

Abstract

We have previously reported the discovery and preliminary structure activity relationships of a series of β-aminoketones that disrupt the binding of coactivators to TR. However, the most active compounds had moderate inhibitory potency and relatively high cytotoxicity, resulting in narrow therapeutic index. Additionally, preliminary evaluation of in vivo toxicology revealed a significant dose related cardiotoxicity. Here we describe the improvement of pharmacological properties of thyroid hormone receptor coactivator binding inhibitors. A comprehensive survey of the effects of substitutents in key areas of the molecule was carried out, based on mechanistic insight from the earlier report. This study revealed that both electron withdrawing and hydrophobic substituents on the aromatic ring led to higher potency. On the other hand, moving from an alkyl to a sulfonyl alkyl side chain led to reduced cytotoxicity. Finally, utilization of amine moieties having low pKa’s resulted in lowered ion channel activity without any loss of pharmacological activity.
Introduction
The thyroid hormone receptors (TRs), belonging to the superfamily of nuclear receptors (NRs), regulate development, growth and metabolism.1–3 The thyroid hormone (T3) induces the majority of transcriptional responses mediated by TR’s in vivo.4 The TR’s have two isoforms, TRα and TRβ, encoded by two genes, with each isoform having two distinct subtypes due to alternative splicing.5 Although these isoforms are widely expressed, there are distinct patterns of expression that vary with tissue and developmental stage. TRα or TRβ knockout mice display unique phenotypes, suggesting that the different TR isoforms have unique regulatory roles. 4, 6
TR has three functional domains: a N-terminal ligand independent transcription activation domain (AF-1), a central DNA binding domain (DBD), and a carboxy terminal ligand binding domain (LBD) containing a thyroid hormone inducible coactivator binding domain (AF-2).7 Upon binding of thyroid hormone, TR undergoes a conformational change, releasing corepressor proteins and recruiting coactivator proteins, such as steroid receptor coactivators (SRC’s), to regulate gene transcription.8 Although two structurally distinct antagonists of T3 have been reported, no antagonist drugs are yet approved to treat thyroid receptor malfunctions. 9–12
We have previously reported the discovery and preliminary structure activity relationships of a series of β-aminoketones that disrupt the binding of coactivators to TR without affecting T3 binding.13–15 These time-dependent irreversible inhibitors were proposed to work by in situ generation of an enone followed by a reaction between the electrophilic enone and a nucleophilic cysteine in the coactivator binding pocket. The recently reported X-ray structure of TRβ bound to an enone derived by elimination from one of these aminoketones supports this hypothesis.16
TRβ is unique among the nuclear receptors in having three cysteine residues (C294, C298, and C306) located in or near the coactivator binding site. Active site mutagenesis and mass spectroscopy revealed that the enones derived from this series of β-aminoketones selectively attack C298, even in the presence of 10 mM β-mercaptoethanol. A preliminary SAR study of β-aminoketones and the various electrophilic compounds confirmed important features of these selective small molecule inhibitors.15 However, the most active compounds emerging for this study had apparent IC50’s in the low micromolar range with relatively high cytotoxicity. Additionally, preliminary evaluation of in vivo toxicology revealed a significant dose-related cardiotoxicity, which is consistent with the original use of these β-aminoketones as sodium channel targeted local anesthetics.17, 18 Reduction of cytotoxicity, lowering of ion channel activity, and potentially improvement of potency – as long as selectivity could be maintained – would increase the therapeutic window and enable us to utilize these compounds in vivo to study the role of coactivator recruitment in thyroid hormone endocrine action.
Herein, we present the synthesis and characterization of β-aminoketones with improved properties with an emphasis on (i) the orientation of carbonyl group, (ii) substitution of phenyl core structure (iii) substitution on α-position of the aminoketone, (iv) alternate β-amino moieties, and (v) functionality of alkyl side chain (Figure 1). These chemical features were optimized in order to provide the best balance between maximal potency, efficacy, and selectivity; minimal cellular toxicity; favorable physiochemical properties; and minimal activity at cardiac ion channels.
Figure 1.

Structure of β-aminoketone 1 and five parts of SAR modification.
Results and Discussion
Chemistry
The synthesis of the β-aminoketone compounds was accomplished by two different routes (Scheme 1): A) Friedel-Crafts acylation or B) Mannich reaction. Activated aromatic compounds like 2 were acylated under Friedel-Crafts conditions resulting in the formation of ortho and para substituted ketones. These reactions proceeded in high yield and with generally high selectivities depending on the substituent. In general, analogs of 2 were alkylated and subsequently reacted with 3-chloropropionyl chloride in the presence of AlCl3 at 0 °C generating ortho- and para-acylated compounds 3 and 4. Subsequent treatment with dimethylamine (2 M in THF) resulted in the formation of the desired β-aminoketones 5 and 6 in high yield.
Scheme 1.

General synthetic routes for β-aminoketones.a
A) Synthetic route using Friedel-Crafts acylation
B) Synthetic Route using Mannich reaction
aReagents and conditions. (a) hexyl bromide, K2CO3, DMF, 80–90 °C, 12 h; (b) 3-chloropropionyl chloride, AlCl3, DCM, 0 °C; (c) 10 eq. dimethylamine (2 M in THF), THF, rt, 30 min; (f) paraformaldehyde, Me2NH•HCl, H2O/MeCN (1/9, v/v), MW, 120 °C, 2 h.
Derivatives of 2 with strong electron withdrawing substituents (e.g. R = NO2, CF3 and SO2Me) could not be made using this general route. Additionally, steric hindrance slowed reaction rates for the 3-MeO, 3-MeS, and most disbustituted rings resulting in low yield of acylated products. In these cases, the desired β-aminoketones were obtained using a Mannich reaction19. In general, this three-component reaction utilizes harsh reaction conditions and suffers from low yields. The application of microwave assisted heating increased the yields and enabled us to synthesize β-aminoketones that were inaccessible using the Friedel-Crafts acylation route.20, 21 Most of the hydroxyl acetophenones starting materials were commercially available or were synthesized from the corresponding phenol.22 Heating these compounds in the microwave at 120°C in water/acetonitrile (1:9) in the presence of paraformaldehyde and dimethylamine hydrochloride salt resulted in the formation of desired β-aminoketones in good yields.
