

Abstract
Neurotoxic β-amyloid (Aβ) peptides participate in Alzheimer’s disease (AD); therefore, reduction of Aβ generated from APP may provide a therapeutic approach for AD. Gene knockout studies in transgenic mice producing human Aβ may identify targets for reducing Aβ. This study shows that knockout of the cathepsin B gene in mice expressing human wild-type APP (hAPPwt) results in substantial decrease of Aβ40 and Aβ42 by 67% in brain, and decreases levels of the C-terminal β-secretase fragment (CTFβ) derived from APP. In contrast, knockout of cathepsin B in mice expressing hAPP with the rare Swedish (Swe) and Indiana (Ind) mutations had no effect on Aβ. The difference in reduction of Aβ in hAPPwt mice, but not in hAPPSwe/Ind mice, shows that the transgenic model can affect cathepsin B gene knockout results. Since most AD patients express hAPPwt, these data validate cathepsin B as a target for development of inhibitors to lower Aβ in AD.
Keywords: Beta-amyloid, cathepsin B, gene knockout, amyloid precursor protein, protease
Introduction
Alzheimer’s disease (AD) is an age–related neurodegenerative disorder that results in progressive loss of memory and accumulation of neurotoxic β–amyloid (Aβ) peptides in brain [1–3]. The abnormal accumulation of Aβ peptides has been demonstrated as a major factor in the disease, based largely on studies of transgenic mouse models which overproduce Aβ, resulting in memory deficit and amyloid plaque brain pathology similar to that observed in AD patients [4–6]. Aβ peptides are generated from the amyloid precursor protein (APP) by proteases known as β– and γ–secretases, which release the N- and C-termini of Aβ, respectively. Several forms of Aβ are produced, including Aβ40 (1-40) and Aβ42 (1-42), which have the same N-termini but differ in their C-terminal residues.
The accumulation of brain Aβ can be reduced by inhibiting Aβ production, via reduction of β–secretase processing of APP. The vast majority of AD patients express wild–type (wt) APP (hAPPwt) which contains the wt β–secretase cleavage site sequence [7–9]. The aspartyl protease BACE 1 is a β–secretase [10–14], but it is relatively inefficient at cleaving the wt β–secretase site [15–17]. Inhibitors of BACE 1 have not yet been shown to reduce brain Aβ or improve memory deficit in AD patients. Thus, there remains a need to identify other potential targets that may also reduce production of brain Aβ from APP [17, 18].
The cysteine protease cathepsin B has been proposed as an alternative candidate β–secretase, which resides in the regulated secretory pathway of neurons, where it produces Aβ by efficient cleavage of the wt β–secretase site of APP [19–21]. In transgenic mice expressing human APP with the wt β-secretase site, inhibitors of cathepsin B significantly reduce brain levels of Aβ40 and Aβ42, and also reduce brain levels of C-terminal β–secretase fragment (CTFβ) derived from APP by β–secretase [21]. Importantly, these inhibitors provide substantial improvement in memory deficit, and reduce brain amyloid plaque load [21]. Furthermore, in normal guinea pigs, which express APP containing the human wt β–secretase site sequence ([22, 23], the cathepsin B inhibitors result in significant decreases in brain Aβ and CTFβ [24, 25]. The conclusion drawn from these inhibitor data is that cathepsin B has potential as a target for developing inhibitors that lower brain levels of Aβ for possible therapeutic treatment of AD.
However, another study using a genetic knockout approach finds that deletion of the cathepsin B gene in transgenic mice expressing hAPP containing the rare Swedish (Swe) and Indiana (Ind) mutations at the β–secretase and γ-secretase sites, respectively, results in no change in brain Aβ or CTFβ [26]. The conclusion drawn from the knockout data is that cathepsin B may not be a useful target for developing inhibitors to reduce brain Aβ as a therapeutic approach for AD.
