval between mitosis and S phase is <5 min. The labeling patterns were also consistent with the notion that the duration of S phase 15 is roughly constant, at 35–45 min, in all cells.
Figure 2. S Phase 15 Follows Directly after Mitosis 14.

(A), (B), and (C) show cycle 14 and early cycle 15 embryos of increasing age stained with antibodies to cyclin A. The cyclin staining patterns indicate cell cycle states; cells in late interphase are brightly labeled, whereas dark cells have just passed through mitosis. (D), (E), and (F) show embryos pulse labeled with BrdU for 20 min immediately prior to fixation. Embryos in (D), (E), and (F) are the same age as embryos in (A), (B), and (C), respectively. The cyclin and BrdU patterns are almost exactly inverse to each other. This indicates that cyclin degradation, which occurs at metaphase (Lehner and O’Farrell, 1989), occurs a constant number of minutes before DNA replication. From comparing distributions of metaphase figures and BrdU-labeled nuclei in single embryos (not shown) and using the map of mitotic times (Foe, 1989), we estimate that the metaphase to S phase time interval is <5 min. All embryos show a 3/4 ventrolateral view; anteriors are to the left and dorsal surfaces uppermost.
A similar analysis, using cyclin A patterns as a mitotic map, showed that as cells exit mitosis 15 they enter S phase 16 virtually immediately. While we are confident that all cells entered S phase 15 directly following mitosis 14, the complexity of the later division and replication patterns precludes us from stating that all of the embryonic cells enter S phase 16 immediately after mitosis 15. Although most cells appear to retain coupling between mitosis and S phase initiation during M15/S16, our analysis cannot exclude the possibility that small subsets of cells, particularly in the ventral neurogenic region, aquire a G1 period at this stage.
stg Induces Division in G2 Cells
The DNA labeling experiments described above indicated that differential control of cell cycle progression is executed in G2 during cycle 14. The G2 arrest of stg mutants, the stg mRNA expression pattern, and stg’s homology to a known G2/M regulator (cdc25+) in turn suggest that stg is the rate limiting regulator that executes this differential control. To test this hypothesis, we constructed a chimeric gene containing the Drosophila hsp70 promotor, which is heat inducible, fused to a stg cDNA fragment containing the entire open reading frame. Germline transformants carrying this chimeric gene were generated, and two independent lines were used for our studies. We monitored the expression of the hsp70-stg (hs-stg) gene by in situ hybridization and found that a 25 min heat pulse at 37°C during midcycle 14 (160–185 min AED, 25°C) was sufficient to induce high concentrations of hs-stg mRNA distributed uniformly throughout the embryo. Quantification of this RNA on a Northern blot (not shown) indicated that the hs-stg gene produced 2- to 4-fold the amount of maternal stg mRNA normally present in a syncytial blastoderm (cycle 13) embryo. Like the normal stg mRNA, the hs-stg mRNA was unstable and was undetectable by in situ hybridization 30 min after the end of the heat pulse.
We monitored the effect of heat-induced stg expression by fixing embryos at intervals after the end of this heat pulse (160–185 min AED) and then staining them with the DNA stain Hoechst 33258 and with antibodies to tubulin or cyclin A. Five to ten minutes after the end of the heat pulse, cells began entering metaphase in what appeared to be a random pattern, and virtually all of the embryonic cells passed through anaphase and telophase and into interphase during a period of 10–20 min (Figure 3). Slightly longer (30 or 40 min) heat pulses caused a more rapid and more uniform mitotic response, whereas shorter pulses (10 or 15 min) caused an even more random pattern. Five minute heat pulses had no effect. The stochastic appearance of the mitotic pattern (Figure 3) may be attributed to stochastic variations in the pattern of activation of the hs-stg gene, which we confirmed by in situ hybridization (not shown). Most importantly, mitotic times of all the embryonic cells were advanced. In particular, the cells along the ventral surface, most of which do not normally divide until 250–300 min AED, entered mitosis up to 100 min too early. We found that mitoses in two regions, the amnioserosa and mitotic domain B, consistently lagged 10–15 min behind the rest of the embryo. The cells in these regions are normally nondividing (Foe, 1989). All aspects of the hs-stg–induced mitosis, including chromosome and spindle morphology, cyclin degradation, chromosome separation, and cytokinesis, appeared to be normal. Wild-type embryos subjected to identical heat pulses showed no abnormalities in the normal division pattern. After the hs-stg–induced mitosis, there was a quiescent period during which no new mitoses occurred, even though cells in normal embryos would have been dividing at this time. As discussed below, cells enter a refractory period after mitosis when they cannot be driven into mitosis by stg. This period is roughly the same length as S phase (35–45 min).
Figure 3. stg Expression in G2 of Cycle 14 Induces Mitosis.

(A), (B), and (C) show ventral views of hs-stg embryos that were fixed and stained with anti-cyclin A antibodies 0, 5, and 15 min after a 25 min heat pulse (37°C), initiated at 175 min AED. Cyclin degradation, which occurs at metaphase, occurs in a stochastic pattern that encompasses virtually all of the embryonic cells within 15 min of the heat pulse. See Figures 2A, 2B, and 2C for normal cyclin A expression patterns during cycle 14. (D) shows anaphase chromosomes in a hs-stg–induced mitosis. Note the absence of abnormalities such as chromosome bridges.
stg Induces Division Only in G2 Cells
Heat pulses applied to hs-stg embryos at a series of times during interphase 14 showed that mitosis could only be induced by heat pulses beginning at or after 160 min AED. This is 30 min after mitosis 13 and corresponds to the beginning of G2 (Edgar and Schubiger, 1986). Since we never observed chromosome bridges in even the earliest divisions induced in hs-stg embryos, we suspected that only cells that had completed DNA replication were induced to divide. In situ hybridization to embryos heat pulsed at a series of times in interphase 14 showed, however, that heat pulses before 160 min AED (during S phase) resulted in much lower accumulations of stg mRNA than heat pulses after 160 min AED. Thus, we could not evaluate whether the failure of early heat pulses to drive divisions was due to insufficient induction of stg mRNA or to insensitivity of cells to stg during S phase.