Evaluation of β-aminoketones
Preliminary evaluation of all compounds included assessment of biochemical activity and cytotoxicity. Additionally, the solubility and passive permeability of the small molecules were investigated to support biochemical and cell based assay results and to guide the earliest stages of probe discovery towards tractable compounds.23–25 The ability of the synthesized β-aminoketones to bind to both TR isoforms and inhibit the interaction with coregulator peptide SRC2-2 (second NID box of SRC2) was evaluated using a previously reported competitive fluorescence polarization (FP) assay.13 The cytotoxicity of β-aminoketones was evaluated in human liver cancer cells (HepG2) using a commercially available luminescence cell viability assay (CellTiter-Glo, Promega Corp.). The solubility of each β-aminoketone was determined in PBS buffer containing 5% DMSO, reflecting the conditions of the fluorescence polarization assay. The permeability of each compound across a membrane bilayer was measured using a parallel artificial membrane permeation assay (PAMPA). The assay was carried out at neutral pH (pH = 7.4) reflecting the conditions required for activity in cell-based assays. Finally, as a marker for cardiac ion channel activity, each compound was evaluated its effects on the hERG channel using a potassium induced cellular depolarization assay.
The effects of substitution patterns of the aromatic ring on activity
Our original studies showed that the hexyl side chain could be successfully replaced by substitutents that were roughly the same size and hydrophobic character. In order to simplify exploring the effects of ring substitutions, we replaced the original hexyl substituent with a hexyloxy group. This substitution enhanced solubility and suppressed potential metabolic oxidation by cytochrome 450’s at the benzylic methylene. 26–28
The prior SAR study showed that a para relationship between the hydrophobic substituent and the aminoketone appeared to be an essential feature of potent β-aminophenylketones.15 However, one ortho-substituted compound, hexyl 2-(acryloyloxy)benzoate, showed high affinity for TR. To investigate this SAR relationship more carefully, β-aminophenylketones with hexyloxy substituents in ortho, meta, and para positions were synthesized and characterized as described above. The results are summarized in Table 1.
Table 1.
β-Aminophenylketone Regioisomers
| Compound Number | Structure | SRC Binding Inhibition, TRβ (IC50, µM)a | Cell viability HepG 2 (EC50, µM) b |
|---|---|---|---|
| 9 | 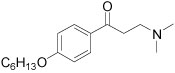 |
16.5±1.4 | 65±19 |
| 10 | 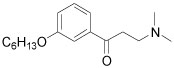 |
7.1±0.8 | 45±5 |
| 11 | 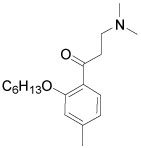 |
>50 | >100 |
Values are the mean of three independent experiments in triplicate.
Values are means of two independent experiments in triplicate. The general error limits are ±5%.
The meta-substituted β-aminophenylketone 10 showed the highest potency (IC50 = 7.1 µM) in the SRC binding assay with TRβ while the para-substituted 9 is less potent, with an IC50 = 16.5 µM. Ortho- substituted compound 11 showed no measurable activity. We examined other ortho-substituted compounds, with ethyl, methoxy, chloro, bromo, and iodo substituents, but none had measurable activity in either biochemical or cytotoxicity assays (data not shown). Although the meta-substituted β-aminophenylketone was more potent, careful exploration of its SAR was hindered by synthetic difficulties in producing a diverse set of analogs. Therefore, we performed the SAR analysis of the ring substitutions using para-substituted compounds.
We explored the effects of other substitutents on the central aromatic ring using a modified Topliss scheme in which all substitutents were simultaneously explored and the substituent set expanded according to Hansch to give an understanding of the interlocking effects of sterics, hydrophobics, and electrostatics. 29, 30 The substituent set used is shown in Figure 2. Suitable starting materials were converted into the corresponding β-aminophenylketone using a Friedel-Crafts acylation (A) or Mannich reaction (B). Details of the synthetic procedures and supporting data are provided in the Supporting Information. All compounds were evaluated in the assays discussed above using independent, replicate dose-response methods to generate IC50 or EC50 constants which are summarized as a heat map in Figure 3. (tabulated data with errors can be found in the Supporting Information).
Figure 2.

Diversity of aromatic substitutents.
Figure 3.

Activity summary for ring-substituted β-aminophenylketones: Heatmap shows IC50 values for the inhibition of coregulatory peptide binding (SRC2-2) by TRβ, EC50 values for cellular proliferation inhibition (HepG2), and calculated therapeutic index (TI, ratio of two prior values). Compound order is sorted by therapeutic index. aValues are the mean of three independent experiments in triplicate. bValues are means of two independent experiments in triplicate. cValues are EC50 over IC50.
