

Abstract
It has been recently shown that the Alzheimer's disease (AD) pathogenic peptide amyloid β1–42 (Aβ1–42) binds to the α7 nicotinic acetylcholine receptor (α7nAChR) with high affinity and the α7nAChR and Aβ1–42 are both found colocalized in neuritic plaques of human brains with AD. Moreover, the intraneuronal accumulation of Aβ1–42 was shown to be facilitated by its high-affinity binding to the α7nAChR, and α7nAChR activation mediates Aβ-induced tau protein phosphorylation. To test the hypothesis that α7nAChRs are involved in AD pathogenesis, we used a transgenic mouse model of AD overexpressing a mutated form of the human amyloid precursor protein (APP) and lacking the α7nAChR gene (APPα7KO). We have shown that, despite the presence of high amounts of APP and amyloid deposits, deleting the α7nAChR subunit in the mouse model of AD leads to a protection from the dysfunction in synaptic integrity (pathology and plasticity) and learning and memory behavior. Specifically, APPα7KO mice express APP and Aβ at levels similar to APP mice, and yet they were able to solve a cognitive challenge such as the Morris water maze test significantly better than APP, with performances comparable to control groups. Moreover, deleting the α7nAChR subunit protected the brain from loss of the synaptic markers synaptophysin and MAP2, reduced the gliosis, and preserved the capacity to elicit long-term potentiation otherwise deficient in APP mice. These results are consistent with the hypothesis that the α7nAChR plays a role in AD and suggest that interrupting α7nAChR function could be beneficial in the treatment of AD.
Introduction
Alzheimer's disease (AD), with ∼26,000,000 patients worldwide, constitutes the most common neurodegenerative disorder. By 2050, ∼106,000,000 people are expected to suffer from AD if no preventive treatments become available (Brookmeyer et al., 2007).
Evidence suggests that accumulation of the amyloid β protein (Aβ) plays an important and potentially causal role in AD pathogenesis (Selkoe, 1991; Hardy and Higgins, 1992). However, although Aβ is itself cytotoxic, cell death in human brains is not well correlated with disease progression (Terry et al., 1991). Instead, changes in the density of the presynaptic marker synaptophysin are better correlated with disease progression than Aβ plaque load or cell death (Terry et al., 1991). Moreover, deficits in synaptic communication and plasticity in transgenic mouse and rat models with Aβ deposition (Rowan et al., 2003; Saganich et al., 2006; Liu et al., 2008) support the idea of the importance of synaptic health in the development of the disease. Interestingly, these synaptic changes occur before plaque accumulation, evidence that gives strength to the idea that the soluble nonfibrillary species of Aβ plays a crucial role in AD pathogenesis.
Currently, little is known about the mechanistic pathway leading downstream from Aβ to synaptic pathology and cognitive decline. The finding that Aβ1–42 binds to the α7 nicotinic acetylcholine receptor (α7nAChR) with picomolar affinity (Wang et al., 2000a,b) has led to the hypothesis that the α7nAChR subunit plays a role in AD. This is supported by evidence that (1) Aβ1–42 and α7nAChR are both colocalized in neuritic plaques in human brains with AD; (2) human neuroblastoma cells overexpressing α7nAChR are killed by Aβ1–42, whereas α7nAChR agonists protected the cells; (3) Aβ inhibits α7nAChR-dependent calcium activation and acetylcholine release (Wang et al., 2000a); (4) internalization of Aβ1–42 is facilitated by its binding to the α7nAChR on neuronal cell surfaces, followed by endocytosis of the resulting complex and its accumulation within the lysosomal compartment (Nagele et al., 2002); and (5) synaptic transmission is impaired by Aβ-dependent endocytosis of NMDA receptors after α7nAChR stimulation (Snyder et al., 2005). Findings from Dineley et al. (2001, 2002) further demonstrate that derangement of hippocampus signal transduction cascades in AD arises when increased Aβ burden chronically activates the ERK MAPK cascade in an α7nAChR-dependent manner, and Wang et al. (2003) demonstrated in hippocampal synaptosomes and human neuroblastoma cells that α7nAChR activation mediates Aβ-induced tau protein phosphorylation. Moreover, although Small et al. (2007) presented some evidence that Aβ1–42 binds to membrane lipids but does not bind to the α7nAChR, both increases and decreases in α7nAChR number have been reported in AD animal models and human patients (Bednar et al., 2002; Dineley et al., 2002; Teaktong et al., 2003; Yu et al., 2005; Counts et al., 2007), making a stronger case for a relationship between AD pathology and α7nAChR. Finally, a recent finding strongly supports α7nAChR as a mediator of Aβ-induced pathology in AD by demonstrating that both α7nAChR agonists and antagonists can modulate Aβ1–42-induced tau phosphorylation, possibly by involving GSK-3β (Hu et al., 2008).
To test the hypothesis that α7nAChRs are involved in AD pathogenesis, we used a transgenic mouse model of AD overexpressing a mutated form of the human APP (Hsia et al., 1999) on the background of a knock-out (KO) mouse for the α7nAChR (Orr-Urtreger et al., 1997). Our results support this hypothesis and pinpoint α7nAChR as a potentially important modulator of AD pathogenesis.
Materials and Methods
Animals
We generated a new line of transgenic mice by mating two established transgenic lines, PDAPP (J9) and the α7nAChR-null mutant (α7KO) mouse. PDAPP (J9) has been described previously (Hsia et al., 1999). Briefly, these mice carry human APP with two familial AD mutations, Swedish (K670N, M671L) and Indiana (V717F), downstream from the platelet-derived growth factor β promoter. For genotyping simplicity and because familial AD caused by APP mutations is inherited in autosomal dominant manner, the transgenic animals used were heterozygous with respect to the transgene. α7KO mice lack exons 8–10 of the gene coding for α7nAChR (Orr-Urtreger et al., 1997). Thus, our mouse line was derived by crossing mice heterozygous for the mutated human APP transgene and for the α7 null mutation with mice heterozygous for the α7 null mutation. We obtained six different genotypes from these crosses. We used the following four genotypes for this study: WT (no human APP, no α7 mutation); α7KO (homozygous for the α7 mutation, no human APP); APP (heterozygous for human APP, no α7 mutation); and APPα7KO (heterozygous for human APP, homozygous for the α7 mutation). Mice heterozygous for the α7 mutation alone and heterozygous for both human APP transgene and α7 mutation were used as breeders but were not subjected to the experimental manipulations performed in this study. All mice used for this study had >90% C57BL/6J background and <10% of the background derived from the original hybrid founders (DBA/2 and 129/SvEv, used to create APP and α7KO mice, respectively). To ensure comparable variability in background genes across genotypes, we included mice from 5–10 different litters per genotype. Genotypes were determined by PCR of tail DNA obtained at weaning and again after euthanasia for confirmation of genotype. At weaning, mice were also ear tagged with small metal earrings for identification. Animals were 13–16 months old at the beginning of testing. Both males and females were used. The same mice used for behavior were used for neuropathological analysis and electrophysiology but due to unexpected technical problems, behavioral tests were conducted at 13–16 months, whereas electrophysiology and neuropathology were performed at 19–22 months of age. In a few cases, for neuropathology assessment and long-term potentiation (LTP) recordings, we included age-matched naive mice (not used for behavior). Moreover, the only behavior recorded using an independent age-matched group of mice was visual cliff avoidance due to the need to shave the whiskers and to avoid any influence in other behaviors or postmortem evaluations from this transient sensory deprivation. Animals were housed in groups of 2–5 mice in standard cages located in individually ventilated racks with ad libitum access to regular food pellets and water in a room with controlled temperature (21 ± 1°C) and humidity (45–55%) and a 12:12 h light–dark cycle. All animal procedures were in compliance with National Institutes of Health (NIH) animal care guidelines, and all protocols were approved by the Salk Institute animal committee.
General evaluation
To verify the normal development of the novel APPα7KO mouse line, we assessed sensory, autonomic, neuropsychiatric, spinocerebellar, muscle, and lower motor neuron functions using a partial list of the test battery described in the SHIRPA protocol (Rogers et al., 1997). At 3–4 months of age, we measured body weight, body length, body tone, body position (pelvic elevation, tail elevation), spontaneous activity, transfer arousal (freezing, initiation of movement, gait, piloerection), startle response, touch escape, corneal reflex, pinna reflex, toe pinch, provoked biting, grip strength, wire maneuver, urination, defecation, tremor, and lacrimation as described by Rogers et al. (1997).
Behavior
Learning and memory.