To address this question we applied a heat pulse at 210–235 min AED, a time when roughly half of the embryonic cells are in G2 of cycle 14, and most of the others are in S phase of cycle 15. We confirmed by in situ hybridization that all of the cells in these embryos produced large quantities of stg mRNA. However, we found that only cells still in G2 at the time of the heat pulse (mitotic domains 21–25, M, N, A, and B of Foe, 1989) were induced to divide (Figure 4). Cells in the early mitotic domains, which had already divided and were in S phase during the heat pulse, were not induced to divide again. Instead, these cells remained in interphase for greater than 30 min after the end of the heat pulse and then entered mitosis 15 with their normal timing and spatial pattern.
Figure 4. stg Induces Division Only in G2 Cells.

(A) The cyclin A pattern in a normal embryo at about 230 min AED. Brightly labeled cells on the ventral surface are in G2 of cycle 14. The other, darker cells have divided and are in S phase 15.
(B) Cyclin A pattern in a hs-stg embryo heat pulsed between 210 and 240 min AED and fixed at about 250 min AED. The G2 cells along the ventral surface have divided and degraded their cyclin A. Cells in other regions have not been induced to divide, as evidenced by their relatively high levels of cyclin accumulation.
(C) A hs-stg embryo treated as in (B), but labeled with BrdU for 20 min after the heat shock. Cells in the ventral region, which have been induced to divide, have entered S phase 15 and are heavily labeled. Cells in other regions show only spots of late replicating heterochromatin, indicating that they were in mid-S phase during the heat shock and in late S phase during the labeling period that followed. A normal, substantially different BrdU labeling pattern for this stage can be seen in Figure 2E. All embryos are 3/4, ventrolateral views, with anterior to the left and dorsal uppermost.
Many cell types appear to have a “replication feedback” mechanism that actively prevents the initiation of mitosis before DNA replication is completed (for review see Hartwell and Weinert, 1989). In Drosophila embryos, blocking DNA synthesis before mitosis 13 does not block mitoses, suggesting that such a mechanism does not operate in cleavage stage embryos (Raff and Glover, 1988). However, blocking S phase 14 does inhibit mitosis 14, indicating that replication feedback may begin functioning sometime in interphase 14 (B. A. E. and V. E. Foe, unpublished data). Thus, the failure of hs-stg to induce mitosis in S phase cells could be attributed to the overriding action of a replication feedback mechanism. Alternatively, stg’s inability to induce mitosis in S phase cells might indicate that it takes longer than S phase (about 40 min) to synthesize sufficient quantities of other products required for mitosis, such as cyclins. To distinguish between these two possibilities, we performed the following experiment: S phase 14 was blocked by injecting aphidicolin (a DNA synthesis inhibitor) into hs-stg embryos during mitosis 13 (at 130 min AED). Following the injection, stg mRNA was induced by a heat pulse from 160 to 185 min AED. The embryos were observed and finally fixed for analysis at 215 min AED. We found that large amounts of stg mRNA were expressed (assayed by in situ hybridization) and that levels of cyclin A were high (assayed by immunofluorescence). Nevertheless, 59 of 66 aphidicolin-injected embryos were arrested in interphase, whereas 71 of 83 controls (hs-stg embryos injected with a control buffer) showed many mitotic figures. The seven aphidicolin-injected embryos that were not arrested in interphase probably received too little of the drug, or received it too late, to sustain a complete inhibition of DNA replication. This result suggests that it is the failure to complete DNA synthesis, rather than an insufficient elapsed time since the previous mitosis, that makes S phase cells insensitive to stg product.
Mitoses 14 and 15 Trigger S Phases 15 and 16, Respectively
The DNA labeling experiments described earlier, as well as some elegant experiments done with early Xenopus embryos (Harland and Laskey, 1980; Blow and Laskey, 1988), suggest that S phase initiation in early embryos is tightly coupled to mitosis and is not differentially regulated. To test this idea, we labeled hs-stg embryos with BrdU following a heat pulse in G2 of cycle 14 (170–195 min AED). In these embryos, all of the nuclei entered S phase and became labeled within 5 min after the hs-stg–induced mitosis (Figure 5B). Even nuclei that normally do not divide, such as those in the amnioserosa, were induced to initiate DNA replication after division. A second normal division, exhibiting no chromosome bridges, could be induced in these embryos 45 min after the first induced division (not shown). The absence of chromosome bridges, which are readily observed in Drosophila embryos when mitosis occurs before the completion of DNA replication (B. A. E., unpublished data), allows us to conclude that the induced S phase goes to completion rapidly. Like our description of S phases in normal embryos, this result is consistent with the idea that G2/M is the only differentially regulated transition in the cell cycle at this stage of development. Once a G2/M transition is triggered, subsequent aspects of cycle progression appear to proceed automatically, in an orderly sequence, until the next G2 is achieved. Similar experiments showed that cells driven through mitosis 15 by hs-stg entered S phase 16 directly following the induced mitosis (not shown).
Figure 5. Mitosis 14 Triggers S Phase 15.