Most of the substituted β-aminophenylketones investigated were more potent than unsubstituted compound 9 except compounds 12{4}, 12{7}, and 12{28}. The 3-methoxy substituted compound 12{4} showed reduced potency while both 3-dimethylamino substituted 12{7} and 2-benzyl substituted 12{28} failed to inhibit the TRβ-coactivator interaction. β-Aminophenylketones bearing strong electron withdrawing substituents including mono-halogens 12{9} – 12{15}, 3-sulfone 12{17}, nitro 12{20}, and trifluoromethyls 12{18} and 12{19} showed affinities in the low-micro molar range (1–5 µM). However, the 2-methylsulfone 12{16} gave low inhibitory potency (8 µM) even though it has strong electron withdrawing group. This implies that a hydrophobic electron withdrawing group gives better potency. More hydrophobic substitutents such as alkyl groups 12{1}–12{3}, 12{8}, and thioether 12 {5–6} gave slightly improved potency (4–10 µM) compared to compound 9. This implies that electron donation genereally is more driving than hydrophobicity. For disubstituted compounds, compounds 12{22} – 12{26} showed potent inhibition (0.8–1.8 µM) in comparison with their monosubstituted counterparts, regardless of their electronic properties. Cell viability in the presence of β-aminoketones was also investigated. Most of compounds exhibited weak inhibition of cell proliferation (EC50 > 20 µM). Notable exceptions were the 3,5-dimethyl 12{22}, 3-trifluoromethyl 12{19}, 2-tert-butyl 12{8}, and 2-phenyl 12{27} compounds. The halogen substituted compounds were all weakly growth inhibitory among active compounds (IC50 > 10 µM) with the exception of 2,6-dichloro compound 12{26}. The therapeutic index shows that 2,3- and 2,5-dichloro compounds 12{24} and 12{25} were not only the most potent compound but also the least toxic compounds. Compounds 12{24} and 12{25} also had acceptable solubility and permeability (SI, Table 1, 10 µM and 600 ×10−6 cm/s for 12{24} and 20 µM and 1040 ×10−6 cm/s for 12{25}). Generally solubility of this series of compounds was good (between 2–185 µM, SI Table 1). However, β-Aminophenylketones with 2-tert-butyl 12{8}, 2-trifluoromethyl 12{18}, 2-phenyl 12{27}, and 2-benzyl 12{28} substituents were poorly soluble (solubility < 10 µM). Most of β-aminophenylketones exhibited good permeability at pH=7.4 (SI, Table 1).
Replacement of the Phenyl Ring
A general trend correlating increased core hydrophobicity with increased potency suggested exploring alternate core scaffolds. From the initial screening data we observed an increase of efficacy inhibition of SRC2-2 recruitment by TRβ from 12% to 33% at a fixed 10 µM dose with exchange of the phenyl core structure by a naphthalene structure.{Arnold, 2007 #83} Based on this observation several alternate cored β-aminoketones were synthesized and subjected to the same array of assays discussed earlier. The results are summarized in Table 2.
Table 2.
Summary of β-Aminoarylketones with Alternate Aromatic Rings
 | |||
|---|---|---|---|
| Compound Number | Aryl Ring | SRC Binding Inhibition, TRβ (IC50, µM)a | Cell viability HepG 2 (EC50, µM) b |
| 9 | 16.5±1.4 | 65±19 | |
| 13{1} | 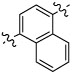 |
4.0±0.4 | 38±6 |
| 13{2} | 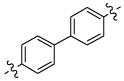 |
>50 | >130 |
| 13{3} | 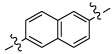 |
>50 | >65±14 |
Values are the mean of three independent experiments in triplicate.
Values are means of two independent experiments in triplicate. The general error limits are ±5%.
β-Aminonaphthylketone 13{1} showed an improved inhibitory potency in the SRC binding competition for TRβ (IC50 = 4 µM) but was more toxic in comparison with β-aminophenylketone 9. In addition compound 13{1} was less soluble than compound 9, although it retained high permeability (SI, Table 2). Biphenyl β-aminoketone 13{2} and 1,6-substituted β-aminonaphthylketone 13{3} were both failed to inhibit the interaction between TRβ and SRC2-2 under the assay conditions. These studies indicated that no significant advantage would arise from using a larger, polyaromatic, core structure.
Substitution of α-Carbon
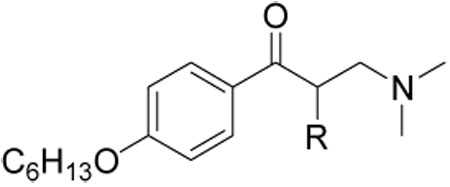
Initial evaluation of lead compounds had indicated that they were ion channel active cardiotoxins that exhibited QT prolongation. For this reason it was desired to lower interactions with the hERG and other ion channels. Mitigation of ion channel effects of substituted amines generally follows two strategies: increased steric bulk around the amine and/or modulation of the basicity of the amine.{Jamieson, 2006 #88; Aronov, 2006 #89; Meyers, 2007 #86} To examine the effect of α-carbon substituents of β-aminophenylketones we synthesized the compounds depicted in Table 3. All compounds were analyzed by the same array of assays discussed earlier.
Table 3.
Summary of α-Substituted β-Aminophenylketones
 | |||
|---|---|---|---|
| Compound Number | R | SRC Binding Inhibition, TRβ (IC50, µM)a | Cell viability HepG 2 (EC50, µM)b |
| 9 | H | 16.5±1.5 | 65±19 |
| 14{1} | Me | >50 | 56±18 |
| 14{2} | iPr | >50 | 62±27 |
| 14{3} | Ph | >50 | 27±19 |
| 14{4} | 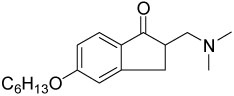 |
1.7±0.3 | 14±2 |
| 14{5} | 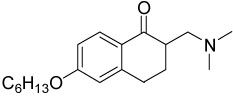 |
2.3±0.2 | 12±2 |
| 14{6} | 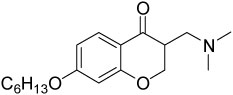 |
1.6±0.1 | 12±1 |
Values are the mean of three independent experiments in triplicate.
Values are means of two independent experiments in triplicate. The general error limits are ±5%.
α-Substituted compounds 14{1}–14{3} (tested as racemates) were not able to inhibit the interaction between TR and SRC2-2. In contrast, cyclic compounds 14{4}–14{6} were highly potent, exhibiting IC50 values of 1–2 µM. However, they carried the liability of an increased growth inhibitory potency for the HepG2 cell line, giving poor therapeutic index in comparison with compound 9. These studies indicated that increased steric bulk and/or conformational restriction of the β-aminoketone moiety were both deleterious with respect to functional inhibition of thyroid signaling due to loss of efficacy or increased growth inhibition, respectively.