Learning and memory was evaluated using the Morris water maze. Animals were trained to locate an escape platform in a circular pool (1.16 m diameter) of water made opaque by addition of nontoxic paint. Performance was evaluated as the time used to reach and climb onto the platform (latency). Mice were trained for a total of 8 d. To acclimate the animals to the test conditions, on days 1–3 mice were trained to locate a visible (cued) platform, a transparent platform submerged 1 cm from the water surface but indicated by a black and white striped flag attached to a pole such that it was 7 cm above the platform. Mice received 3 trials per day (10–15 min intertrial rest interval). The platform was located in a different position for each of these trials, and the animals were released from the same position with their heads facing the wall of the pool. When a mouse found the platform, it was allowed to stay on it for 15 s. If a mouse did not find the platform within the allotted 60 s time, it was guided gently with a “rescue” black spoon to the platform and allowed to stay on it for 15 s. After completing each trial, mice were removed from the pool using the rescue black spoon, dried with a soft towel, and returned to their home cage. On days 4–8, animals were trained to locate a hidden platform (same as before but with the flag cue removed), using extramaze visual cues (different-shaped posters hanging on the walls, a bench, and a computer). Each mouse was given three 60 s trials per day (10–15 min intertrial rest interval). In this case, the platform was always located in the same position [the center of the northeast (NE) quadrant], and the animals were released from different positions in each trial (the center of the SE, SW, and NW quadrants). Again, when mice found the platform, they were allowed to stay on it for 15 s. If a mouse did not find the platform within the allotted 60 s, it was guided gently with the spoon to the platform and allowed to stay on it for 15 s. After completing each trial, mice were removed from the pool using the spoon, dried, and returned to their home cage.
One day after completing this training, each animal was given a 60 s probe trial to evaluate retention of the task. During the probe test, the platform was removed, and searching behavior in the target quadrant (quadrant where the platform was located during hidden training) was measured. Animals were released from the center of the quadrant opposite to the target quadrant. A video-tracking system with Ethovision software (Noldus) was used to collect mouse movement (latency, swim path, distance, and speed).
Animals were excluded from the analysis if they were consistent floaters or if they were unable to follow the rescue black spoon. This happened more frequently in the APP mice [APP = 6/16 = 37.5%; WT = 4/17 = 23.5%; APPα7KO = 4/18 = 22.2%; and α7KO = 1/17 = 5.9%; p = 0.168 using a Fisher's test).
Motor coordination and balance.
Motor coordination and balance were evaluated by placing mice on a Rotarod (Economex, Columbus Instruments). After 30 s acclimating to the rod in a stationary mode, animals were trained to stay on for 90 s with the rod rotating at a constant 5 rpm speed. After this training, the actual test consisted of measuring the latency to fall from the rod rotating at an accelerated speed (from 4 to 40 rpm over a 6 min period). Mice were given three trials with a 45–60 min intertrial rest interval. The average latency to fall from these three trials was used for comparisons. Mice were returned to their home cage after each trial.
Ambulatory activity.
Ambulatory activity was evaluated by placing mice into an open-field arena for 5 min. The open-field apparatus consisted of a 43.2 × 43.2 × 30 cm arena with two 16-infrared beam arrays, connected through an interface to a PC running MED Activity program (Med Associates). The two arrays were located 1 cm from the arena floor, to record horizontal activity as distance traveled. The arena was located in a room with dim light. Animals were placed in the center of the arena to initiate the experiment and returned to their home cage at the end of the 5 min evaluation period.
Visual perception.
Visual perception was evaluated using a visual cliff avoidance test. The visual cliff apparatus evaluates the ability of the mouse to see the drop-off at the edge of a horizontal surface. We used a Plexiglas box that was built with a horizontal plane, connected to a vertical drop (50 cm high) connected to a second horizontal plane at a lower level. Contact paper with a black and white checkerboard pattern covered all three planes which accentuated the vertical drop-off. A sheet of clear Plexiglas was placed across the top horizontal plane (30 cm long by 50 cm wide), extending across the cliff (50 × 50 cm). Thus there was the visual appearance of a cliff, but in fact the Plexiglas provided a solid horizontal surface. A small platform (9 × 6 cm) was located 1 cm above the Plexiglas surface halfway over the “cliff” side and halfway over the “safe” side (horizontal checkered surface). Mice were placed on the platform at the start of each trial until completing eight successive (stepping down from platform) trials. We scored as positive (+1) when the mouse chose to step down onto the “safe” side and negative (0) when the mouse chose to step down onto the “cliff” side. Mice were returned to their home cage after each trial. To eliminate the confounding factor of sensory information from the whiskers, they were shaved off before starting this experiment (no further behavior test or postmortem evaluation was performed in mice in which whiskers were shaved off).
Neuropathology
Tissue processing.
Mice were killed by deep anesthesia with isoflurane gas and decapitation. Brains were removed and divided sagittally. One hemibrain was postfixed in phosphate-buffered 4% paraformaldehyde, pH 7.4, at 4°C for 48 h and sectioned at 40 μm with a Vibratome 2000 (Leica) for neuropathological analysis, while the other hemibrain was snap frozen in an isopentane/dry ice slurry and stored at −70°C for later protein analysis.
Analysis of neurodegeneration and levels of human APP and Aβ.
To evaluate the integrity of the neuronal structure, briefly as previously described (Mucke et al., 1995; 2000; Rockenstein et al., 2005a), blind-coded 40-μm-thick vibratome sections from mouse brains fixed in 4% paraformaldehyde were immunolabeled with the mouse monoclonal antibodies against synaptophysin (synaptic marker, 1:20, Millipore Bioscience Research Reagents), microtubule-associated protein 2 (MAP2, dendritic marker, 1:40, Millipore Bioscience Research Reagents), NeuN (neuronal marker, 1:1000, Millipore Bioscience Research Reagents), or glial fibrillary acidic protein (GFAP, astroglial marker, 1:500, Millipore Bioscience Research Reagents). To evaluate the levels of human APP and amyloid plaque load, briefly as previously described (Rockenstein et al., 2005a), additional sections were immunostained with a mouse monoclonal antibody against human APP (8E5 clone courtesy of ELAN) or the mouse monoclonal antibody against Aβ (4G8, 1:600, Senetek), respectively. GFAP primary antibodies were detected using biotinylated anti-mouse IgG (1:100, Vector Laboratories) and avidin–biotin–peroxidase complex (Vectastain Kit, Vector Laboratories) used according to the manufacturer's recommendations with 3′3′-diaminobenzidine (Vector Laboratories) as the chromogen. For all the other primary antibodies, after overnight incubation, sections were incubated with fluorescein isothiocyanate (FITC)-conjugated horse anti-mouse IgG secondary antibody (1:75, Vector Laboratories), transferred to SuperFrost slides (Fisher Scientific), and mounted under glass coverslips with anti-fading media (Vector Laboratories). All sections were processed under the same standardized conditions. The immunolabeled blind-coded sections were serially imaged with the Quantimet 570C system (Leica) or LSCM (MRC1024, Bio-Rad), and the digitized images were analyzed with the NIH Image 1.43 program, as previously described (Toggas et al., 1994; Mucke et al., 1995, 2000). For each mouse, a total of three sections were analyzed, and for each section, four fields in the frontal cortex and hippocampus were examined. For synaptophysin, MAP2, and Aβ, results were expressed as percentage area of the neuropil occupied by immunoreactive (IR) terminals, dendrites, or deposits, respectively; for GFAP and human APP immunostaining, levels were expressed as pixel intensity, and for NeuN, the mean neuronal density was estimated using a modified dissector method essentially as described (Hsia et al., 1999; Masliah et al., 2000).
We also determined levels of APP, APP products, and Aβ immunoreactivity in homogenates containing dissected cortex plus hippocampus, by Western blot, as previously described (Rockenstein et al., 2005a,b, 2007). Briefly, 20 μg per lane of cytosolic and particulate fractions, assayed by the BCA method (Pierce Biotechnology), were loaded into 4–12% SDS-PAGE gels and blotted onto polyvinylidene fluoride (PVDF) membranes. Blots were incubated with antibodies against full-length (FL) APP (mouse monoclonal, clone 22C11, 1:500, Millipore Bioscience Research Reagents), Aβ (mouse monoclonal, clone 6E10, 1:1000, Signet Laboratories), and APP C-terminal fragments (CTF-APP, rabbit polyclonal CT15, 1:1000, courtesy of Dr. E. Koo, University of California, San Diego, San Diego, CA), followed by secondary antibodies tagged with horseradish peroxidase (HRP, 1:5000, Santa Cruz Biotechnology) and visualized by enhanced chemiluminescence and analyzed with a Versadoc XL imaging apparatus (Bio-Rad). Analysis of actin levels was used as a loading control.
Finally, we determined Aβ levels by ELISA in dissected cortex plus hippocampus samples, which were sequentially extracted in a two-step procedure as previously described (Suzuki et al., 1994; Kawarabayashi et al., 2001). Briefly, samples were sonicated in 2% SDS with protease inhibitors and centrifuged at 100,000 × g for 1 h at 4°C. The resultant supernatant was harvested, constituting the SDS-soluble fraction. The resultant pellet was then extracted in 70% formic acid (FA) and centrifuged, and the resultant supernatant was collected (the FA-soluble fraction). For the sandwich capture ELISA, the following antibodies against Aβ were used: polyclonal 3160 (anti-Aβ1–40) was used for capture, and BA-27 (specific for Aβ1–40) and BC-05 (specific for Aβ1–42) were used for detection.