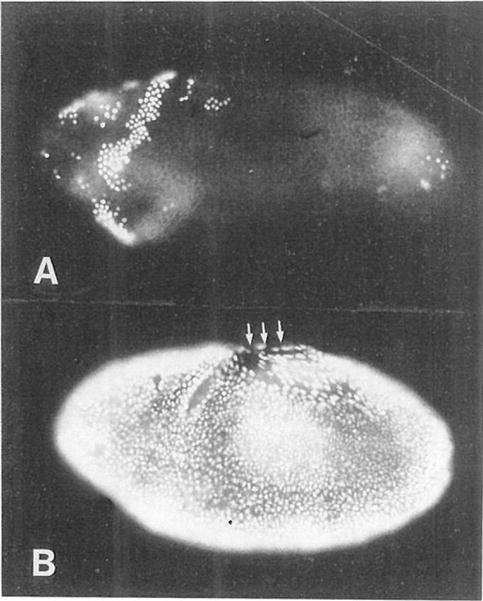
(A) A wild-type embryo that was heat pulsed between 165 and 190 min AED (during G2 of interphase 14) and then labeled for 20 min with BrdU. The labeling pattern is normal; nuclei in mitotic domains 1–4 have progressed through mitosis and into S phase 15.
(B) A hs-stg embryo that was similarly treated. All of the nuclei have been induced to divide and have progressed into S phase 15 prematurely. Note the labeled nuclei in the amnioserosa (upper middle region, arrows), which is normally nondividing and nonreplicating at this time. Both embryos are 3/4, ventrolateral views, with anterior to the left and dorsal uppermost.
Obligatory M/S Coupling Is Terminated during G2 of Cycle 16
Mitosis 16 occurs between 340 and 470 min AED, and, like mitoses 14 and 15, it begins in the mesoderm, then traverses the dorsolateral regions, and finally spreads ventrally, such that cells in the ventral neurogenic region are the last to divide. Mitosis 16 is the final embryonic division for most of the epidermal cells and includes essentially all of the dorsolateral epidermal cells, as judged by the abrupt degradation of cyclin A and the frequency of metaphase figures. In experiments on cycle 16, we have focused on these dorsolateral cells because they are readily identified and undergo mitosis in a narrow time window with a relatively simple spatial pattern. Pulse labelings with BrdU following mitosis 16 showed that only a small fraction of these dorsolateral cells enter S phase 17 after dividing. Most of those that do enter S appear to be precursor cells for the central and peripheral nervous systems (Bodmer et al., 1989). Thus, the majority of the dorsolateral epidermal cells enter their first G1 period after mitosis 16.
We found that induction of hs-stg mRNA by a heat pulse from 350–380 min AED, a time when most of the dorsolateral epidermal cells are in G2 of interphase 16, could induce these cells to go through mitosis 16 prematurely (as assayed by cyclin A degradation). Ventral cells, which are in S phase at this time, seemed to be unaffected. When these embryos were labeled with BrdU following the heat pulse, we were surprised to find that many of the dorsolateral cells (which normally enter G1 after mitosis 16) entered an abnormal S phase (S phase 17) after the hs-stg–induced mitosis 16 (Figure 6). Wild-type embryos heat pulsed at the same time showed no alterations in the normal patterns of S phases. By varying the timing of the heat pulses, we found that only cells induced to divide very early in G2 of interphase 16 entered this abnormal S phase 17. This experiment suggests that the mechanism that couples S phases to mitoses is still active in the early part of G2 of cycle 16 and is inactivated just before mitosis 16.
Figure 6. The Decision to Enter G1 of Cycle 17 Is Executed in G2 of Cycle 16.
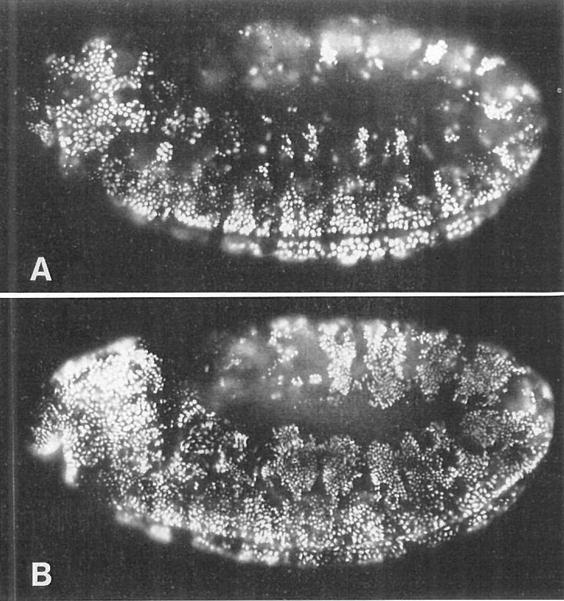
(A) A wild-type embryo heat pulsed from 350–380 min AED and then labeled with BrdU for 30 min. This pattern is identical to that seen in wild-type embryos that were not heat pulsed. Ventral cells are completing S phase 16 and are heavily labeled. Dorsolateral cells have progressed through mitosis 16, but only a few of them (one small cluster per segment) have entered S phase 17 and become labeled. The majority of these dorsolateral cells do not label with BrdU after mitosis 16, but instead enter their first G1 period.
(B) A hs-stg embryo that was similarly heat pulsed and BrdU labeled. In this embryo, the dorsolateral cells have been induced to go through mitosis 16 prematurely, and many of them have become heavily labeled with BrdU. More than the normal number of labeled cells can also be seen in the head, to the left. Thus, premature entry into mitosis 16 triggers an unscheduled S phase 17. Mitotic and replication patterns in the ventral region are essentially unaffected. Both embryos are lateral views, with anterior to the left. These are germ-band extended embryos, so the dorsal-most tissue (the amnioserosa) is the region in the center of the embryo, and strips of dorsolateral tissue curl around it. Ventral tissues are at the periphery of the embryo, on both the top and the bottom.