To resolve the conflicting conclusions drawn from the cathepsin B inhibitor [21] and knockout data [26], this study directly compares the effects of cathepsin B gene knockout on Aβ in transgenic mice expressing hAPPwt, present in most Alzheimer’s disease patients, and in mice expressing mutant hAPPSwe/Ind which was used in the previous cathepsin B knockout study [26]. Results from this study demonstrate that deletion of the cathepsin B gene in the hAPPwt mice significantly reduces brain Aβ40 and Aβ42 by ~67%, and reduces APP-derived CTFβ by 41%. And, as previously reported [26], deletion of the cathepsin B gene in the hAPPSwe/Ind mice has no effect on brain levels of Aβ40 or Aβ42, and no effect on CTFβ. The striking difference in the consequences of cathepsin B gene deletion on Aβ brain levels in hAPPwt transgenic mice, compared to mice expressing mutant hAPPSwe/Ind, highlight the importance of selecting a mouse model that most closely resembles the majority of Alzheimer’s patients who express hAPPwt. These findings are consistent with the hypothesis that cathepsin B participates in Aβ production from APP containing the wild-type β-secretase site, but not from APP with the Swe mutant β–secretase site [21]. Significantly, because the majority of AD patients express hAPPwt, the knockout data of this study validates cathepsin B as a target for developing inhibitors to reduce brain Aβ generated from wt APP for potential therapeutic treatment of AD.
Materials and Methods
Transgenic cathepsin B knockout mice
The cathepsin B deficient mice (CatB−/−) were generated as previously described [27] and backcrossed to the C57BL/6 background for more than 10 generations. Transgenic mice were obtained from the Jackson Laboratory in which either the hAPPwt, or hAPP containing the Swedish mutations (K670N/M671L) at the β–secretase site combined with the Indiana mutation (V717F) at the γ–secretase site (hAPPSwe/Ind), were expressed under the platelet–derived growth factor beta promoter [28]. All mice were maintained on a C57BL/6 background. The hAPPwtXCatB−/− and hAPPSwe/IndXCatB−/− mice were generated by crossing the indicated strains. PCR analysis was utilized to determine the genotype of the animals, performed as previously described [27, 28]. Animal studies were conducted according to regulations by the NIH and as approved by the IACUC at the Medical University of South Carolina.
Preparation of Brain Extracts
Animals were sacrificed and brain extracts were prepared as previously described [21]. Briefly, brain extracts were homogenized (1:3 weight/volume of buffer) in buffer of 5 M guanidine HCl in 50 mM Tris-HCl, pH 7.6, 150 mM NaCl, plus protease inhibitors (Sigma). Homogenates were diluted to 0.5 M guanidine and centrifuged (200,000 × g for 20 min.), and supernatant and pellet fractions were collected. Protein content was determined by the Bradford method.
Aβ Analyses
Enzyme-linked immunosorbent assays (ELISAs) measured Aβ peptides by methods previously described [21]. The pellet from the brain extract procedure was sonicated in 6 M guanidine and centrifuged at 200,000 × g for 20 min at 4 °C, and the supernatant was diluted to 0.5 M guanidine. The two supernatants were combined, and Aβ40 and Aβ42 were determined using ELISA kits specific for each peptide (KHB3481/KHB3441; Biosource International). The mean levels of Aβ peptides in CatB−/− and CatB+/+ mice were compared
CTFβ and sAPPα Analyses
Western blot assays measured CTFβ and sAPPα as previously described with the same amounts of protein in each lane [21]. CTFβ was determined in the pellet fraction from the brain extract (antibody 8717, Sigma). In addition, sAPPα was assessed in the supernatant fraction from the brain extract (antibody GE10, Signet Laboratories). Relative amounts of CTFβ and sAPPα were measured by densitometry and results were expressed as percentage of the mean CTFβ and sAPPα compared to the control group. As control, β-actin western blots (anti-β-actin from Cell Signaling Technology) monitored equal loading of the same amounts of samples (20 μg protein) in gel lanes.