Electrophysiology
We performed in vitro slice electrophysiology in horizontal hippocampal slices (400 μm). We studied the Schaffer collateral–CA1 synapse using methods that we and others have published extensively (Swanson and Heinemann, 1998; Contractor et al., 2003; Armstrong et al., 2006; Saganich et al., 2006). All recordings were done with the experimenter blind to the genotype. Slices were prepared using standard methods (Contractor et al., 2003). Briefly, animals were anesthetized with isoflurane and decapitated. Tail biopsies were taken from all mice to confirm genotypes. To minimize the trauma associated with isolating hippocampal slices, the brains were removed under ice-cold oxygenated sucrose-slicing artificial CSF (ACSF) containing the following: 85 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 25 mm glucose, 75 mm sucrose, 0.5 mm CaCl2, and 4 mm MgCl2, equilibrated with 95% O2/5% CO2, which also contained the NMDA blockers d,l-APV (10 μm) and kynurenic acid (100 μm). Slices were incubated at 28°C for 30 min, followed by exchange of the sucrose-ACSF for an oxygenated sodium incubation-ACSF solution containing the following: 125 mm NaCl, 2.4 mm KCl, 1.2 mm NaH2PO4, 25 mm NaHCO3, 25 mm glucose, 1 mm CaCl2, and 2 mm MgCl2, in which slices remained for at least 2 h before recording. Individual slices were then transferred to the recording chamber and continuously perfused with an oxygenated sodium recording-ACSF containing 2 mm CaCl2 and 1 mm MgCl2. Extracellular field EPSPs (fEPSPs) were recorded at room temperature in the stratum radiatum of the CA1 region of hippocampus with a glass recording pipette filled with oxygenated sodium recording-ACSF with 2–3 MΩ tip resistance, using a MultiClamp 700A amplifier (Molecular Devices). fEPSPs were evoked using a concentric bipolar electrode (Frederick Haer Company) positioned in the Schaffer collateral/commissural pathway. Stimuli were controlled by pClamp 9 software (Molecular Devices) and were generated with an A310 Accupulser coupled to an A360 stimulation isolation unit (Warner Instruments). Data collection and analysis were performed with pClamp 9 (Molecular Devices), Excel, and SigmaPlot (Microsoft) programs. Basal synaptic transmission was assessed by comparing the input and output relationship of the fEPSPs recorded. Input was the peak amplitude of the fiber volley, and the output was the initial slope of the fEPSP. For each animal, we measured the fiber volley amplitude and initial slope of the fEPSP responses to a range of stimulation from 200 to 800 μA. The input–output relationship was then calculated by dividing the slope of the fEPSP by the fiber volley amplitude (from each point along the linear portion on the response curve) and taking the average value. This input–output value for each stimulation level was then averaged to give a single measure of basal synaptic transmission for each slice. For early-phase LTP (E-LTP) recordings (Sweatt, 1999), 400 μm hippocampal slices were prepared as described above but were incubated at 32°C for 30 min in sucrose-ACSF followed by a 2 h recovery period in the incubation-ACSF solution before recording. After a 20 min baseline period, LTP was induced by four tetani delivered 5 min apart, each at 100 Hz for 1 s (Walsh et al., 2005; Townsend et al., 2006a,b, 2007). The slope of the fEPSP was monitored for 60 min and compared with the slope of the field response during the control period. For late-phase LTP (L-LTP) recordings (Sweatt, 1999), the slices were incubated at 32°C for 1 h in sucrose-ACSF followed by a 3 h recovery period in the incubation-ACSF solution before recording. After a 30 min baseline period, LTP was induced as described for early-phase LTP above. The slope of the fEPSP was monitored for 150 min and compared with the slope of the field response during the baseline period.
Statistical analysis
For all experiments, experimenters were blind with respect to genotype. All data were expressed as mean ± SEM. To amplify the group size and increase reproducibility of the behavioral data, the testing was performed in two groups with genotype-balanced number and age-matched mice at different time points, and the data were pooled for analysis. Statistical analyses were performed with the GB-Stat 9.0 program (Dynamic Microsystems). For water maze analysis of latencies and speed measurements, differences among means were assessed using a two-way ANOVA with repeated measures for the “session” factor. In this case, “genotype” was the other factor. For water maze analysis of the probe test, we used a one-way ANOVA to compare probe measures between the target quadrant and all the other three quadrants within each genotype group. For all other experiments, differences among means were assessed by genotype using a one-way ANOVA. Post hoc Fisher's PLSD, Dunn's (Bonferroni) correction, or Dunnett's tests were performed when appropriate. Null hypotheses were rejected at the 0.05 level.
Results
All progeny generated from the cross between the original PDAPP (J9) line and Acra7KO line, named here as APP, APPα7KO, α7KO, and the WT siblings, appear normal at young ages, as shown previously for the parental lines (Orr-Urtreger et al., 1997; Paylor et al., 1998; Hsia et al., 1999). The progeny showed a Mendelian distribution and normal survival at the embryonic, perinatal, and adult stages. Furthermore, these mice grow normally, and showed a normal development as deduced from a general evaluation made at 3–4 months of age to assess sensory, autonomic, neuropsychiatric, spinocerebellar, muscle, and lower motor neuron functions by testing several parameters as reported in the SHIRPA protocol (Rogers et al., 1997). The progeny showed normal body weight, body length, body tone, body position (pelvic elevation, tail elevation), spontaneous activity, transfer arousal (freezing, initiation of movement, gait, piloerection), startle response, touch escape, some reflexes (corneal reflex, pinna reflex, toe pinch, provoked biting), grip strength, wire maneuver, urination, defecation, tremor, and lacrimation (data not shown).
Deficits in learning and memory in APP mice are improved in APPα7KO mice
Several studies have shown behavioral learning deficits in transgenic mice models of AD. The PDAPP J9 line was used to generate the mice for this study. Raber et al. (2000) reported a learning deficit by 6 months of age when this line was tested in the Morris water maze probe trial after the mice learned to find a visible and hidden platform. We further characterized the learning abilities of these APP-overexpressing mice at later ages and compared performances with APP littermates lacking functional α7nAChR (APPα7KO). α7KO and WT mice littermates were used as controls. As expected, 13- to 16-month-old APP mice performed very poorly in all versions of the Morris water maze tested: visible, hidden, and probe trials. However, interestingly, APPα7KO mice solved the different versions of this test significantly better than the APP mice (Fig. 1A, B), reaching levels similar to α7KO and WT controls. We initiated the training using a visible platform to give the animals a chance to acclimate to test conditions: swimming, climbing to the platform, handling by introducing and retrieving the animals from the water tank and guiding the animals to the platform when they do not find it themselves. As shown in Figure 1A, all groups learned to locate the visible platform in the course of 3 d (3 trials per day) measured as the reduction in the latency to climb the visible platform (F(2,106) = 14.899; p < 0.0001; repeated measures for session factor of a two-way ANOVA; Fisher's LSD post hoc test revealed the following significant differences for each individual group: both APP and APPα7KO day 3 vs day 1, p < 0.05; WT day 3 vs day 1, p < 0.01; α7KO day 2 and day 3 vs day 1, p < 0.05 and p < 0.01 respectively). Conversely, when comparing the performances among the different genotypes, there is a significantly poorer performance by the APP mice than by all three other genotypes: WT, α7KO, and most importantly, APPα7KO (Fig. 1A) (F(3,52) = 10.316; p < 0.0001; genotype factor of a two-way ANOVA; p < 0.01 vs α7KO and p < 0.05 vs WT or APPα7KO in the last session day by post hoc Fisher's LSD test).
Figure 1.

Deletion of the α7nAChR prevents learning and memory deficits observed in APP-overexpressing animals. Water maze performance was evaluated in mice at 13–16 months of age. Mice were tested for a total of 9 d. A, From days 1 to 3, visible platform training was performed to acclimatize the animals to the test conditions and from days 4 to 8; the animals were trained with a hidden platform to test spatial learning abilities. We measured latency to reach the platform. APP mice showed significant spatial learning impairments as a lack of improvement in five consecutive hidden platform sessions (p > 0.05), compared with APP mice lacking functional α7nAChR at levels similar to controls, WT and α7KO mice. For simplification, significant differences between groups are only shown for the last day of spatial learning (**p < 0.01 vs all 3 other groups). Note also that although APP mice significantly improved their performance in the visible platform version of the Morris water maze test (p < 0.05, day 3 vs day 1), they needed more time to reach the platform than all the other three genotypes (**p < 0.01 vs α7KO and *p < 0.05 vs WT or APPα7KO in the last session day). B, On day 9, memory was evaluated with a probe test as the frequency the target quadrant was visited in relation to the other three quadrants. Deletion of the α7nAChR rescued the memory deficit observed in APP-overexpressing animals and reached values similar to controls, WT and α7KO, as shown by a significant increase in frequency to enter the target quadrant (**p < 0.01 vs all 3 other quadrants). Note the inverse frequency in quadrant preference observed in APP mice (##p < 0.01 vs target quadrant and #p < 0.05 vs left and right quadrants). C–F, Motor (C, swim speed; D, rotarod; E, open field) and visual (F, visual cliff test) abilities are not significantly affected in APP mice, suggesting that performance deficits in all aspects of the Morris water maze test are consequence of affected learning and memory mechanisms (see Results for details on each of these four evaluations, especially the complex swim speed analysis). Differences among means were assessed by two-factor (training session and genotype) ANOVA with repeated measures for the training block factor or one-factor (genotype or quadrant) ANOVA followed by post hoc multiple-comparisons Fisher's test. Mean values ± SEM are shown.