The Role of Amine Substitutions
The majority of ion-channel modulators bear amine functionalities with pKa values between 8.0–9.0, ensuring high permeability of the uncharged amine and binding of the protonated amine at the receptor site. A strategy to develop compounds that are active towards their primary target but not binding hERG has been to reduce the pKa value of the amine. {Jamieson, 2006 #88; Aronov, 2006 #89; Meyers, 2007 #86} The identification of β-aminoketones with low amine pKa’s that still inhibited cofactor binding to TR in the initial hit exploration indicated that this strategy might afford compounds with better in vivo toxicology profiles. Prior to careful exploration of amine substitutions with respect to ion channel effects a more broad structure-activity relationship needed to be established. TR inhibition by β-aminoketones is dependent on elimination rate of the pro-drug into the active enone. In Mannich bases, this elimination rate is pKa dependent. {Bundgaard, 1981 #67; Davioud-Charvet, 2003 #49}
In order to establish the range of this component of the SAR, β-aminophenylketones with pKa values between 3 to 10 were synthesized and tested. (Figure 4) The compounds were synthesized using a Friedel-Crafts reaction to afford the halo-ketone followed by treatment with different amines in the presence of DBU. The pKa values of each compound were calculated using the imbedded calculator in Pipeline Pilot (Accelrys Software Inc.). The compounds were evaluated with the same array of assays described earlier. In addition, the compounds’ abilities to block KCl-stimulated depolarization of HEK293-hERG cells (at 10 µM concentration) was assessed using a Vm-sensitive fluorescent dye-based assay (Molecular Devices). A comprehensive description of the assay (performed by the National Institute of Mental Health Psychoactive Drug Screening Program), including a detailed protocol, sample data, and analysis procedures, is available on-line (http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=376&loc=ea_ras or http://pdsp.med.unc.edu/UNC-CH%20Protocol%20Book.pdf). The results are summarized in Table 4.
Figure 4.

Amine substituents examined.
Table 4.
Summary of β-Aminophenylketones Bearing Different Nitrogen Substituents
 | ||||
|---|---|---|---|---|
| Compound Number | SRC Binding Inhibition, TRβ (IC50, µM)a | Cell viability HepG 2 (EC50, µM) b | hERG inhibition (% terfenadine effect)c | Calculated pKa |
| 9 | 16.5±1.5 | 65±19 | 61±9 | 9.12 |
| 15{1} | 2.7±0.4 | 48±20 | 48±15 | 9.95 |
| 15{2} | 9.1±1.2 | 66±34 | 75±2 | 9.61 |
| 15{3} | 18.9±6.8 | 59±17 | 69±9 | 9.28 |
| 15{4} | 22.0±2.4 | 67±22 | 35±4 | 6.45 |
| 15{5} | 5.6±0.9 | 54±13 | 60±7 | 8.1 |
| 15{6} | 3.8±0.3 | 81±32 | 29±12 | 6.13 |
| 15{7} | 4.1±0.5 | 56±19 | 32±13 | 6.5 |
| 15{8} | >50 | >130 | 8±11 | 5.26 |
| 15{9} | >50 | >130 | 22±1 | 4.38 |
| 15{10} | >50 | >130 | 18±3 | 3.67 |
| 15{11} | >50 | 54±31 | 32±3 | -d |
| 15{12} | >50 | >130 | −11±6 | -d |
Values are the mean of three independent experiments in triplicate.
bValues are means of two independent experiments in triplicate. The general error limits are ±5%.
Values are the mean of two independent experiments in quadruplicate. Depolarization inhibition data in HEK293-hERG cells are expressed as % inhibition of KCl-stimulated depolarization as measured using a Vm-sensitive dye (Molecular Devices), scaled as follows: % inhibition for compound = 100 × (value for test compound – value for negative control)/(value for 10 µM terfenadine – value for negative control)
Not available.
All β-aminophenylketones bearing alkyl amines that were tested inhibited the interaction between TR and SCR2-2 (Table 4, 15{1}–15{7}). In contrast, β-aminophenylketones bearing anilines were inactive (Table 4, 15{8}–15{10}). These compounds possess amines with pKa values lower than 5.3 that are unlikely to be protonated at pH 7.4, and thus probably do not readily generate the active enone form of the compounds. The piperazine and morpholine analoges (15{5} – 15{7}), with intermediate pKa values (8.1, 6.13, and 6.45, respectively), showed relatively potent inhibition (3.8–5.6 µM). We hypothesize that this is due to a β-atom effect raising the pKa. The β-aminophenylketones 15{11} and 15{12}, bearing imidazole and succinimide substituents, respectively, were inactive. Unlike activity against the target, the cytotoxicity of this series of compounds showed little change (48–81 µM), regardless of amino group. This series of compounds was tested to see if KCl-stimulated depolarization of HEK293-hERG cells was affected by the amino group. Most of the compounds active against TR also inhibited KCl-stimulated depolarization of HEK293-hERG cells, in a manner similar to a prototypical hERG blocker (terfenadine). Among the active compounds, aziridine 15{4}, phenylpiperazine 15{6}, and morpholine compound 15{7} showed the lowest inhibition of KCl-stimulated depolarization of HEK293-hERG cells (35%, 29%, and 32% of the inhibitory effect of terfenadine, respectively). In contrast, all of the inactive compounds showed no or poor blockade of KCl-stimulated depolarization of HEK293-hERG depolarization. Examining the relationship between pKa and antagonism of KCl-stimulated depolarization of HEK293-hERG cells (Figure 5) revealed that increasing basicity correlates fairly strongly with inhibition of depolarization responses in HEK293-hERG cells.(R2 = 0.8, P = 0.0002). The two most notable outliers were 15{1}and 15{8}, which carry significantly more bulky substitutents around the amine.
Figure 5.

Relationship between the pKa of the amino group of β-aminophenylketones and inhibition of KCl-stimulated depolarization of HEK293-hERG cells.