Statistical Evaluation
Experiments consisted of ten mice in each group. Each biochemical analysis consisted of two or three replicates. Statistical analyses utilized software designed for scientific data analysis by Student’s t-tests (Prism 4 GraphPad).
Results
Deletion of the cathepsin B gene reduces brain Aβ and CTFβ, and increases sAPPβ, in transgenic mice expressing hAPPwt
Deletion of the cathepsin B gene (CatB−/−) significantly reduced brain levels of Aβ40 and Aβ42 levels by 66% and 68%, respectively, in mice expressing hAPPwt, compared to control hAPPwt mice (CatB+/+) (figure 1).
Figure 1. Deletion of the cathepsin B gene in transgenic mice expressing human wild-type APP (hAPPwt) reduces brain Aβ40 and Aβ42.
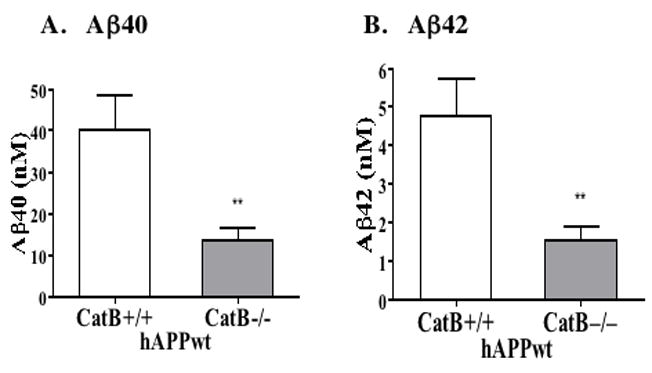
A. Aβ40 levels in brain. The Cat B+/+ and Cat B−/− mice expressing hAPPwt contained mean brain Aβ40 levels of 40.0 ± 21% and 13.6 ± 22% nM, respectively (significant, **p < 0.007). Knockout of the cathepsin B gene resulted in a 66% reduction in brain Aβ40 in mice expressing hAPPwt.
B. Aβ42 levels in brain. The Cat B+/+ and Cat B−/− mice expressing hAPPwt contained mean brain Aβ42 levels of 4.8 ± 21% and 1.5 ± 24% nM, respectively (significant, **p < 0.007). Knockout of the cathepsin B gene resulted in a 68% reduction in brain Aβ42 in animals expressing hAPPwt.
The effects of cathepsin B knockout on the C-terminal β-secretase fragment (CTFβ) derived from APP by β-secretase processing was then examined. Deletion of the cathepsin B gene (CatB−/−) resulted in significant reduction of brain levels of CTFβ by 41% in hAPPwt transgenic mice compared to controls (CatB+/+), illustrated by quantitation of CTFβ western blots (figure 2A, B). These data are consistent with a role of cathepsin B in Aβ production from hAPPwt by β-secretase processing.
Figure 2. Deletion of the cathepsin B gene in transgenic mice expressing human wild-type APP (hAPPwt) reduces brain CTFβ, and increases sAPPα, derived from APP.
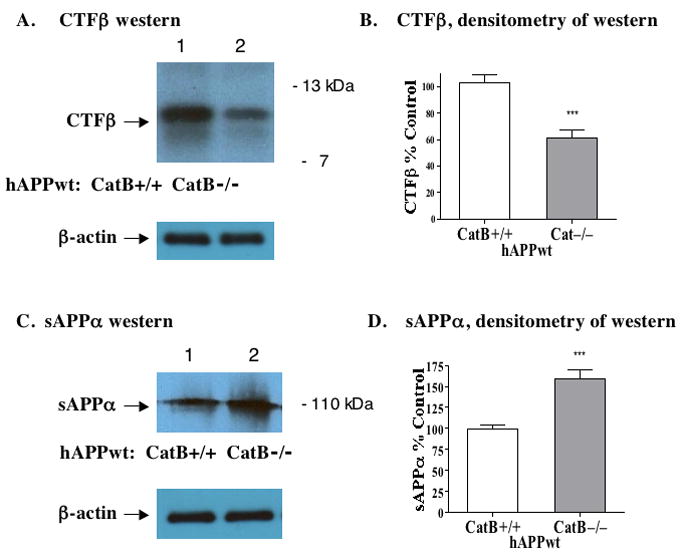
A. CTFβ western blot images. Western blot of the CTFβ band (~12kDa) shows its reduced levels in the Cat B−/− mice compared to the Cat B+/+ mice. The control blot for β-actin illustrates equal loading of sample, 20 μg protein, per gel lane.