We continued the training using the hidden platform version of the test. The animals must use spatial navigation abilities to locate this hidden escape platform by using extramaze cues. In this case, contrary to the previous visible platform training, APP mice did not show improvement in the time needed to find the hidden platform over successive sessions (Fig. 1A). Conversely, APPα7KO mice showed a significant reduction in the latency to reach the platform in the last two sessions of the test that was comparable to values obtained by the control WT and α7KO mice (Fig. 1A) (F(4,212) = 12.532; p < 0.0001; repeated measures for session factor of a two-way ANOVA; Fisher's LSD post hoc test revealed nonsignificant progress for APP, while it showed significant improvement for the other three genotypes: APPα7KO days 4 and 5 vs day 1, p < 0.01; WT day 5 vs day 1, p < 0.01; and α7KO day 2 vs day 1, p < 0.05 and days 3, 4, and 5 vs day 1, p < 0.01). When assessing differences between genotypes for this hidden version of the Morris water maze, APP-overexpressing mice lacking functional α7nAChR (APPα7KO mice), similar to control groups, showed a striking rescue of the performance deficits observed in APP animals (F(3,52) = 14.482; p < 0.0001; genotype factor of a two-way ANOVA; p < 0.01 for sessions 3 and 5 and p < 0.05 for session 4 between APPα7KO and APP by Fisher's LSD post hoc test).
We also performed a probe trial 24 h after completing the visible and hidden platform training to evaluate retention of the learning. As expected by the observation that APP animals were not able to learn the spatial version of the test, they also showed no retention of the target quadrant (the quadrant where the platform was located during the hidden version of training); instead, they showed preference for the quadrant where they were released for the probe trial (opposite quadrant) (Fig. 1B). On the other hand, APPα7KO mice similar to WT and α7KO mice showed a significant retention of the target quadrant, evaluated as the frequency to enter in the target quadrant versus the frequency to enter any of the other three quadrants (p < 0.01; Fisher's LSD test after significant one-way ANOVA for each individual group) (Fig. 1B).
Because motor and visual functions are crucial for performance in the Morris water maze task, we evaluated several related parameters. Figure 1C shows the swim speed across the nine water maze sessions for all groups. During the spatial version of the task (hidden platform and probe trials, days 4–9), the differences in speed between genotypes were not significant (hidden: F(3,52) = 1.171; p = 0.33; and probe: F(3,52) = 2.61; p = 0.062). During the visible platform version of the test, swim speed was similar between APP and WT mice, but higher speeds were observed for APPα7KO and α7KO mice (APP vs APPα7KO mice, p < 0.01 for sessions 1 and 2, and p < 0.05 for session 3; and APP vs α7KO mice, p < 0.01 for all three sessions). Curiously, the only significant differences in swim speed were observed in the visible version of the test, in which all the genotypes showed significant learning (Fig. 1A), but conversely, no significant differences were detected in the hidden and probe versions of the task, in which the APP mice were the only genotype that did not learn or retain the task (Fig. 1A, B). Thus, it seems unlikely that swim speed variation accounts for the learning and memory disparities displayed by these groups of mice. In addition, we challenged the animals on the rotarod as an independent measure of motor coordination and balance (a crucial ability for swimming), and in the open field for locomotor activity. All the genotypes performed similarly in the rotarod (Fig. 1D) (latency to fall, F(3,50) = 1.464; p = 0.24; one-way ANOVA) and open-field tests (Fig. 1E) (distance traveled, F(3,50) = 1.640; p = 0.19; one-way ANOVA), suggesting their general motor functions are not impaired. Furthermore, although all genotypes learned the visible version of the Morris water maze, some performance differences were still observed (Fig. 1A); thus, we further analyzed visual perception using an independent task. Using the visual cliff avoidance test, we did not detect any difference between genotypes in the ability to detect a virtual drop-off (F(3,24) = 0.336; p = 0.80) (Fig. 1F). This test was done using an age-matched group of mice.
These behavioral results demonstrate that the α7nAChR deletion in the background of APP overexpression in mice rescues the cognitive deficits observed in the APP-overexpressing mouse model of AD, consistent with the hypothesis that the α7nAChR play a role in AD.
Neuropathology in the APP mice model of AD is ameliorated by deleting the α7nAChR subunit despite similar levels of APP, Aβ expression, and plaque formation
Consistent with previous reports on the PDAPP mouse model of AD (Games et al., 1995; Hsia et al., 1999; Esposito et al., 2006), we found a marked reduction in the synaptic terminal marker synaptophysin both in frontal cortex (F(3,25) = 21.558; p < 0.0001) and hippocampus (F(3,25) = 28.442; p < 0.0001) in 19- to 22-month-old APP mice (p < 0.01 vs both controls, WT and α7KO, in both areas) (Fig. 2A). However, in APPα7KO mice, a significant preservation of synaptophysin-positive terminals was observed in both brain regions (p < 0.01) (Fig. 2A) (see also supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Consistent with previous publications (Games et al., 1995; Hsia et al., 1999; Esposito et al., 2006), we found a marked reduction in the MAP2-immunoreactive dendrites in both frontal cortex (F(3,31) = 11.145; p < 0.0001) and hippocampus (F(3,31) = 11.954; p < 0.0001) in APP mice (p < 0.01 vs both controls, WT and α7KO, in both areas) (Fig. 2B). However, in APPα7KO mice, a significant preservation of MAP2 expression was observed in both brain regions (p < 0.01) (Fig. 2B) (see also supplemental Fig. 2, available at www.jneurosci.org as supplemental material), although the levels of MAP2 immunoreactivity were still lower than those of the control mice in the hippocampus (p < 0.05 vs controls, WT and α7KO) (Fig. 2B). We also measured astrogliosis in frontal cortex and hippocampus using GFAP immunoreactivity as a marker. As shown in Figure 2C, GFAP immunoreactivity levels were significantly elevated in frontal cortex in APP mice (F(3,31) = 4.047; p = 0.0165; with p < 0.01 vs either control, WT or α7KO) (see also supplemental Fig. 3, available at www.jneurosci.org as supplemental material). GFAP immunoreactivity levels detected in the frontal cortex of APPα7KO mice were between the normal GFAP immunoreactivity values observed in WT and α7KO controls and the elevated GFAP immunoreactivity detected in APP mice, not reaching statistical significance (p > 0.05 vs any other group) (Fig. 2C). In the hippocampus, APPα7KO mice showed GFAP immunoreactivity values similar to the control groups, while GFAP immunoreactivity levels in the APP group tended (F(3,31) = 2.726; p = 0.063) to be higher although not statistically significant (Fig. 2C).
Figure 2.
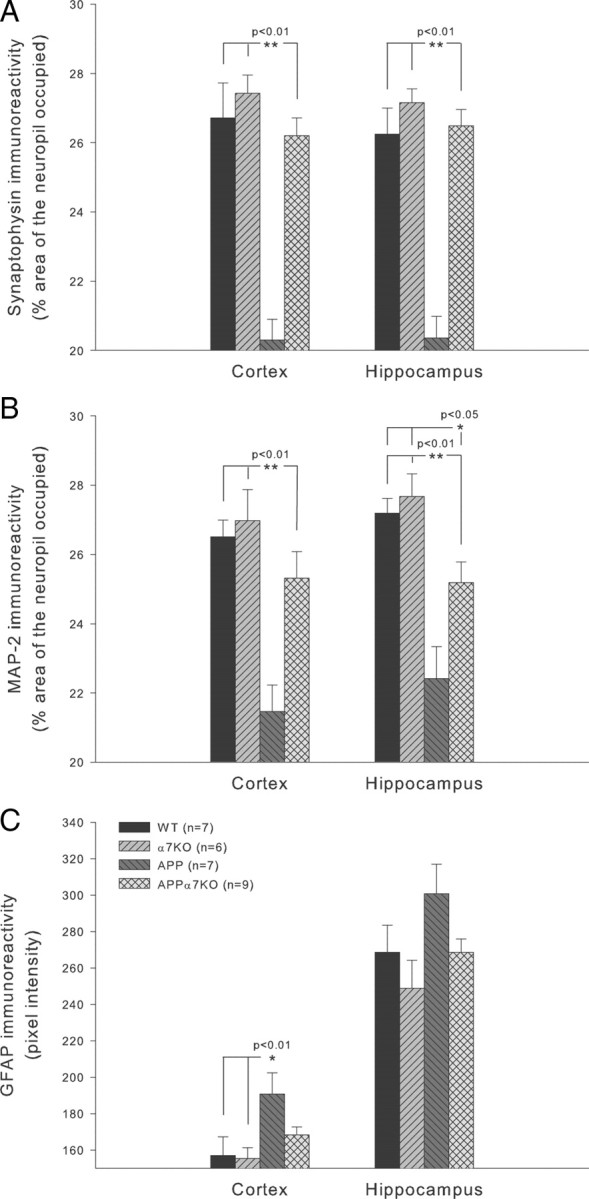
Deletion of the α7nAChR ameliorates the neuropathology observed in APP-overexpressing animals. Immunohistochemistry for the synaptic terminal marker synaptophysin, the dendritic marker MAP2, and the astrogliosis marker GFAP was evaluated in the frontal cortex and hippocampus of 19- to 22-month-old mice. A, A significant reduction in synaptophysin was observed in both cortex and hippocampus of APP mice compared with APPα7KO mice (p < 0.01). B, MAP2 was also markedly reduced in APP mice in both cortex and hippocampus compared with the normal or nearly normal levels in the cortex (p < 0.01) and hippocampus (p < 0.01 vs APP but also p < 0.05 vs controls, WT and α7KO) of APPα7KO mice, respectively. C, We also measured astrogliosis in frontal cortex and hippocampus using GFAP immunoreactivity as a marker. Immunoreactivity levels were significantly elevated in frontal cortex in APP mice (p < 0.01 vs controls, WT or α7KO). In contrast, GFAP immunoreactivity levels were only partially increased in the APPα7KO mice, not reaching statistical significance compared with WT or α7KO controls or APP mice (p > 0.05). In the hippocampus, APPα7KO mice showed GFAP immunoreactivity values similar to the control groups, while GFAP immunoreactivity levels in the APP group tended (p = 0.063) to be higher. Differences among means were assessed by one-way (genotype) ANOVA followed by post hoc multiple-comparisons Fisher's test. Mean values ± SEM are shown.