Functionality of Hydrophobic Side Chain
In our preliminary SAR studies of β-aminophenylketones we established that para-alkyl substitutents between 6–8 carbon atoms were essential for highly active inhibitors of the TR-coactivator interaction.{Arnold, 2007 #83} Such freely rotable, highly hydrophobic side chains carried a correlated liability of poor solubility. To balance these issues the incorporation of more polar groups in the side chain, incorporated relatively closely to the ring, was explored. All compounds were subjected to the same array of assays discussed earlier and the results are summarized in Table 5.
Table 5.
Summary the Activities of Hydrophobic Side Chain Modified β-Aminophenylketones
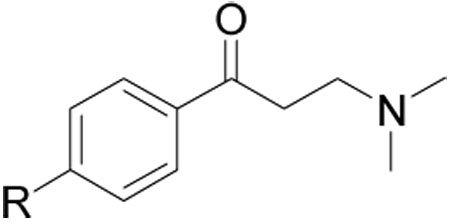 | ||||
|---|---|---|---|---|
| Compound Number | R | SRC Binding Inhibition, TRβ (IC50, µM)a | Cell viability HepG 2 (EC50, µM) b | hERG inhibition (% terfenadine effect)c |
| 1 | 9.6±1.1 | 54±9 | 68±12 | |
| 9 | 16.5±1.5 | 65±19 | 61±9 | |
| 16{1} | >50 | >100 | 3±1 | |
| 16{2} | >50 | >100 | 16±10 | |
| 16{3} | 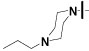 |
>50 | >100 | 35±10 |
| 16{4} | 5.9±0.6 | 34±15 | 48±4 | |
| 16{5} | 4.9±0.7 | >100 | 36±9 | |
| 16{6} | 8.1±1.4 | >100 | 15±7 | |
| 16{7} | 12.4±3.2 | >100 | 38±7 | |
| 16{8} | >50 | >100 | 15±20 | |
| 16{9} | >50 | >100 | 23±15 | |
Values are the mean of three independent experiments in triplicate.
Values are means of two independent experiments in triplicate. The general error limits are ±5%.
Values are the mean of two independent experiments in quadruplicate.
All compounds bearing heteroatoms within the hydrophobic chain in any position other than immediately adjacent to the phenyl ring were inactive in TR receptor binding assay (Table 5, 16{1–3}). This is not unexpected given that our co-crystal structure revealed that this group threads through a narrow hydrophobic cleft on the protein surface. All of these compounds were less toxic in comparison with compound 1, although this might be due to the reduced permeability exhibited by most (SI, Table 5). The thioether 16{4} and sulfone 16{5} compounds showed reasonable potency (5.9 µM and 4.9 µM, respectively for TRβ). Interestingly, sulfone 16{5} was non-toxic to HepG2 cells at high concentrations, while thioether 16{4} showed moderate toxicity (40.7 uM). The amide 16{6} and reverse amide 16{7} also showed reasonable inhibitory potency for TRβ (8.1 µM, and 12.4 µM, respectively) with low toxicity. However, sulfonamide 16{8} and the urea 16{9} were inactive in the SRC2-2 binding assay. Aside from placement of polar groups being highly important, no clear structure-activity relationships were evident for this part of the molecule. This compound series was also tested for inhibition of KCl-stimulated depolarization of HEK293-hERG cells. Most of compounds showed moderate inhibitory activity (3–68%), consistent with the other substitution patterns of the aryl ring and aminoketone. Among the active compounds, sulfone 16{5}, and amides 16{6} and 16{7} showed fairly weak inhibition of KCl-stimulated depolarization of HEK293-hERG. All of active compounds had good solubility (58–224.5 µM) and permeability (565–1926 × 10−6 cm/s). This study suggested that utilization of a heteroatom attachment for this side chain might prove fruitful as a strategy to circumvent both toxicity and inhibition of KCl-stimulated depolarization of HEK293-hERG cells without substantial loss of activity.
Selectivity between the Isoforms TRα and TRβ
In addition to testing all compounds against the TRβ-LBD, all compounds were also tested against TRα to determine if this series could afford selective antagonists. Most compounds exhibited similar potency of antagonism towards both TRα and TRβ
Modeling of Physiochemical Behaviors
Having prepared a range of derivatives that systematically varied steric and electronic factors, quantitative structure-activity and structure-property relationships were explored. The importance of generation of an active conjugated enone in the mechanism of action implied that Hammett analysis {Hansch, 1991 #94; Jaffe, 1953 #95} might help in explaining the SAR of substituted β-aminoketones. Meanwhile, the importance of both solubility and permeability in activity indicated that Hansch constants (π) might be critical in predicting both biochemical and cellular activity.{Chiu, 2004 #97; Hansch, 1995 #96}. Compounds outlined in Figure 3 and Table 5 were used for this analysis and the results are presented in Figure 6.
Figure 6.

Hammett and Hansch analysis of β-aminoketones in dependency of inhibition of TRβ. (A) electronic effect (meta and para substituents); (B) hydrophobicity (meta substituents); (C) hydrophobicity (ortho substituents
The IC50 values were plotted against literature σ- and π- constants of substituents. In general, we observed that electron withdrawing substituents (positive σ-values) improved the ability of β-aminophenylketones to inhibit the interaction between SRC2-2 and TR, regardless of substitution location whereas β-aminophenylketones with electronic donating groups (positive σ-values) exhibit weaker activities (Figure 6A). While this correlation was significant, the trend was not particularly strong (slope = 9.6, p = 0.003). The substituent effect of β-aminophenylketones with regard to their hydrophobicity was also analyzed (Figure 6B and 6C). While no effect was evident with meta substitutents, a relatively strong substituents effect could be found with ortho substituted compounds (Figure 6C, slope = 19.7, p = 0.002). These studies indicated that the major effector of potency is the hydrophobicity of the ortho-substituent, relative to the ketone. Overall, this analysis served to focus future work on incorporation of ortho hydrophobic, electron withdrawing (halogens in particular) on the ring.