B. CTFβ levels quantitated by densitometry. Densitometry of CTFβ western blots (from figure 2A) provided quantitation of CTFβ. The Cat B+/+ and Cat B−/− mice had mean brain CTFβ levels of 103% ± 6% and 61% ± 10%, respectively (significant, ***p < 0.0002). Knockout of the cathepsin B gene reduced CTFβ by about 41% in animals expressing hAPPwt.
C. sAPPα western blot images. Western blot shows higher levels of the sAPPα band (~110 kDa) in the CatB−/− mice compared to the Cat B+/+ mice. The control blot for β-actin illustrates equal loading of sample, 20 μg protein, per gel lane.
D. sAPPα levels quantitated by densitometry. Densitometry of sAPPα western blots (from figure 2C) provided quantitation. The Cat B+/+ and Cat B−/− mice had mean brain sAPPα levels of 99% ± 5% and 160% ± 5%, respectively (significant, ***p < 0.0001). Deletion of cathepsin B increased sAPPα by about 61% in hAPPwt mice.
In addition, cathepsin B deficiency increased brain levels of sAPPα by 61% in cathepsin B knockout mice (CatB−/−) relative to controls (CatB+/+) (illustrated by western blots and quantitation in figure 2C, D). The sAPPα fragment is derived from APP by cleavage at the α-secretase site within the Aβ domain, which precludes Aβ production. The increase in sAPPα occurs during decreased production of Aβ in the cathepsin B knockout condition.
In transgenic mice expressing mutant hAPPSwe/Ind, deletion of the cathepsin B gene has no effect on brain Aβ, CTFβ or sAPPα
Deletion of the cathepsin B gene (CatB−/−) in mice expressing hAPPSwe/Ind did not change the mean Aβ40 and Aβ42 brain levels, compared to control hAPPSwe/Ind mice expressing cathepsin B (CatB+/+) (Table 1) The cathepsin B deletion also had no effect on brain levels of CTFβ or sAPPα in hAPPSwe/Ind transgenic mice compared to the hAPPSwe/Ind controls expressing cathepsin B (CatB+/+) (Table 2).
Table 1.
Deletion of the Cathepsin B Gene in hAPPSwe/Ind Mice has No Effect on Aβ40 and Aβ42 in Brain.
| Aβ peptide levels in brain extracts |
||
|---|---|---|
| Aβ peptide |
Control, CatB +/+ |
Knockout, CatB−/− |
| Aβ40 | 104 ± 14 nM | 115.5 ± 16 nM |
| Aβ42 | 16.6 ± 12 nM | 19.8 ± 9 nM |
Aβ40 and Aβ42 peptides in brain extracts from CatB+/+ and CatB−/− of hAPPSwe/Ind mice were measured as described in the methods.
Table 2.
Deletion of the Cathepsin B Gene in hAPPSwe/Ind Mice has No Effect on CTFβ or sAPPα in Brain.
| Relative levels in brain extracts of hAPPSwe/Ind Mice, % Control |
||
|---|---|---|
| APP-derived fragment |
Control, CatB +/+ |
Knockout, CatB−/− |
| CTFβ | 99 ± 8 % | 103 ± 7 % |
| sAPPα | 100 ± 8 % | 98 ± 6 % |
CTFβ and sAPPα were evaluated in hAPPSwe/Ind mice with CatB+/+ or CatB−/− conditions, by western blots and densitometry, as described in the methods.
All mice appeared healthy
Animals remained healthy and were observed to have normal eating, body weight, grooming, and general appearance. None of the mice prematurely died or became ill.