In addition, we evaluated the possibility of neuronal loss in frontal cortex and hippocampus. This is a controversial issue with conflicting results among the different AD transgenic mouse models studied (for review, see Duyckaerts et al., 2008). We measured NeuN immunoreactivity, and although there is a trend for a reduction in the number of NeuN+ neurons in APP mice, the differences did not reach statistical significance (data not shown).
Moreover, we compared the levels of glutamate receptor subunits in hippocampus plus cortex since it has been previously described that activation of α7nAChR is necessary for Aβ-induced NMDA receptor endocytosis (Snyder et al., 2005). Although no change in the total glutamate receptor subunit NR1 and NR2b proteins was reported in Snyder's paper (2005), the experiments there were performed under an acute exposure (1 h) of Aβ1–42 peptide, a situation extremely different from a chronic exposure to Aβ as it occurs in our animal model. Thus, we thought relevant to measure total NR1 and NR2b receptor proteins in our mice. We hypothesized that chronic in vivo exposure to Aβ species in our experimental animals would lead to differential degree of glutamate receptor subunit internalization with subsequent differences in the turnover rate of glutamate receptor subunits. This could be reflected as a change in total receptor protein when comparing mice with and without α7nAChRs. Therefore, we measured levels of NR1 and NR2b glutamate receptor subunits in brain homogenates of hippocampus plus cortex by Western blot, and we found no differences for either of the subunits between the four experimental groups used here. These results are shown in supplemental Figure 5 (available at www.jneurosci.org as supplemental material).
Although this result suggests that changes in NMDA receptor populations are not responsible for the beneficial effects of the α7nAChR loss of function in our AD mouse model, it will still be important to know whether NMDA receptor surface expression is increased in APPα7KO mice and whether such an increase might underlie the synaptoprotective effect of α7nAChR deletion in APP transgenic mice. Unfortunately at this moment, technical restrictions preclude us from accurately measuring proportions of intracellular/extracellular NMDA receptor proteins or NMDA-mediated currents in aged brain slices; thus, this potential mechanism warrants further exploration.
Finally, we assessed the levels of Aβ and APP expression using immunohistochemistry, Western blot, and ELISA methods, and the results revealed no differences between APP and APPα7KO mice (p > 0.05) (Fig. 3) (see also supplemental Figs. 4, 5, available at www.jneurosci.org as supplemental material), suggesting that the amelioration in behavioral performance and partial neuroprotection are independent of Aβ levels, plaque load, or APP processing.
Figure 3.

APP and APPα7KO mice express same levels of Aβ and APP in cortex and hippocampus. We assessed the levels of Aβ and APP expression using Western blot (A, B), immunohistochemistry (C, D), and ELISA (see supplemental Fig. 5, available at www.jneurosci.org as supplemental material) methods, and the results revealed no differences between APP and APPα7KO mice (p > 0.05). In A and B, results are shown for dissected cortex plus hippocampus. Mean values ± SEM are shown.
Taking together, these pathology results demonstrate that deletion of the α7nAChR in the APP-overexpressing mouse model of AD prevents synaptic loss observed in the APP mice, consistent with the hypothesis that the α7nAChR subunit plays a role in AD.
Deficits in LTP in APP mice are absent in APPα7KO mice
LTP in the CA1 region of the hippocampus is extensively studied and is thought to underlie the experience-dependent modification of behavior (Malenka and Bear, 2004). There is now substantial evidence showing deficits in synaptic plasticity in many different AD mouse models (Chapman et al., 1999; Larson et al., 1999; Giacchino et al., 2000; Oddo et al., 2003; Saganich et al., 2006), albeit not all (Hsia et al., 1999). We found that aged APP mice had significant deficits in E-LTP at the Schaffer collateral–CA1 synapse compared with WT control mice [p < 0.05 by Dunn's (Bonferroni) correction post hoc test after a significant one-way ANOVA: F(3,25) = 3.291; p = 0.0396]. Conversely, APPα7KO and α7KO mice groups did not show significant differences from WT mice (p > 0.05). Analysis of the last 10 data points recorded (one every 40 s) corresponding to the interval going from 60 to 66 min after LTP induction, demonstrated a 226 ± 35% (n = 5/3, 5 slices from 3 animals) increase in the average fEPSP slope in WT animals compared with a 146 ± 9% (n = 6/3) increase in APP mice (Fig. 4A). In contrast, we did not see a significant reduction in LTP in APPα7KO (172 ± 13%; n = 8/4) or α7KO (179 ± 11%; n = 7/4) mice compared with control WT mice (Fig. 4A). Although the 26 and 33% increase in the LTP measured in APPα7KO and α7KO mice, respectively, was not significant compared with APP mice in this E-LTP experiment, the magnitude of the effect may be an underestimate since the data include only those animals in which acceptable recordings were obtained. A strikingly high proportion of aged APP mice did not produce good brain slices for recordings and analysis of the data. Only 3 of 7 APP mice (42.9%) yielded slices with recordings that could be included in the data analysis. Conversely, APPα7KO mice had a high proportion of good slices and recordings (4 of 5 mice = 80.0%), similar to the slice recordings from α7KO (4 of 4 mice = 100.0%) or WT mice (3 of 4 mice = 75.0%) (Fig. 4A, table inset). This larger variability in the ability to obtain quality recordings from APP mice is likely a reflection of the larger neurodegeneration observed in this group (Fig. 2), which might lead to less viable brain tissue slices and/or more numerous “dead spots” [silent areas between recordable areas; see Larson et al. (1999)].
Figure 4.
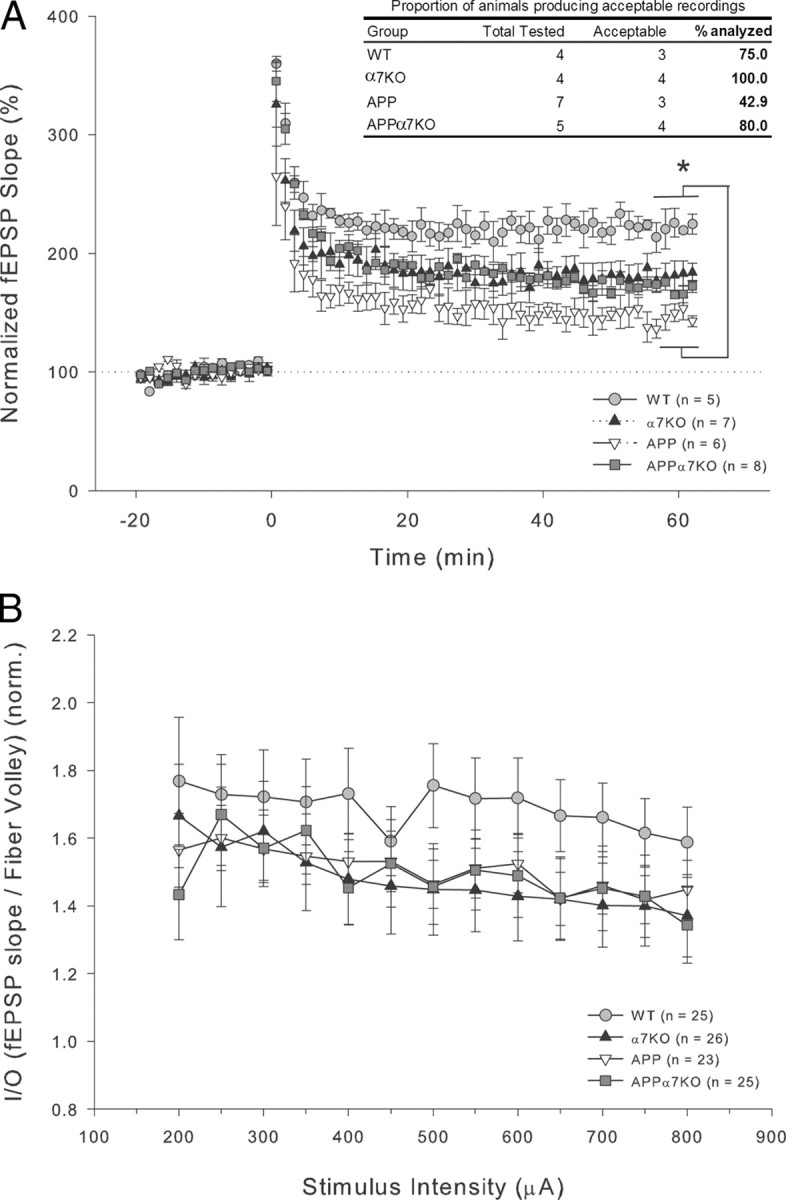
Deletion of α7nAChR prevents LTP deficits observed in APP-overexpressing mice at the Schaffer collateral to CA1 synapse. A, After a 20 min baseline, LTP was induced by four tetani delivered 5 min apart, each at 100 Hz for 1 s. fEPSPs were monitored for 60 min after tetanus. LTP in APP animals was significantly lower than in the control WT group (*p < 0.05). In contrast to APP mice, LTP was normal (no significant differences vs WT) in APPα7KO and α7KO mice groups. The magnitude of this effect may be an underestimate since the data include only those animals in which we could obtain acceptable recordings, and a strikingly high proportion of APP mice did not produce acceptable recordings (see table inset). Differences among means were assessed by one-way (genotype) ANOVA followed by post hoc Dunn's (Bonferroni) correction. B, Basal synaptic transmission was also recorded at the same synapse in a different set of age-matched mice, but no significant differences in the I/O slopes (fEPSP vs fiber volley) between all the four different genotypes were found. Mean values ± SEM are shown.