Finally, we looked at the effects of stg expression during the G1 period that follows mitosis 16 in the dorsolateral epidermis. Although hs-stg mRNA could be induced to high levels during this period, we did not find the ectopic mitotic figures or increased cell densities that would indicate a division was induced. We also saw no alteration in the normal pattern of S phases (data not shown). Therefore, we suspect that stg expression during this G1 period had no effect.
Is the Division Pattern Essential?
The normal pattern of cell divisions in the embryo is complex, highly ordered, and synchronized precisely with other developmental processes such as cell determination, morphogenetic movements, and cell differentiation. What are the consequences of altering this division pattern in a developing embryo? We have addressed this question by introducing abnormally timed divisions using the hs-stg gene.
In one experiment, we introduced a single synchronous division by a heat pulse during interphase 14 (170–195 min AED). Inspection of the division patterns at intervals after the heat pulse showed that these embryos underwent one synchronous division, and then resumed an essentially normal cell division program about 45 min after the end of the heat pulse (or about 35 min after the induced division). When cell division resumed, it followed a program appropriate for the age of the embryo: divisions occurred in a pattern characteristic of mitosis 15, rather than of mitosis 14. Thus, for most of the embryonic cells, the heat pulse probably did not cause an extra round of division. Rather, a patterned mitosis 14 was simply replaced by a synchronous mitosis 14. We were surprised to find that the effect of this treatment was less severe than expected. Although 60% of the hs-stg embryos failed to hatch after 36 hr, these showed no pattern defects in the differentiated cuticle. Forty percent of the hs-stg embryos did hatch, and 62% of these developed into viable, fertile adults.
Single 25 min heat pulses at progressively later times in development, which altered the division timing of fewer and fewer cells, resulted in progressively lower rates of death (Table 1). Curiously, heat pulses after 7 hr AED, which might be expected to affect cell cycle patterns in the developing nervous system, had no significant effect on viability. In an attempt to perturb division patterns more profoundly, we delivered twenty 25 min heat pulses over a 24 hr period beginning at 170 min AED. This treatment killed 90% of hs-stg embryos (Table 1), and we suggest that those that did survive were overaged (perhaps >7 hr AED) at the start of the first heat pulse.
Table 1.
Viability After Inducing hs-stg at Different Times in Development
| Strain | Heat Pulse at (hr AED) | Mitotic Stage at Heat Pulse | % Unhatched at 36 hr AED | (N) |
|---|---|---|---|---|
| Wild Type | — | — | 8 | 96 |
| Wild Type | 3.0 | G2 of cycle 14 | 6 | 63 |
| Wild Type | 6.0 | Cycle 16 | 7 | 75 |
| Wild Type | Multiplea | 3.0–24.0 | 5 | 120 |
| HS-STG3 | — | — | 9 | 241 |
| HS-STG3 | 3.0 | G2 of cycle 14 | 59 | 297 |
| HS-STG3 | 4.0 | Cycles 14 and 15 | 60 | 149 |
| HS-STG3 | 5.0 | Cycle 15 | 53 | 210 |
| HS-STG3 | 6.0 | Cycle 16 | 45 | 116 |
| HS-STG3 | 7.0 | Cycles 16 and neurogenesis | 16 | 207 |
| HS-STG3 | 10.0 | Neurogenesis | 13 | 119 |
| HS-STG3 | 13.0 | Neurogenesis | 9 | 244 |
| HS-STG3 | 18.0 | Neurogenesis (?) | 9 | 204 |
| HS-STG3 | Multiplea | 3.0–24.0 | 90 | 143 |
Eggs were collected for 20 min and allowed to develop at 25°C. Heat pulses were for 25 min at 37°C, and the remainder of development was at 25°C. Hatching normally occurs at 24 hr AED at 25°C. The 6%–9% unhatched eggs in control populations are mostly unfertilized. Tests for lethality caused by heat pulse–induced divisions were done using eggs collected from HS-STG3/+ parents, since HS-STG3 is a recessive lethal. Seventy-five percent of the embryos derived from these parents carried one or two hs-stg genes, and we confirmed by cyclin A staining patterns that 75% of these embryos underwent heat-induced divisions. The fractions of unhatched eggs listed are normalized to reflect hatch rates in hs-stg embryos only. Thus, % unhatched listed = actual % unhatched/.75.
In the multiple heat pulse experiments the temperature cycled between 37°C (for 25 min) and 25°C (for 50 min). Cycling commenced with a 37°C heat pulse at 170 min AED, and ended at 24 hr AED.