Discussion
The significant result of this study is that deletion of the cathepsin B gene results in substantial reduction of brain Aβ40 and Aβ42 by ~67% in mice expressing hAPPwt. This finding demonstrates that the cathepsin B gene has a major role in production of Aβ peptides from hAPPwt in brain. The key feature of this novel finding is that the decrease in Aβ occurs in transgenic mice expressing hAPPwt, which is present in the majority of AD patients. Thus, these data validate cathepsin B as a target for development of inhibitors to reduce Aβ for AD therapeutics.
CTFβ is a proteolytic product derived from APP by β–secretase cleavage, and is a measure of β–secretase activity. The 41% reduction in CTFβ resulting from eliminating the cathepsin B gene in mice expressing hAPPwt suggests that deletion of the cathepsin B gene decreases β–secretase activity, and that the reduced brain Aβ in these mice may be due to a reduction in β-secretase.
The sAPPα fragment is a proteolytic product of APP generated by α–secretase cleavage, which produces a non-amyloidgenic peptide product and competes with β–secretase for cleavage of APP. The 61% increase in sAPPα resulting from deletion of the cathepsin B gene in mice expressing hAPPwt indicates that the deletion affects factors which influence α–secretase activity. The concomitant reduction in β–secretase activity in these animals may increase the availability of the APP substrate and thereby allow α–secretase to produce greater amounts of sAPPα.
These cathepsin B knockout data are consistent with results from in vivo cathepsin B inhibitor studies. Compounds which inhibit cathepsin B significantly reduce brain Aβ peptides (Aβ40 and Aβ42) and CTFβ, and increase sAPPα in transgenic mice expressing hAPP containing the wt β–secretase site sequence [21]. Most importantly, the cathepsin B inhibitors improve memory deficit and reduce amyloid plaque load [21]. In normal guinea pigs, inhibitors of cathepsin B also reduce brain Aβ peptides (Aβ40 and Aβ42) and CTFβ, and increase sAPPα [24, 25]; guinea pigs are a model of human APP processing [29] and possess APP containing the human wt β–secretase site sequence [22, 23, 29].
The cathepsin B knockout data are further supported by cellular studies. Both siRNA or chemical inhibition of cathepsin B reduce the amount of Aβ secreted from rat hippoocampal neurons in culture [30], which express APP containing the human wt β–secretase site sequence [22]. Also, an inhibitor of cathepsin B reduces levels of Aβ secreted from bovine chromaffin cells, accompanied by reduced CTFβ [19]; these chromaffin cells express bovine APP containing the human wt β–secretase site sequence [22].
Thus, studies of genetic knockout, chemical inhibition, and siRNA silencing of cathepsin B in several animal species and cells expressing APP with the wt β-secretase site all result in a significant reduction of Aβ.
In contrast, in mice expressing mutant hAPPSwe/Ind, deletion of the cathepsin B gene does not reduce Aβ40 or Aβ42, and has no effect on CTFβ or sAPPα, as shown in this study. These findings confirm the data previously reported for cathepsin B gene knockout mice expressing hAPPSwe/Ind [26].
The dramatically different results of cathepsin B knockout that occur in the hAPPwt compared to hAPPSwe/Ind mice are consistent with the ability of cathepsin B to cleave the wt β-secretase site, but not the Swe mutant β-secretase site of APP [20, 21]. In hAPPwt mice, knockout of the cathepsin B gene reduces Aβ and CTFβ, because cathepsin B has been shown to be highly efficient at cleaving the wt β–secretase site of hAPPwt [21]. Therefore, the absence of cathepsin B results in decreased amounts of Aβ. Similar to knockout of cathepsin B, inhibitors of cathepsin B reduce Aβ in mice expressing the wt β-secretase site of hAPP [21]. On the other hand, in hAPPSwe/Ind mice, deletion of the cathepsin B gene has no effect on Aβ or CTFβ, because cathepsin B cannot cleave the Swe β–secretase site of hAPPSwe/Ind [21]; therefore, the absence of cathepsin B has no effect on the production of Aβ in hAPPSwe/Ind mice. Similarly, inhibitors of cathepsin B have no effect on Aβ in mice expressing mutant hAPP with the Swe β-secretease site of APP [21, 31].