We also recorded basal synaptic transmission at the Schaffer collateral to CA1 synapse in a different set of age-matched mice, but found no significant differences in the baseline input–output slopes (I/O; fEPSP vs fiber volley) between all the four different genotypes (Fig. 4B).
Last, we examine L-LTP because the APPα7KO and the α7KO mice appear to be returning to baseline quicker than the WT mice and with a more parallel slope to APP mice in the LTP traces in Figure 4A (E-LTP). Thus, we wanted to examine LTP at a time point (longer than 2 h) that may engage synaptic processes involved in long-term memory storage (Sweatt, 1999) and may be a better correlate to the long-term memory processes examined by the Morris water maze task. To this end, we used a different set of age-matched mice that were naive to behavioral testing. In addition, we took great care in handling the tissue to obtain slices that remained healthy for the longer recording time. By increasing the incubation time at heated temperature, and the overall recovery time after slicing (see Materials and Methods section), we were able to record from 1 slice per animal for 150 min following a longer baseline period (30 min). Figure 5 shows that APP mice were the only group incapable of maintaining LTP at the late phase. After a significant induction of posttetanic potentiation), a weak LTP was holding for the first 30 min (p < 0.05; by Dunnett's post hoc test after a significant one-way repeated-measures ANOVA: F(4,25) = 2.761; p = 0.047) but then gradually and steadily returned to baseline. Conversely, APPα7KO mice maintained an appreciable potentiation for the entire 2.5 h of recording with no signs of decay, similar in potency and shape to WT and α7KO controls. To determine the effects of genotype and potentiation over time, the data were pooled into six 30 min blocks for statistical analysis.
Figure 5.

Deletion of α7nAChR in APP-overexpressing mice elicits a normal sustained late-phase LTP compared with the decay observed in APP-overexpressing littermates at the Schaffer collateral to CA1 synapse. After a 30 min baseline, LTP was induced by four tetani delivered 5 min apart, each at 100 Hz for 1 s. fEPSPs were monitored for 150 min after tetanus. LTP in APP animals was short lived during an early phase and rapidly decayed to baseline levels after 1 h. In contrast to APP mice, early and late LTP are normal in APPα7KO mice with no signs of decay and with potency similar to controls, WT and α7KO mice, for the entire 150 min recorded. Mean values ± SEM are shown.
This remarkable finding, namely, the absence of significant deficits in both early- and late-phase LTP in APPα7KO mice, even in the presence of levels of Aβ and plaques as high as those of APP mice, is consistent with the conclusion that inactivating functional α7nAChR reverses the deficits in the mouse model of AD.
Discussion
We have shown that despite the presence of high amounts of Aβ and amyloid deposits, deletion of the α7nAChR subtype in the mouse model of AD leads to protection from the dysfunction in synaptic integrity (pathology and plasticity) and learning and memory behavior. Specifically, APPα7KO mice express APP and Aβ at levels similar to APP mice and yet were able to solve a cognitive challenge such as the Morris water maze test significantly better than APP mice and reach comparable performance as control groups. Moreover, deletion of the α7nAChR subtype protected the brain from loss of the synaptic markers synaptophysin and MAP2, reduced gliosis, and preserved LTP that is otherwise defective in APP mice. These results are consistent with the hypothesis that the α7nAChR plays a role in AD and suggests that interrupting α7nAChR function could be beneficial in the treatment of AD.
Whether the beneficial effects of the α7nAChR loss of function is attributable to decreased intracellular Aβ or other mechanisms cannot be ascertained from the experiments in this study; however, in the last few years, several groups have provided evidence in transgenic rat (Echeverria et al., 2004) and mouse (Oddo et al., 2003; Casas et al., 2004; Billings et al., 2005; Zerbinatti et al., 2006; Knobloch et al., 2007) AD models and human patients with AD (D'Andrea et al., 2001; D'Andrea et al., 2002; Nagele et al., 2002) and Down syndrome (Gyure et al., 2001; Mori et al., 2002) implicating intracellular Aβ in the pathogenesis of AD, including its relationship to extracellular amyloid plaques as well as deleterious effects on synaptic function and learning and memory [for review, see Gouras et al. (2005), D'Andrea and Nagele (2006), and LaFerla et al. (2007)]. Specifically, the work of Nagele et al. (2002), suggested that the internalization of Aβ1–42 is facilitated by its high-affinity binding to the α7nAChRs on neuronal cell surfaces, followed by endocytosis of the resulting complex and its accumulation within the lysosomal compartment provoking intracellular toxicity. Our results showing that in the presence of similar amounts of amyloid plaques and extracellular Aβ, deletion of the α7nAChR subtype improved many aspects of cognitive, pathological, and neurophysiological deficits otherwise seen in APP mice, are consistent with the work of Nagele et al. (2002).
Another potential mechanism underlying the beneficial effects of the α7nAChR loss of function in APP-overexpressing mice is a possible Aβ-induced NMDA receptor endocytosis mediated by the activation of α7nAChR, as was shown in cortical neuron cultures by Snyder et al. (2005). Similar to Snyder's paper, we found no change in the total glutamate receptor subunits NR1 and NR2b proteins even though the Snyder experiments were performed under an acute exposure (1 h) of Aβ1–42 peptide, a situation extremely different from the chronic exposure to Aβ in our animal model. Although this result suggests that changes in NMDA receptor population are not responsible for the beneficial effects of the α7nAChR loss of function in our AD mouse model, it will still be important to ascertain whether NMDA receptor surface expression is increased in APPα7KO mice and whether such an increase might underlie the synaptoprotective effect of α7nAChR deletion in APP transgenic mice. Unfortunately, at this moment, technical restrictions preclude us from accurately measuring the proportions of intracellular versus extracellular NMDA receptor proteins or NMDA-mediated currents in aged brain slices; thus, this potential mechanism warrants further exploration.
Fodero et al. (2004) recently reported that α7nAChRs mediate an Aβ1–42-induced increase in the level of acetylcholinesterase in primary cortical neurons, an effect opposite to the current rationale for the use of the first class of drugs, only moderately effective but approved for the treatment of AD symptoms, inhibitors of acetylcholinesterase. Moreover, the other class of drugs approved for the treatment of AD represented by memantine, a use-dependent calcium channel blocker of the glutamate NMDA receptor, was recently shown to more potently block α7nAChRs than NMDA receptors in rat hippocampal neurons (Aracava et al., 2005; Banerjee et al., 2005), providing some extra evidence supporting the potential beneficial effect of antagonizing α7nAChRs.
Together, our data suggest that blocking the α7nAChR with an antagonist could lessen some of the symptoms of AD. This proposal of blocking α7nAChR in a disease characterized for cognitive decline is controversial in view of the evidence of cognitive improvement using α7nAChR agonists (Mazurov et al., 2006; Cincotta et al., 2008; Tietje et al., 2008). However, some effects of α7nAChR agonists can be mimicked by selective α7nAChR antagonists (Fujii et al., 2000; Ferchmin et al., 2003; Hu et al., 2007). The fast desensitization of α7nAChRs after its activation (Fenster et al., 1999; Mike et al., 2000; Quick and Lester, 2002; Gay et al., 2008) makes it difficult to distinguish between truly agonistic and antagonistic effects of an α7nAChR-targeted drug. In fact, it is not clear whether the cognitive enhancing effects are the result of receptor activation per se or activation-induced receptor desensitization (Banerjee et al., 2005).