In another experiment, we attempted to rescue stg7B (formerly stg7B69) embryos using mitoses driven by the hs-stg gene (Figure 7). Homozygous stg7B embryos have no mitoses after mitosis 13, and although they develop substantially, they secrete a cuticle that is virtually devoid of differentiated pattern elements (Figure 7B). They fail to involute the head region, form no head structures, very few thoracic or abdominal denticles, and only rudimentary tail structures. We generated stg7B/stg7B; hs-stg embryos by crossing stg7B/+; hs-stg/+ flies to each other. Even without a heat shock, these embryos differentiated significantly more cuticular structure than stg7B/stg7B embryos (Figure 7C). This may be attributed to leaky expression of the hs-stg gene. In situ hybridization showed that this gene (RK-STG2) is expressed in the abdomen during cycle 14 without a heat pulse, and cyclin A staining patterns showed that it drives one randomly patterned division there. To achieve a greater degree of rescue, the stg/stg; hs-stg embryos were subjected to several 25 min heat pulses, beginning at 160 min AED. Each 37°C heat pulse was followed by a 60 min period at 25°C. Inspection of the division patterns in these embryos at various intervals using cyclin A immunofluorescence revealed that the heat-induced divisions occurred essentially synchronously and bore no resemblance to the normal mitotic pattern. Increasing the number of hs-stg–driven divisions gave a progressive increase in the extent of normal cuticular differentiation. Embryos heat pulsed once showed a normal number of differentiated segments, but had fewer than normal numbers of ventral denticles per segment and incomplete head and tail structures (Figure 7D). Some embryos that were heat pulsed twice, and nearly all of those heat pulsed three times, developed essentially normal cuticles (Figure 7E). This is a pleasing correlation, since the epidermal cells that secrete the cuticle normally have two or three postblastoderm divisions. Despite their normal cuticles, the embryos heat pulsed three times failed to hatch, and staining with a neuron-specific antibody (44C11; Bier et al., 1988) revealed that they had too few central and peripheral nervous system cells. This is not surprising since the central and peripheral nervous system cells normally have up to six postblastoderm divisions (Hartenstein et al., 1987; Bodmer et al., 1989), whereas our protocol would have delivered only three divisions.
Figure 7. The Normal Cell Division Pattern Is Not Essential for Pattern Formation in the Epidermis.

In this experiment, we attempted to rescue stg− embryos using cell divisions driven by the hs-stg gene.
(A) Cuticle preparation of a wild-type embryo just before hatching (about 23 hr AED).
(B) Cuticle of a stg7B/stg7B embryo. Note the lack of head involution and denticles.
(C–E) stg7B/stg7B embryos carrying the hs-stg gene, which were heat pulsed 0, 1, or 2 times, respectively. The hs-stg gene we used in this experiment (RK-STG2) is leaky, and induces one postblastoderm division in the abdomen without a heat pulse. This accounts for the partial rescue of the abdomen without a heat shock (C). The cuticle of the embryo heat pulsed once (D) has the proper number and polarity of segments, but each segment has fewer denticles than normal (typically 4 rows rather than the normal 6 or 7). Embryos heat pulsed two (E) or three times (not shown) developed essentially normal cuticles. Unhatched embryos (about 36 hr AED) were dechorionated, devitellinized in methanol/heptane, rehydrated, mounted in 1:1 lactic acid: Hoyer’s mountant, and baked at 60°C for 12 hr. Heads are to the left and dorsal surfaces uppermost.
We found that significant rescue of the cuticular pattern could be achieved by heat pulses as late as 7 hr AED, but not by heat pulses at 9 hr AED or later. This suggests that inducing the hs-stg gene after 7 hr AED does not trigger epidermal cell divisions in stg/stg embryos. Interestingly, the time interval when rescue was effective (roughly 2.5–7.5 hr AED) corresponds to the period when epidermal cell proliferation normally takes place. Other products required for mitosis, such as cyclins, are constitutively expressed in the epidermis during this period and are subsequently turned off (Lehner and O’Farrell, 1989, 1990). Each of the experiments we describe here suggests that a normal number of cell divisions is important to achieve proper cuticular differentiation, whereas the relative timing of these divisions is less critical.
Discussion
Modes of Embryonic Cell Cycle Regulation
In an earlier report, we presented the cell cycle arrest phenotype of stg mutant embryos and data describing the embryonic expression pattern of stg mRNA (Edgar and O’Farrell, 1989). We hypothesized that stg mRNA was the rate limiting factor for initiation of mitosis in the embryo, and that patterns of stg transcription thus controlled the patterns of embryonic mitoses. Here we corroborate this hypothesis by showing that ectopic expression of stg mRNA can trigger ectopic, but mechanically normal, cell divisions in the embryo. By inducing stg ectopically at different times during embryogenesis, we find that stg mRNA can induce mitosis in virtually all of the embryonic cells during the G2 periods of cell cycles 14, 15, and 16. This demonstrates that other factors required for mitosis are present at functionally sufficient levels throughout the embryo during these cycles (e.g., cyclins, cdc2, and other factors; see Lehner and O’Farrell, 1989, 1990; C. F. Lehner and P. H. O., submitted; Gatti and Baker, 1989). Since stg mRNA expression normally occurs in each cell at the end of G2, just prior to each of these mitoses, we believe that stg mRNA levels do indeed cause the mitotic pattern seen during these cycles.
Our analysis also revealed that stg has no detectable effect when expressed during S phases or G1 periods. This is consistent with the G2 arrest phenotypes of both stg and cdc25 mutants in indicating that this gene product executes its function at the G2/M transition, and is rate limiting for cell cycle progression only during this transition (Edgar and O’Farrell, 1989; Russell and Nurse, 1986; Russell et al., 1989). In addition to G2/M, other cell cycle transitions in the early embryo might be regulated differentially. Our comparison of patterns of mitosis and DNA synthesis in the embryo, however, suggests that other transitions, such as the M/S and S/G2 transitions of cycles 14 and 15, are automatic. We have confirmed this idea by showing that expressing stg in G2 always leads to a complete normal cell cycle: a mitosis followed immediately by an S phase, and finally arrest in the subsequent G2. This indicates that factors controlling S phases are constitutively active during cycles 14 and 15.