These data resolve the apparent conflicting conclusions drawn from the inhibitor [21] and previous knockout data [26] regarding cathepsin B as a useful target for developing AD treatments. Since deletion of the cathepsin B gene reduces Aβ in hAPPwt mice, cathepsin B is likely to be a useful drug target for development of inhibitors to reduce Aβ in the majority of AD patients expressing hAPPwt. However, inhibition of cathepsin B is not likely to be useful for reducing Aβ in the few AD patients expressing hAPPSwe [32], since deleting the cathepsin B gene does not effect Aβ in animals expressing Swe mutant hAPP.
In conclusion, deletion of the cathepsin B gene significantly reduces Aβ40 and Aβ42, as well as CTFβ, in transgenic mice expressing hAPPwt, which is expressed in the majority of AD patients. Therefore, the key point of this study is that results of the cathepsin B gene knockout in hAPPwt transgenic mice validates cathepsin B as a target for developing inhibitors to lower brain Aβ levels for the treatment of most AD patients.
Acknowledgments
We thank Dr. M. D. Pierschbacher for helpful review of the manuscript. This research was supported by the NIA/NIH R21 Grant AG027446 and 1R44AG032784 (to American Life Science Pharmaceuticals (ALSP)). V.H. holds equity in ALSP and serves on the Scientific Advisory Board of ALSP. The terms of this agreement have been reviewed by the University of California, San Diego, in accordance with its conflict of interest policies. G.H. holds equity in and is employed by ALSP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sisodia SS. Alzheimer’s disease: perspectives for the new millennium. J Clin Invest. 1999;104:1169–1170. doi: 10.1172/JCI8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 3.Gandy S, Martins RN, Buxbaum J. Molecular and cellular basis for anti-amyloid therapy in Alzheimer disease. Alzheimer Dis Assoc Disord. 2003;17:259–266. doi: 10.1097/00002093-200310000-00011. [DOI] [PubMed] [Google Scholar]
- 4.McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Dodart JC, Mathis C, Bales KR, Paul SM. Does my mouse have Alzheimer’s disease? Genes Brain Behav. 2002;1:142–155. doi: 10.1034/j.1601-183x.2002.10302.x. [DOI] [PubMed] [Google Scholar]
- 6.van Leuven F. Single and multiple transgenic mice as models for Alzheimer’s disease. Prog Neurobiol. 2000;61:305–312. doi: 10.1016/s0301-0082(99)00055-6. [DOI] [PubMed] [Google Scholar]
- 7.Turner RS. Alzheimer’s disease. Semin Neurol. 2006;26:499–506. doi: 10.1055/s-2006-951622. [DOI] [PubMed] [Google Scholar]
- 8.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 9.Dewachter I, Van Leuven F. Secretases as targets for the treatment of Alzheimer’s disease: the prospects. Lancet Neurol. 2002;1:409–416. doi: 10.1016/s1474-4422(02)00188-6. [DOI] [PubMed] [Google Scholar]
- 10.Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 11.Sinha S, Lieberburg I. Cellular mechanisms of beta-amyloid production and secretion. Proc Natl Acad Sci U S A. 1999;96:11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 13.Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 14.Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gruninger-Leitch F, Schlatter D, Kung E, Nelbock P, Dobeli H. Substrate and inhibitor profile of BACE (beta-secretase) and comparison with other mammalian aspartic proteases. J Biol Chem. 2002;277:4687–4693. doi: 10.1074/jbc.M109266200. [DOI] [PubMed] [Google Scholar]