Moreover, the suggestion driven by our data that deleting the α7nAChR could lessen some of the symptoms of AD may sound controversial with recent data from Octavio Arancio's group suggesting that α7nAChR activation is necessary for LTP enhancement by picomolar concentration of Aβ (Puzzo et al., 2008). However, the involvement of α7nAChRs in the enhancing effects of Aβ on synaptic plasticity and memory were only performed using the low dose of Aβ preparation (∼200 pm), which is close to the concentration found in the normal brain (Pawlik et al., 2004; Yao et al., 2004; Schmidt et al., 2005). That report also demonstrated the well established opposite effect caused by high levels of Aβ1–42 (∼200 nm), namely an impairment of LTP and memory, but unfortunately, the involvement of α7nAChRs in the detrimental effects of Aβ on synaptic plasticity and memory was not explored. Results published by Snyder et al. (2005) represent an example of the detrimental effects of high concentration of Aβ (1 mm), which are also thought to be mediated by α7nAChRs. Therefore, together these results by Puzzo et al. (2008) and Snyder et al. (2005) and our findings seem to confirm a close interaction between Aβ and α7nAChRs that could turn from positive effects in a normal physiological state (low concentration of Aβ) to detrimental effects in pathological conditions (high concentration of Aβ) like Alzheimer's disease.
In summary, the results presented in this paper demonstrate that the α7nAChR knock-out mutation rescues a significant part of the human APP-overexpressing phenotype. Thus, the α7nAChR contributes to both the pathology and behavioral deficits observed in the APP-overexpressing mouse. These results are consistent with the hypothesis that the α7nAChR plays a role in AD and suggests that blocking the α7nAChR could alleviate some symptoms of AD.
Footnotes
This work was supported in part by National Institutes of Health/National Institute of Aging Grant 5 P01 AG010435-16 (S.F.H. and E.M.), Bundy Foundation (G.D.), and the Ellison Medical Foundation (S.F.H). We thank Dr. Lennart Mucke for providing the PDAPP (J9) transgenic mice line, Dr. Jim Patrick for providing the α7nAChRKO mice, and Dr. Edward Koo for performing the capture ELISA for analysis of biochemically extracted Aβ. We also thank Dr. Michael Saganich and Dr Edward Han for their kind input on setting up the electrophysiology experiments and data interpretation; Dr. Lida Becerra for her help with some statistical analysis; Dr. T. K. Booker and Brian Glogowski for proofreading this manuscript; and Vanessa Ferraro, Conny Maron, and Amanda Vlasveld for extensively genotyping our mice colony.
References
- Aracava et al., 2005.Aracava Y, Pereira EF, Maelicke A, Albuquerque EX. Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than N-methyl-d-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 2005;312:1195–1205. doi: 10.1124/jpet.104.077172. [DOI] [PubMed] [Google Scholar]
- Armstrong et al., 2006.Armstrong JN, Saganich MJ, Xu N-J, Henkemeyer M, Heinemann SF, Contractor A. B-Ephrin reverse signaling is required for NMDA-independent long-term potentiation of mossy fibers in the hippocampus. J Neurosci. 2006;26:3474–3481. doi: 10.1523/JNEUROSCI.4338-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee et al., 2005.Banerjee P, Samoriski G, Gupta S. Comments on “Memantine blocks {alpha}7* nicotinic acetylcholine receptors more potently than N-methyl-d-aspartate receptors in rat hippocampal neurons.”. J Pharmacol Exp Ther. 2005;313:928–929. doi: 10.1124/jpet.104.081976. author reply 930–933. [DOI] [PubMed] [Google Scholar]
- Bednar et al., 2002.Bednar I, Paterson D, Marutle A, Pham TM, Svedberg M, Hellström-Lindahl E, Mousavi M, Court J, Morris C, Perry E, Mohammed A, Zhang X, Nordberg A. Selective nicotinic receptor consequences in APP(SWE) transgenic mice. Mol Cell Neurosci. 2002;20:354–365. doi: 10.1006/mcne.2002.1112. [DOI] [PubMed] [Google Scholar]
- Billings et al., 2005.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Brookmeyer et al., 2007.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- Casas et al., 2004.Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman et al., 1999.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TVP, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Cincotta et al., 2008.Cincotta SL, Yorek MS, Moschak TM, Lewis SR, Rodefer JS. Selective nicotinic acetylcholine receptor agonists: potential therapies for neuropsychiatric disorders with cognitive dysfunction. Curr Opin Investig Drugs. 2008;9:47–56. [PubMed] [Google Scholar]
- Contractor et al., 2003.Contractor A, Sailer AW, Darstein M, Maron C, Xu J, Swanson GT, Heinemann SF. Loss of kainate receptor-mediated heterosynaptic facilitation of mossy fiber synapses in KA2−/− mice. J Neurosci. 2003;23:422–429. doi: 10.1523/JNEUROSCI.23-02-00422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts et al., 2007.Counts SE, He B, Che S, Ikonomovic MD, DeKosky ST, Ginsberg SD, Mufson EJ. {alpha}7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch Neurol. 2007;64:1771–1776. doi: 10.1001/archneur.64.12.1771. [DOI] [PubMed] [Google Scholar]
- D'Andrea and Nagele, 2006.D'Andrea MR, Nagele RG. Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer's disease pyramidal neurons. Curr Pharm Des. 2006;12:677–684. doi: 10.2174/138161206775474224. [DOI] [PubMed] [Google Scholar]
- D'Andrea et al., 2001.D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–134. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- D'Andrea et al., 2002.D'Andrea MR, Nagele RG, Wang HY, Lee DH. Consistent immunohistochemical detection of intracellular beta-amyloid42 in pyramidal neurons of Alzheimer's disease entorhinal cortex. Neurosci Lett. 2002;333:163–166. doi: 10.1016/s0304-3940(02)00875-3. [DOI] [PubMed] [Google Scholar]
- Dineley et al., 2001.Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley et al., 2002.Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- Duyckaerts et al., 2008.Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverria et al., 2004.Echeverria V, Ducatenzeiler A, Dowd E, Jänne J, Grant SM, Szyf M, Wandosell F, Avila J, Grimm H, Dunnett SB, Hartmann T, Alhonen L, Cuello AC. Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience. 2004;129:583–592. doi: 10.1016/j.neuroscience.2004.07.036. [DOI] [PubMed] [Google Scholar]
- Esposito et al., 2006.Esposito L, Raber J, Kekonius L, Yan F, Yu G-Q, Bien-Ly N, Puoliväli J, Scearce-Levie K, Masliah E, Mucke L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenster et al., 1999.Fenster CP, Hicks JH, Beckman ML, Covernton PJ, Quick MW, Lester RA. Desensitization of nicotinic receptors in the central nervous system. Ann N Y Acad Sci. 1999;868:620–623. doi: 10.1111/j.1749-6632.1999.tb11335.x. [DOI] [PubMed] [Google Scholar]
- Ferchmin et al., 2003.Ferchmin PA, Perez D, Eterovic VA, de Vellis J. Nicotinic receptors differentially regulate N-methyl-d-aspartate damage in acute hippocampal slices. J Pharmacol Exp Ther. 2003;305:1071–1078. doi: 10.1124/jpet.102.048173. [DOI] [PubMed] [Google Scholar]
- Fodero et al., 2004.Fodero LR, Mok SS, Losic D, Martin LL, Aguilar MI, Barrow CJ, Livett BG, Small DH. Alpha7-nicotinic acetylcholine receptors mediate an Abeta(1–42)-induced increase in the level of acetylcholinesterase in primary cortical neurones. J Neurochem. 2004;88:1186–1193. doi: 10.1046/j.1471-4159.2003.02296.x. [DOI] [PubMed] [Google Scholar]
- Fujii et al., 2000.Fujii S, Ji Z, Sumikawa K. Inactivation of alpha7 ACh receptors and activation of non-alpha7 ACh receptors both contribute to long term potentiation induction in the hippocampal CA1 region. Neurosci Lett. 2000;286:134–138. doi: 10.1016/s0304-3940(00)01076-4. [DOI] [PubMed] [Google Scholar]
- Games et al., 1995.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Gay et al., 2008.Gay EA, Giniatullin R, Skorinkin A, Yakel JL. Aromatic residues at position 55 of rat alpha7 nicotinic acetylcholine receptors are critical for maintaining rapid desensitization. J Physiol. 2008;586:1105–1115. doi: 10.1113/jphysiol.2007.149492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacchino et al., 2000.Giacchino J, Criado JR, Games D, Henriksen S. In vivo synaptic transmission in young and aged amyloid precursor protein transgenic mice. Brain Res. 2000;876:185–190. doi: 10.1016/s0006-8993(00)02615-9. [DOI] [PubMed] [Google Scholar]
- Gouras et al., 2005.Gouras GK, Almeida CG, Takahashi RH. Intraneuronal A[beta] accumulation and origin of plaques in Alzheimer's disease. Neurobiology of Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Gyure et al., 2001.Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001;125:489–492. doi: 10.5858/2001-125-0489-IAAPDO. [DOI] [PubMed] [Google Scholar]
- Hardy and Higgins, 1992.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hsia et al., 1999.Hsia AY, Masliah E, McConlogue L, Yu G-Q, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu et al., 2007.Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li J. High content screen microscopy analysis of A[beta]1–42-induced neurite outgrowth reduction in rat primary cortical neurons: Neuroprotective effects of [alpha]7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 2007;1151:227–235. doi: 10.1016/j.brainres.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Hu et al., 2008.Hu M, Waring JF, Gopalakrishnan M, Li J. Role of GSK-3beta activation and alpha7 nAChRs in Abeta(1–42)-induced tau phosphorylation in PC12 cells. J Neurochem. 2008;106:1371–1377. doi: 10.1111/j.1471-4159.2008.05483.x. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi et al., 2001.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch et al., 2007.Knobloch M, Konietzko U, Krebs DC, Nitsch RM. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging. 2007;28:1297–1306. doi: 10.1016/j.neurobiolaging.2006.06.019. [DOI] [PubMed] [Google Scholar]