An often quoted cell cycle paradigm postulates a “basic” cell cycle oscillator that is universal to eukaryotic cells and is modified in different organisms and cell types by different kinds of accessory mechanisms (for reviews see Murray and Kirschner, 1989; Hartwell and Weinert, 1989). In this context, Drosophila embryos appear to have an essentially unregulated basic cell cycle oscillator during cycles 1–13. These cycles consist of a rapid progression of M and S phases, and are driven by maternally supplied products using exclusively posttranscriptional mechanisms (Edgar et al., 1986; O’Farrell et al., 1989). Following mitosis 13, this basic cycle is modified by the addition of G2 phases of variable length. The first G2 occurs just after maternal stg mRNA is degraded, and subsequent G2 phases are correlated with the absence of zygotically expressed stg mRNA. The aquisition of G2 phases appears to be the consequence of a transition from constitutively supplied stg function to regulated expression. After mitosis 16, the basic cycle is modified further in many cells by the addition of a G1 phase. The mechanism responsible for this is unknown, but one plausible explanation is that the constitutive function of S phase factors is terminated at this stage. In addition to S phase factors, the constitutive expression of mitotic factors such as cyclins also ends after mitosis 16 (Lehner and O’Farrell, 1989, 1990). Thus, the cell cycles following mitosis 16 (which are predominantly in the nervous system) could conceivably be controlled through regulated expression of stg, cyclins, S phase regulatory factors, or by different combinations of these factors in different cells.
Replication Feedback in the Embryo
One accessory that may be added to the basic cycle is a feedback mechanism that prevents initiation of mitosis before the completion of DNA replication (see Hartwell and Weinert, 1989). Although cleavage stage Drosophila embryos (cycles 1–13) appear to lack such a mechanism (Raff and Glover, 1988), replication feedback does seem to operate during cycle 14 (B. A. E. and V. E. Foe, unpublished data). Here we show that the inhibition of mitosis caused by blocking S phase 14 cannot be overcome by overexpressing stg mRNA. This implies the existence of a stg-independent replication feedback mechanism. A similar experiment performed in fission yeast with cdc25+ overexpression has yielded the opposite result, suggesting that replication feedback in yeast is cdc25+ dependent (Enoch and Nurse, 1990). The reasons for this difference are unclear, but may relate to the level of overexpression. Although replication feedback is commonly thought of as a safety mechanism used in response to environmental stress, it may have a developmental significance as well. Our studies show that cells in at least two tissues in the postblastoderm Drosophila embryo (the mesoderm and the dorsolateral epidermis, mitotic domains 10 and 11) appear to express stg mRNA constitutively through cycles 14 and 15, rather than having transient pulses of stg expression during G2, as is generally the case (B. A. E., unpublished data). These cells lack a prolonged G2 phase in cycle 15, suggesting that S phase feedback, rather than stg mRNA abundance, may limit their rate of cycle progression.
Cell Division and Development
The postblastoderm cell divisions in Drosophila occur in an exquisitely complex spatiotemporal pattern (Hartenstein and Campos-Ortega, 1985; Foe, 1989). This pattern is essentially invariant, evolutionarily conserved, and precisely coordinated with other aspects of morphogenesis (Foe and Odell, 1989). Moreover, the cell division patterns are highly correlated with patterns of cell fate determination and are one of the earliest visible markers of such (Foe, 1989). An example of this is the striking segmental iteration in division and DNA replication patterns, which can be seen in Figures 2 and 6. Such correlations suggest that division timing might play a role in morphogenetic processes, such as the formation of segments. Experimental observations, however, indicate the contrary. Embryos homozygous for null stg mutations, for instance, differentiate a surprisingly complete set of larval structures, including segments, without having any postblastoderm cell divisions (Hartenstein and Posakony, 1990). In this paper, we test the role of cell division in pattern formation by altering the relative timing and/or number of divisions. Interestingly, the pattern of differentiated structures made by the epidermal cells (the larval cuticle) is normal when the correct number of divisions is supplied with the wrong timing. Thus, the process of determining epidermal cell identities, which requires normal function of the segment polarity and homeotic genes, is not dependent on division timing. In contrast, reducing the number of divisions did have deleterious effects on the cuticle pattern. However, the pattern deletions caused by reduced numbers of divisions were random; they probably resulted directly from reduced cell numbers rather than from perturbations of cell fates.
If complexity in the division pattern is dispensible, why does it exist? First, we must note that altering the timing of a single patterned mitosis was lethal to 60% of embryos, and that perturbing mitotic timing on a continuous basis (by inducing stg expression every 75 min) was nearly completely lethal (Table 1). This indicates that altering the division pattern does introduce some (as yet undefined) developmental defects. We speculate that the embryos that survive perturbations of the division pattern may compensate for such developmental defects by regulating subsequent patterns of cell division or cell death. Most importantly, the fact that altering the division pattern is frequently lethal means that the division program that has been conserved by evolution has a significant selective advantage over randomly patterned or synchronous divisions.
Nevertheless, many embryos do survive drastic perturbations of the division program, and an essentially normal embryonic body pattern can be generated when divisions are synchronous, rather than patterned. This suggests that much of the complexity in division patterns is not absolutely required. One plausible explanation for “empty” complexity in the division pattern stems from our hypothesis that stg transcription is regulated by a battery of segmentation and homeotic (“selector”) genes (Edgar and O’Farrell, 1989; O’Farrell et al., 1989). These genes encode spatially regulated transcription factors that determine, in addition to cell cycle patterns (Foe and Odell, 1989), virtually all other aspects of cell fate. Since selector genes regulate a variety of downstream functions, many of the details of their expression patterns may have little significance with regard to their roles as regulators of stg. Nevertheless, stg may respond to functionally irrelevant details in selector gene expression patterns in cases where there has been no selective pressure to prevent it. In essence, the mode of regulation that the stg gene seems to have adapted (control by multiple regulators that have multiple targets) might necessarily generate expression patterns that are more complex than is actually required.