We hypothesized that this arise from a steric effect that alters electrophilicity of the eneone. An examination of minimized structures of unsubstituted compound 9 and 3,5-dimethyl substituted compound 12{22} by MOE (Forecefield-MMFF94) showed that compound 9 has small predicted dihedral angle (0 °) and low energy barrier for rotation of the ketone (6 kcal/mol) while 3,5-disubstituted compound 12{22} has large predicted dihedral angle (~90 °) and high energy barrier for rotation (>50 kcal/mol). This distorted dihedral angle disturbs the π−π overlap between the carbonyl group and the aromatic ring thus decreasing the resonance stability of the elimination intermediate and giving both an increased rate of elimination and increased reactivity of the resulting enone for Michael addition.
Second Generation Compounds
Based on the evaluation of the initial suite of β-aminophenylketones we designed and synthesized a set of second generation compounds to test hypotheses about SAR. These focused on a substitution pattern containing: i) para-orientation between hydrophobic chain and carbonyl group; ii) 3-positioned electron withdrawing substituents, or di-substituted rings; iii) an unsubstituted α-carbon; iv) an amino ketone with an amine having a pKa between 6 and 8; and v) an ether, thioether, or sulfonyl attached side chain. We targeted three chemsets for the second generation as shown Figure 7. A secondary focus was to emphasize groups that gave low hERG activity in the previous work. In order to fully explore potential synergy between substituents, the second generation compounds were produced as a fully combinatorial array (17{X, R1, R2R3NH}, 3×5×6 = 90 compounds). Details of the synthetic procedures and supporting data are provided in the Supporting Information. This set of compounds was evaluated using the suite of assays discussed above and the results are summarized as a hit map in Figure 8.
Figure 7.

Array of compounds targeted for second generation studies.
Figure 8.

Activity summary of the second generation compounds sorted by therapeutic index (TI): Heatmap shows inhibitory constants for inhibition of interaction of coregulatory peptide SRC2-2 and TRβ (IC50), HepG2 proliferation inhibition (EC50), and calculated therapeutic index (ratio of two prior values).aValues are the mean of three independent experiments in triplicate. bValues are means of two independent experiments in triplicate. cValues are EC50 over IC50. dValues are the mean of two independent experiments in quadruplicate.
All of the second generation compounds were evaluated to determine potency for antagonism of TRβ binding to SRC2-2. The majority possessed inhibitory potency in a narrow range (0.4–5 µM). The exception was observed in 3,5-dimethyl substituted oxopiperazine compounds 17{1,5,4} and 17{2,5,4} (SI, Table 6, 18.9 and 11.2 µM, respectively) which were significantly weaker. This trend indicates that the initial SAR models have guided the evolution of these compounds into a potency maxima that is reasonable for the targeted interaction. In contrast, the differences in growth inhibition of HepG2 were considerably more pronounced. The sulfone side chain compounds 17{3, y, z} showed generally significantly less growth inhibition than ethers 17{1, y, z} or thioethers 17{2, y, z}. Thus, lowering of cellular toxicity was the dominant driver for good therapeutic index. With respect to ring substitutions, the 3,5-dimethyl compounds were generally the most toxic (28.1–50.4 µM in sulfone series) while many of the other compounds showed reasonable potency and therapeutic index.
We also explored solubility and permeability of the three series and found significant evident differences. The sulfone series generally possessed moderate, but reasonable, solubility (1–30 µM), whereas both the ether and thioether series showed poor solubility (<10 µM and <5 µM, respectively). As the effective available intracellular concentration is a product of the combination of solubility and permeability we prefer to analyze this data by graphical comparison of these two sets of data (Figure 9). This allows one to focus attention on those compounds with the best mix of properties. Taking these considerations in mind, together with therapeutic index allow one to understand that high therapeutic index is not due to poor solubility or permeability. Indeed aggregate analysis indicates that the compounds that have therapeutic index more than 100 are those most likely to achieve high cellular concentrations.
Figure 9.

The solubility and permeability of the second generation chemsets.
Next we explored the ability of these compounds to antagonize KCl-stimulated depolarization of HEK293-hERG cells as above, focusing predominantly on sulfonyl compounds 17{3, x, y}. The ether 17{1,y,z} and thioether 17{2,y,z} series mostly showed low inhibitiory effect on KCl-treated HEK293-hERG cells but this is most likley due to their poor solubility (data not shown). Most of sulfonyl compounds have no effect on KCl responses from HEK293-hERG cells (<20%) or weak responses (20–40%). The exception was the 3-chloro substituted compounds 17{3,1,3}–17{3,1,6} which have good therapeutic index but moderately active blockers of KCl-stimulated depolarization of HEK293-hERG cells (45–61%) at 10 µM. Among the compounds that have therapeutic index more than 100, the oxypiperazines (17{3,3,4} and 17{3,2,4}) and acylpiperazine (17{3,3,5}) showed no effect on KCl responses from HEK293-hERG cells.
Conclusion
This paper describes the improvement of pharmacological properties of thyroid hormone receptor coactivator binding inhibitors. A major focus of optimization effort was to improve therapeutic index and to reduce cardiac toxicity. Based on the evaluation of the initial SAR study of β-aminophenylketones we designed and synthesized a set of second generation compounds to improve pharmacological properties. Electron withdrawing substituents led to high efficacy and sulfonyl attached side chain led to reduced cytotoxicity. The combination of these elements elevated the small therapeutic index of early hit compounds. In addition, utilization of amine moiety having low pKa’s resulted in lowering ion channel activity without any loss of target activity. The best compounds, based on therapeutic index and ion channel activity, are shown in Figure 10. These four compounds, 17{3,2,4}–17{3,2,5} and 17 {3,3,4}-17{3,3,5}, showed both outstanding thyroid hormone receptor coactivator interaction inhibitory potency and good therapeutic index as well as no observable inhibition of KCl-stimulated depolarization of HEK293-hERG cells. These compounds are currently undergoing evaluation in in vivo models.