- LaFerla et al., 2007.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-[beta] in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Larson et al., 1999.Larson J, Lynch G, Games D, Seubert P. Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res. 1999;840:23–35. doi: 10.1016/s0006-8993(99)01698-4. [DOI] [PubMed] [Google Scholar]
- Liu et al., 2008.Liu L, Orozco IJ, Planel E, Wen Y, Bretteville A, Krishnamurthy P, Wang L, Herman M, Figueroa H, Yu WH, Arancio O, Duff K. A transgenic rat that develops Alzheimer's disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol Dis. 2008;31:46–57. doi: 10.1016/j.nbd.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka and Bear, 2004.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Masliah et al., 2000.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Mazurov et al., 2006.Mazurov A, Hauser T, Miller CH. Selective alpha7 nicotinic acetylcholine receptor ligands. Curr Med Chem. 2006;13:1567–1584. doi: 10.2174/092986706777442011. [DOI] [PubMed] [Google Scholar]
- Mike et al., 2000.Mike A, Castro NG, Albuquerque EX. Choline and acetylcholine have similar kinetic properties of activation and desensitization on the alpha7 nicotinic receptors in rat hippocampal neurons. Brain Res. 2000;882:155–168. doi: 10.1016/s0006-8993(00)02863-8. [DOI] [PubMed] [Google Scholar]
- Mori et al., 2002.Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA. Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid. 2002;9:88–102. [PubMed] [Google Scholar]
- Mucke et al., 1995.Mucke L, Abraham CR, Ruppe MD, Rockenstein EM, Toggas SM, Mallory M, Alford M, Masliah E. Protection against HIV-1 gp120-induced brain damage by neuronal expression of human amyloid precursor protein. J Exp Med. 1995;181:1551–1556. doi: 10.1084/jem.181.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke et al., 2000.Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagele et al., 2002.Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of [beta]-amyloid1–42 in neurons is facilitated by the [alpha]7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- Oddo et al., 2003.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger et al., 1997.Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the α7 neuronal nicotinic acetylcholine receptor lack α-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlik et al., 2004.Pawlik M, Sastre M, Calero M, Mathews PM, Schmidt SD, Nixon RA, Levy E. Overexpression of human cystatin C in transgenic mice does not affect levels of endogenous brain amyloid beta peptide. J Mol Neurosci. 2004;22:13–18. doi: 10.1385/JMN:22:1-2:13. [DOI] [PubMed] [Google Scholar]
- Paylor et al., 1998.Paylor R, Nguyen M, Crawley JN, Patrick J, Beaudet A, Orr-Urtreger A. Alpha7 nicotinic receptor subunits are not necessary for hippocampal-dependent learning or sensorimotor gating: a behavioral characterization of Acra7-deficient mice. Learn Mem. 1998;5:302–316. [PMC free article] [PubMed] [Google Scholar]
- Puzzo et al., 2008.Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick and Lester, 2002.Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Raber et al., 2000.Raber J, Wong D, Yu G-Q, Buttini M, Mahley RW, Pitas RE, Mucke L. Alzheimer's disease: apolipoprotein E and cognitive performance. Nature. 2000;404:352–354. doi: 10.1038/35006165. [DOI] [PubMed] [Google Scholar]
- Rockenstein et al., 2005a.Rockenstein E, Mante M, Alford M, Adame A, Crews L, Hashimoto M, Esposito L, Mucke L, Masliah E. High beta-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-beta levels: implications for the treatment of Alzheimer disease. J Biol Chem. 2005a;280:32957–32967. doi: 10.1074/jbc.M507016200. [DOI] [PubMed] [Google Scholar]
- Rockenstein et al., 2005b.Rockenstein E, Adame A, Mante M, Larrea G, Crews L, Windisch M, Moessler H, Masliah E. Amelioration of the cerebrovascular amyloidosis in a transgenic model of Alzheimer's disease with the neurotrophic compound Cerebrolysin™. J Neural Transm. 2005b;112:269–282. doi: 10.1007/s00702-004-0181-4. [DOI] [PubMed] [Google Scholar]
- Rockenstein et al., 2007.Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3β signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers et al., 1997.Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Rowan et al., 2003.Rowan MJ, Klyubin I, Cullen WK, Anwyl R. Synaptic plasticity in animal models of early Alzheimer's disease. Philos Trans R Soc Lond B Biol Sci. 2003;358:821–828. doi: 10.1098/rstb.2002.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saganich et al., 2006.Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci. 2006;26:13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt et al., 2005.Schmidt SD, Nixon RA, Mathews PM. ELISA method for measurement of amyloid-beta levels. Methods Mol Biol. 2005;299:279–297. doi: 10.1385/1-59259-874-9:279. [DOI] [PubMed] [Google Scholar]
- Selkoe, 1991.Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- Small et al., 2007.Small DH, Maksel D, Kerr ML, Ng J, Hou X, Chu C, Mehrani H, Unabia S, Azari MF, Loiacono R, Aguilar MI, Chebib M. The beta-amyloid protein of Alzheimer's disease binds to membrane lipids but does not bind to the alpha7 nicotinic acetylcholine receptor. J Neurochem. 2007;101:1527–1538. doi: 10.1111/j.1471-4159.2006.04444.x. [DOI] [PubMed] [Google Scholar]
- Snyder et al., 2005.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Suzuki et al., 1994.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- Swanson and Heinemann, 1998.Swanson GT, Heinemann SF. Heterogeneity of homomeric GluR5 kainate receptor desensitization expressed in HEK293 cells. J Physiol. 1998;513:639–646. doi: 10.1111/j.1469-7793.1998.639ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt, 1999.Sweatt JD. Toward a molecular explanation for long-term potentiation. Learn Mem. 1999;6:399–416. doi: 10.1101/lm.6.5.399. [DOI] [PubMed] [Google Scholar]
- Teaktong et al., 2003.Teaktong T, Graham A, Court J, Perry R, Jaros E, Johnson M, Hall R, Perry E. Alzheimer's disease is associated with a selective increase in alpha7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia. 2003;41:207–211. doi: 10.1002/glia.10132. [DOI] [PubMed] [Google Scholar]
- Terry et al., 1991.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tietje et al., 2008.Tietje KR, Anderson DJ, Bitner RS, Blomme EA, Brackemeyer PJ, Briggs CA, Browman KE, Bury D, Curzon P, Drescher KU, Frost JM, Fryer RM, Fox GB, Gronlien JH, Håkerud M, Gubbins EJ, Halm S, Harris R, Helfrich RJ, Kohlhaas KL, et al. Preclinical characterization of A-582941: a novel alpha7 neuronal nicotinic receptor agonist with broad spectrum cognition-enhancing properties. CNS Neurosci Ther. 2008;14:65–82. doi: 10.1111/j.1527-3458.2008.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toggas et al., 1994.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gpl20 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- Townsend et al., 2006a.Townsend M, Cleary JP, Mehta T, Hofmeister J, Lesne S, O'Hare E, Walsh DM, Selkoe DJ. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol. 2006a;60:668–676. doi: 10.1002/ana.21051. [DOI] [PubMed] [Google Scholar]
- Townsend et al., 2006b.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid {beta}-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006b;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend et al., 2007.Townsend M, Mehta T, Selkoe DJ. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- Walsh et al., 2005.Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerization of natural Aβ and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al., 2000a.Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000a;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2000b.Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide Abeta(1–42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem. 2000b;75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2003.Wang HY, Li W, Benedetti NJ, Lee DH. Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J Biol Chem. 2003;278:31547–31553. doi: 10.1074/jbc.M212532200. [DOI] [PubMed] [Google Scholar]
- Yao et al., 2004.Yao J, Petanceska SS, Montine TJ, Holtzman DM, Schmidt SD, Parker CA, Callahan MJ, Lipinski WJ, Bisgaier CL, Turner BA, Nixon RA, Martins RN, Ouimet C, Smith JD, Davies P, Laska E, Ehrlich ME, Walker LC, Mathews PM, Gandy S. Aging, gender and APOE isotype modulate metabolism of Alzheimer's Abeta peptides and F-isoprostanes in the absence of detectable amyloid deposits. J Neurochem. 2004;90:1011–1018. doi: 10.1111/j.1471-4159.2004.02532.x. [DOI] [PubMed] [Google Scholar]
- Yu et al., 2005.Yu WF, Guan ZZ, Bogdanovic N, Nordberg A. High selective expression of alpha7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer's disease and patients carrying Swedish APP 670/671 mutation: a possible association with neuritic plaques. Exp Neurol. 2005;192:215–225. doi: 10.1016/j.expneurol.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Zerbinatti et al., 2006.Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006;281:36180–36186. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]