- 16.Shi XP, Chen E, Yin KC, Na S, Garsky VM, Lai MT, Li YM, Platchek M, Register RB, Sardana MK, Tang MJ, Thiebeau J, Wood T, Shafer JA, Gardell SJ. The pro domain of beta-secretase does not confer strict zymogen-like properties but does assist proper folding of the protease domain. J Biol Chem. 2001;276:10366–10373. doi: 10.1074/jbc.m009200200. [DOI] [PubMed] [Google Scholar]
- 17.Schechter I, Ziv E. Kinetic properties of cathepsin D and BACE 1 indicate the need to search for additional beta-secretase candidate(s) Biol Chem. 2008;389:313–320. doi: 10.1515/BC.2008.025. [DOI] [PubMed] [Google Scholar]
- 18.Hook V, Schechter I, Demuth HU, Hook G. Alternative pathways for production of beta-amyloid peptides of Alzheimer’s disease. Biol Chem. 2008;389:993–1006. doi: 10.1515/BC.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hook V, Toneff T, Bogyo M, Medzihradszky K, Nevenu FJ, Lane W, Hook G, Reisine T. Inhibition of cathepsin B reduces β-amyloid production in regulated secretory vesicles of neuronal chromaffin cells: evidence for cathepsin B as a candidate β-secretase of Alzheimer’s disease. Biol Chem. 2005;386:931–940. doi: 10.1515/BC.2005.108. [DOI] [PubMed] [Google Scholar]
- 20.Hook VYH, Toneff T, Aaron W, Yasothornsrikul S, Bundey R, Reisine T. β-Amyloid peptide in regulated secretory vesicles of chromaffin cells: evidence for multiple cysteine proteolytic activities in distinct pathways for β-secretase activity in chromaffin vesicles. J Neurochem. 2002;81:237–256. doi: 10.1046/j.1471-4159.2002.00794.x. [DOI] [PubMed] [Google Scholar]
- 21.Hook VY, Kindy M, Hook G. Inhibitors of cathepsin B improve memory and reduce Abeta in transgenic Alzheimer’s Disease mice expressing the wild-type, but not the Swedish mutant, beta -secretase APP site. J Biol Chem. 2008;283:7745–7753. doi: 10.1074/jbc.M708362200. [DOI] [PubMed] [Google Scholar]
- 22.Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res Mol Brain Res. 1991;10:299–305. doi: 10.1016/0169-328x(91)90088-f. [DOI] [PubMed] [Google Scholar]
- 23.Beck M, Muller D, Bigl V. Amyloid precursor protein in guinea pigs--complete cDNA sequence and alternative splicing. Biochim Biophys Acta. 1997;1351:17–21. doi: 10.1016/s0167-4781(96)00232-1. [DOI] [PubMed] [Google Scholar]
- 24.Hook G, Hook VY, Kindy M. Cysteine protease inhibitors reduce brain beta-amyloid and beta-secretase activity in vivo and are potential Alzheimer’s disease therapeutics. Biol Chem. 2007;388:979–983. doi: 10.1515/BC.2007.117. [DOI] [PubMed] [Google Scholar]
- 25.Hook V, Kindy M, Hook G. Cysteine protease inhibitors effectively reduce in vivo levels of brain beta-amyloid related to Alzheimer’s disease. Biol Chem. 2007;388:247–252. doi: 10.1515/BC.2007.027. [DOI] [PubMed] [Google Scholar]
- 26.Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron. 2006;51:703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 27.Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, Domschke W, Lippert H, Peters C, Deussing J. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106:773–781. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beck M, Bigl V, Rossner S. Guinea pigs as a nontransgenic model for APP processing in vitro and in vivo. Neurochem Res. 2003;28:637–644. doi: 10.1023/a:1022850113083. [DOI] [PubMed] [Google Scholar]
- 30.Klein DM, Felsenstein KM, Brenneman DE. Cathepsins B and L differentially regulate amyloid precursor protein processing. J Pharmacol Exp Ther. 2009;328:813–821. doi: 10.1124/jpet.108.147082. [DOI] [PubMed] [Google Scholar]
- 31.Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118:2796–2807. doi: 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360:672–74. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]