Figure 10.

The best four compounds based on TI and hERG results.
Experimental section
Chemistry
All materials were obtained from commercial suppliers and used without further purification. All solvents used were dried using an aluminum oxide column. Thin-layer chromatography was performed on pre-coated silica gel 60 F254 plates. Purification of compounds was carried out by normal phase column chromatography (SP1 [Biotage], Silica gel 230–400 mesh) followed by evaporation (HT-4X evaporator [Genevac]. Initiator [Biotage] was used for microwave reaction. Purity determination were performed using UPLC-MS (BEH C18 1.7 µ, 2.1 × 50 mm column, Waters Corp.). Data were acquired using Masslynx v.4.1 and analyzed using the Openlynx software suite. The flow was then split to an evaporative light scattering detector (ELSD) and SQ mass spectrometer. The total flow rate was 1.0 mL/min and gradient program started at 90% A (0.1% formic acid in H2O), changed to 95 % B (0.1% formic acid in ACN), then to 90% A. The mass spectrometer was operated in positive-ion mode with electrospray ionization. All compounds presented were confirmed at 95% purity or better using the method (either ELSD or UV chromatogram) that reported the lowest purity. NMR spectra are recorded on a Bruker 400 MHz and referenced internally to the residual resonance in CDCl3 (δ 7.26 ppm) for hydrogen and (δ = 77 ppm) for carbon atoms. NMR peaks were assigned by MestRec (4.9.9.6) and MestReNova (5.2.2)
General procedure for Friedel-Crafts acylation
To a solution phenol 2 (5.3 mmol, 1 equiv.) in DMF (15 mL) was added K2CO2 (1.47 g, 10.6 mmol, 2 equiv.) and n-hexylbromide (1.12 mL, 8.0 mmol, 1.5 equiv.). The reaction mixture was stirred at 85 °C for overnight, poured into water (30 mL) and extracted with Et2O (2 × 30 mL) . The combined organic layer were washed with brine, dried over MgSO4, and concentrated in vacuo to yield crude hexylphenyl ether intermediate. This crude product was used for next step without purification. To a solution of hexylphenyl ether (2.81 mmol, 1 equiv.) in DCM (10 mL) was added 3-chloropropanoic chloride (0.35 mL, 3.65 mmol, 1.3 equiv.) and AlCl3 (0.49 g, 3.65 mmol, 1.3 equiv.) at 0 °C and stirred for 1 h. The reaction mixture was poured into ice water (50 mL) and extracted with DCM (2 × 30 mL). The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography (SP1, 25M (SiO2), flow rate: 25 mL/min; gradient: 1–30% ethyl acetate in hexanes over 20 CV).
General procedure for 5 and 6
To a solution compound 3 or 4 (1 equiv.) in THF was added dimethylamine (2 M in THF, 10 equiv.) and stirred at rt for 1 h. The reaction mixture was filtered and washed with ether (x2). The combined organic layer was concentrated in vacuo to yield crude aminoketone 5 or 6 and used without further purification.
General procedure for Mannich reaction
To a solution phenol 7 (5.3 mmol, 1 equiv.) in DMF (15 mL) was added K2CO2 (1.47 g, 10.6 mmol, 2 equiv.) and n-hexylbromide (1.12 mL, 8.0 mmol, 1.5 equiv.). The reaction mixture was stirred at 85 °C for overnight, poured into water (30 mL) and extracted with Et2O (2×, 30 mL). The combined organic layer were washed with brine and dried over MgSO4 and concentrated in vacuo to yield crude hexyloxy acetopheon intermediate. This crude product was used for next step without purification. A solution of hexyloxy acetophenon (1 equiv.), dimethylamine hydrochloride (2 equiv.), paraformaldehyde (2 equiv.), c-HCl (cat.), and H2O/actonitrile (1/9) was heated at 120 °C in MW for 2 h. The reaction mixture was concentrated in vacuo, and purified by flash column chromatography (SP1, gradient: 1–10% MeOH in hexanes over 15 CV).
Fluorescence polarization assay (FPA)
This assay has been described in detail previously.13, 14 The concentration of each compound required to inhibit 50% of the binding between TRβ-LBD and SRC2-2 peptide is presented in Table 1 – Table 5. hTRβ1 LBD (His6 T209-D461) was expressed in BL21 (DE3) (invitrogen) and the peptide SRC2-2 (CLKEKHKILHRLLQDSSSPV) was labeled with 5-iodoacetamidofluorescien (Molecular Probes), as described. For each reported value, two independent experiments were carried out in quadruplicate on two separate days using alternate batches of reagents. The competition binding experiments were evaluated using PrismPad and the IC50 values were obtain by fitting data to equation: Sigmoidal dose-response (variable slope) or four parameter logistic equation; Y=Bottom + (Top-Bottom)/(1+10ˆ((LogEC50-X)*HillSlope)); X is the logarithm of concentration; Y is the response. Tabulated data are reported as mean values across all experiments and replicates with associated 95% confidence limits.
Cytotoxicity study
HepG2 cells were at 37 °C in Minimum Essential Medium (MEM), 10% fetal bovine serum, 50 mL penicillin/streptomycin. Cells were grown to 80% confluence, collected, and dispensed in 384 well plates at 50 µl per well (6000 cells). The small molecules were serially diluted from 50000 µM to 2.5 µM in DMSO into a 386-well plate (Costar 3365) and 0.2 µl was transferred into the 384 cell culture plates (Corning 3917) yielding a final compound concentration range of 200-0.01 µM. After 48 h incubation at 37 °C 25 µl of CellTiter-Glo® (Promega) was added and mixed by aspiration and dispense and luminescence was detected (EnVision, PerkinElmer) after 10 minutes.