Experimental Procedures
Constructions, Fly Stocks, and Transformation
hs-stg genes were constructed by cloning the 2.0 kb Xbal–Accl fragment of pcSTG1.6 into two P element vectors containing the Drosophila hsp70 promoter. This fragment was cloned into the Kpnl site of pWH1 (Schneuwly et al., 1987) and into the polylinker of pRK261 (R. Kostriken, personal communication). Stable lines carrying these genes were made by standard P element–mediated transformation of y, Df(w)67 host flies (Rubin and Spradling, 1982; Spradling and Rubin, 1982). One insertion on chromosome 3 (HS-STG3, with the pWH1-stg chimera) and one on chromosome 2 (RK-STG2 with the pRK261-stg chimera) were obtained and used in these studies. HS-STG3 is a recessive lethal, and RK-STG2 causes virtual sterility in homozygous males. Wild-type flies were of the Sevelin strain.
Heat Shock Protocol
Eggs were collected on 140 mm grape juice agar plates for 20 min intervals, after a precollection period of greater than 1 hr. After aging at 25°C, the agar plates were placed in a 37°C incubator, face down to prevent evaporative cooling from the agar surface. Ten minutes at 37°C was the minimum heat pulse sufficient to drive ectopic mitoses, and 25 min heat pulses gave a more uniform expression and mitotic response. In some experiments, multiple heat pulses were delivered using a programmable thermocycling incubator (Bios Corporation). Embryos were fixed and stained with anti-cyclin A antibodies as described (Lehner and O’Farrell, 1989). Embryos stained with anti-tubulin antibodies (Amersham) were pretreated with taxol before fixation as described by Karr and Alberts (1986).
Injections and Labelings
BrdU was delivered by injection of a 50 mM solution {pH 9) as described in Edgar et al. (1986), or by permeabilization essentially as described by Limbourg and Zalokar (1973) and Bodmer et al. (1989). For permeabilization, embryos were placed in a basket with nitex screen, dechorionated for 2 min in 50% bleach, rinsed well, blotted and air dried well, and then swirled in octane (Aldrich gold label) for 5 min. Following this they were swirled into a monolayer on the nitex, air dried briefly, and immersed in Grace’s insect medium (GIBCO) containing 1 mg/ml BrdU for 20 min. The embryos were then swirled in heptane and pipeted into a two-phase fixative as described by Karr and Alberts (1986). BrdU-labeled DNA was detected by immunofluorescence as described by Schubiger and Palka (1987). Aphidicolin (Sigma) was injected at 500 μg/ml in 1% DMSO, a concentration that blocks 95% of DNA synthesis as measured by incorporation of coinjected [35S]TTP into trichloroacetic acid–precipitable material (B. A. E., unpublished data). Control injections were done using 1% DMSO.
In Situ Hybridizations
In situ hybridizations were done using digoxygenin-11-dUTP–labeled DNA probes (Boehringer-Manheim) according to Tautz and Pfeifle (1989), with the following modification. We found that reducing the probe size to <200 bp was necessary to reduce background staining. This was done by first cutting the template DNA into 200–300 bp fragments using restriction enzymes, and then performing the DNA synthesis reaction using 2 mg/ml random primers (pd(N)6; Pharmacia) at 12°C for 12 hr.
Acknowledgments
We would like to thank Christian Lehner for many fruitful discussions and for cyclin A antibodies. Thanks are also due to Rich Kostriken for the forgotten plasmid pRK261, to Charlie Oh for help with in situ hybridizations, to Rolf Bodmer for tips on BrdU labeling, and to J. P. Vincent for advice on transformation. Delia Lakich provided helpful criticism of the manuscript, and helped to quantify the hs-stg mRNA. This work was supported by National Science Foundation and National Institutes of Health grants to P. H. O. and an American Cancer Society Postdoctoral Fellowship to B. A. E.
References
- Bier E, Ackerman L, Barbel S, Jan L, Jan YN. Identification and characterization of a neuron specific antigen in Drosophila. Science. 1988;240:913–916. doi: 10.1126/science.3129785. [DOI] [PubMed] [Google Scholar]
- Blow JJ, Laskey RA. A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature. 1988;332:546–548. doi: 10.1038/332546a0. [DOI] [PubMed] [Google Scholar]
- Blumenthal AB, Kriegstein HJ, Hogness DS. The units of DNA replication in Drosophila melanogaster chromosomes. Cold Spring Harbor Symp Quant Biol. 1974;38:205–223. doi: 10.1101/sqb.1974.038.01.024. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Carretto R, Jan YN. Neurogenesis of the peripheral nervous system in Drosophila embryos: DNA replication patterns and cell lineages. Neuron. 1989;3:21–32. doi: 10.1016/0896-6273(89)90112-8. [DOI] [PubMed] [Google Scholar]
- Ducommun B, Dreatta G, Young P, Beach D. Fission yeast cdc25 is a cell-cycle regulated protein. Biochem Biophys Res Commun. 1990;167:301–309. doi: 10.1016/0006-291x(90)91765-k. [DOI] [PubMed] [Google Scholar]
- Edgar BA, O’Farrell PH. Genetic control of cell division patterns in the Drosophila embryo. Cell. 1989;57:177–187. doi: 10.1016/0092-8674(89)90183-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA, Schubiger G. Parameters controlling transcriptional activation during early Drosophila development. Cell. 1986;44:871–877. doi: 10.1016/0092-8674(86)90009-7. [DOI] [PubMed] [Google Scholar]
- Edgar BA, Kiehle CP, Schubiger G. Cell cycle control by the nucleocytoplasmic ratio in early Drosophila development. Cell. 1986;44:365–372. doi: 10.1016/0092-8674(86)90771-3. [DOI] [PubMed] [Google Scholar]
- Enoch T, Nurse P. Mutation of fission yeast cell cycle control genes abolishes dependence of mitosis on DNA replication. Cell. 1990;60:665–673. doi: 10.1016/0092-8674(90)90669-6. [DOI] [PubMed] [Google Scholar]
- Fantes PA. Isolation of cell size mutants of a fission yeast by a new selective method: characterization of mutants and implications for division control mechanisms. J Bacteriol. 1981;146:746–754. doi: 10.1128/jb.146.2.746-754.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foe VE. Mitotic domains reveal early commitment of cells in Drosophila embryos. Development. 1989;107:1–25. [PubMed] [Google Scholar]
- Foe VE, Alberts BM. Studies of nuclear and cytoplasmic behavior during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J Cell Sci. 1983;61:31–70. doi: 10.1242/jcs.61.1.31. [DOI] [PubMed] [Google Scholar]
- Foe VE, Odell GM. Mitotic domains partition fly embryos, reflecting early cell biological consequences of determination in progress. Am J Zool. 1989;29:617. [Google Scholar]
- Gatti M, Baker BS. Genes controlling essential cell-cycle functions in Drosophila melanogaster. Genes Dev. 1989;3:438–453. doi: 10.1101/gad.3.4.438. [DOI] [PubMed] [Google Scholar]
- Gould KL, Nurse P. Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature. 1989;342:39–45. doi: 10.1038/342039a0. [DOI] [PubMed] [Google Scholar]
- Harland RM, Laskey RA. Regulated replication of DNA microinjected into eggs of X. laevis. Cell. 1980;21:761–771. doi: 10.1016/0092-8674(80)90439-0. [DOI] [PubMed] [Google Scholar]
- Hartenstein V, Campos-Ortega JA. Fate-mapping in wild-type Drosophila melanogaster. I The spatio-temporal pattern of embryonic cell divisions. Roux’s Arch Dev Biol. 1985;194:181–195. [Google Scholar]
- Hartenstein V, Posakony JW. Sensillum development in the absence of cell division: the sensillum phenotype of the Drosophila mutant stg. Dev Biol. 1990;138:147–159. doi: 10.1016/0012-1606(90)90184-k. [DOI] [PubMed] [Google Scholar]
- Hartenstein V, Rudloff E, Campos-Ortega JA. The pattern of proliferation of the neuroblasts in the wild-type embryo of Drosophila melanogaster. Roux’s Arch Dev Biol. 1987;196:473–485. doi: 10.1007/BF00399871. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Karr TL, Alberts BM. Organization of the cytoskeleton in early Drosophila embryos. J Cell Biol. 1986;102:1494–1509. doi: 10.1083/jcb.102.4.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner CF, O’Farrell PH. Expression and function of Drosophila cyclin A during embryonic cell cycle progression. Cell. 1989;56:957–968. doi: 10.1016/0092-8674(89)90629-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner CR, O’Farrell PH. The roles of Drosophila cyclins A and B in mitotic control. Cell. 1990;61:535–547. doi: 10.1016/0092-8674(90)90535-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbourg B, Zalokar M. Permeabilization of Drosophila eggs. Dev Biol. 1973;35:382–387. doi: 10.1016/0012-1606(73)90034-1. [DOI] [PubMed] [Google Scholar]
- McKnight SL, Miller OL., Jr Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophila melanogaster embryo. Cell. 1977;12:795–804. doi: 10.1016/0092-8674(77)90278-1. [DOI] [PubMed] [Google Scholar]
- Moreno S, Hayles J, Nurse P. Regulation of p34cdc2 protein kinase during mitosis. Cell. 1989;58:361–372. doi: 10.1016/0092-8674(89)90850-7. [DOI] [PubMed] [Google Scholar]
- Murray A, Kirschner M. Dominoes and clocks: the union of two views of the cell cycle. Science. 1989;246:614–621. doi: 10.1126/science.2683077. [DOI] [PubMed] [Google Scholar]
- O’Farrell PH, Edgar BA, Lakich D, Lehner C. Directing cell division during development. Science. 1989;246:635–640. doi: 10.1126/science.2683080. [DOI] [PubMed] [Google Scholar]
- Pardee A. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- Rabinowitz M. Studies on the cytology and early embryology of the egg of Drosophila melanogaster. J Morphol. 1941;69:1–49. [Google Scholar]
- Raff JW, Glover DM. Nuclear and cytoplasmic mitotic cycles continue in Drosophila embryos in which DNA synthesis is inhibited with aphidicolin. J Cell Biol. 1988;107:2009. doi: 10.1083/jcb.107.6.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Russell P, Nurse P. cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell. 1986;45:145–153. doi: 10.1016/0092-8674(86)90546-5. [DOI] [PubMed] [Google Scholar]
- Russell P, Moreno S, Reed SI. Conservation of mitotic controls in fission and budding yeasts. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- Sadhu K, Reed SI, Richardson H, Russell P. cdc25Hs, a putative mitotic inducer gene in human cells, is predominantly expressed in G2. Proc Natl Acad Sci USA. 1990 doi: 10.1073/pnas.87.13.5139. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneuwly S, Klemenz R, Gehring WJ. Redesigning the body plan of Drosophila by ectopic expression of the homeotic gene Antennapedia. Nature. 1987;325:816–818. doi: 10.1038/325816a0. [DOI] [PubMed] [Google Scholar]
- Schubiger M, Palka J. Changing spatial patterns of DNA replication in the developing wing of Drosophila. Dev Biol. 1987;123:145–153. doi: 10.1016/0012-1606(87)90436-2. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM. Transposition of cloned P elements into Drosophila germ-line chromosomes. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]