Solubility study
The assay was carried out using the MultiScreen® Solubility assay developed by Millipore. Briefly, 16 test compounds were dispensed into each 96-well polypropylene plate (Costar 3365) as serial diluted concentration ranges from from 10000 µM to 625 µM in DMSO starting from column 1–5 and 7–11. Column 6 and 12 were filled with DMSO. 5 µl of each well was transferred into the 96-well disposable UV-Star™ (Greiner Bio-One). Acetonitrile (97.5 µl) and PBS buffer (97.5 µl) was added to each well and the plate was agitated for 30 minutes (IKA microtiterplate shaker). The UV spectra from 200–500 nm was measured for all wells and subtracted from the background (DMSO). The correlations between concentrations and absorbance at 260, 280 and 300 nm were determined as slopes. Then, 5 µl of each well of the polypropylene plate was added to a MultiScreen Solubility Filter Plate (Millipore) and diluted with 195 µl PBS. The plate was agitated for 2 hours and filtered into a 96-well disposable UV-Star™ plate and the UV absorbance at 260, 280 and 300 nm was measured. The aqueous solubility (Aqueous solubility = Amax filtrate/slope) was determined for all three wavelength and given as average value.
PAMPA procedure
The procedure was conducted using a developed procedure by pION. Briefly, all liquid-handling steps for the PAMPA assay are performed on a Biomek FX Laboratory Automation Workstation (Beckman-Coulter) and analyzed by pION's (London, UK) PAMPA Evolution 96 Command Software. The PAMPA Evolution 96 Permeability Assay Kit includes the Acceptor Sink Buffer (ASB), Double-Sink Lipid Solution and a PAMPA Sandwich plate, preloaded with magnetic disks. 3 µL of lipid were transferred onto the support membrane in the acceptor well, followed by addition of 200 µL of ASB (pH 7.4). Then, 180 µL of diluted test compound (50 µM in system buffer at pH 7.4 starting from a 10 mM DMSO solution) was added to the donor wells. The PAMPA sandwich plate was assembled and placed on the Gut-Box and stirred for 30 minutes. The distribution of the compounds in the donor and acceptor buffers (100 µL aliquot) was determined by measure of the UV spectra from 200 to 500 nm using the SpectraMax reader (Molecular Devices). The permeability coefficient is determined using the maximum absorbance from 200 to 500 nm using the following formula:
VD is the donor well volume (cm3); A is the filter area (cm2); CD (0) is the sample concentration in the donor well at time 0 (mole/cm3); CD (t) is the sample concentration in the donor well at time t (mole/cm3); t is the interval of time (sec); tLAG is the lag time needed to reach steady state conditions (sec) and R is the membrane retention (related to the membrane/water partition coefficient). Standards used were Verapamil (Pe = 1505 × 10−6 cm/s) as a high permeability standard, Carbamazepine (Pe = 150 × 10−6 cm/s) as medium permeability standard and Rantidine (Pe = 2.3 × 10−6 cm/s) as low permeability standard. The compounds were measured in triplets and reported as average values.
hERG binding assay
This screen was performed by the National Institute of Mental Health Psychoactive Drug Screening Program (PDSP), directed by Dr. Bryan L. Roth, at University of North Carolina Chapel Hill (http://pdsp.med.unc.edu/indexR.html). Detailed protocols and data analysis procedures are available on-line (http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=376&loc=ea_ras or http://pdsp.med.unc.edu/UNC-CH%20Protocol%20Book.pdf). Samples and internal controls (known hERG blockers) were submitted in a blind fashion to the PDSP. The assays reliably and reproducibly identified the blinded internal controls as ‘hits’.
Supplementary Material
Acknowledgement
This work was supported by the NIH (DK58080), the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children’s Research Hospital.
Abbreviations
- AF-1
activation domain
- ARO
human anaplastic thyroid cell line
- ATCC
American type culture collection
- ATP
adenosine triphosphate
- CoA
coactivator
- CoR
coregulator
- DBD
DNA-binding domain
- DMSO
dimethyl sulfoxide
- LBD
ligand-binding domain
- LCMS
liquid chromatography-mass spectrometry
- MOE
molecular operating environment software
- NCoR
nuclear receptor corepressor
- NID
nuclear receptor interacting domain
- NR
nuclear hormone receptor
- PAMPA
parallel artificial membrane permeation assay
- PBS
phosphate buffered saline
- PDA
photodiode array
- RXR
retinoid X receptor
- SAR
structure-activity relationship
- SMRT
silencing mediator of retinoic acid
- SRC
steroid receptor coactivator
- SRC2-2
steroid receptor coactivator 2 peptide representing the second NID
- TR
thyroid receptor
- U2OS
human osteosarcoma epithelial cell line
Footnotes
Supporting information
Experimental procedur1es, tabulated activity data, and characterization data for all final compounds. This material is available free of charge via the Internet at http://pubs.acs.org/
References
- 1.Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001;81:1097–1142. doi: 10.1152/physrev.2001.81.3.1097. [DOI] [PubMed] [Google Scholar]
- 2.Malm J. Thyroid hormone ligands and metabolic diseases. Curr Pharm Des. 2004;10:3525–3532. doi: 10.2174/1381612043382873. [DOI] [PubMed] [Google Scholar]
- 3.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–1304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 4.Harvey CB, Williams GR. Mechanism of thyroid hormone action. Thyroid. 2002;12:441–446. doi: 10.1089/105072502760143791. [DOI] [PubMed] [Google Scholar]
- 5.Williams GR. Cloning and characterization of two novel thyroid hormone receptor beta isoforms. Mol Cell Biol. 2000;20:8329–8342. doi: 10.1128/mcb.20.22.8329-8342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brent GA. Tissue-specific actions of thyroid hormone: insights from animal models. Rev Endocr Metab Disord. 2000;1:27–33. doi: 10.1023/a:1010056202122. [DOI] [PubMed] [Google Scholar]
- 7.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ribeiro RC, Apriletti JW, Wagner RL, West BL, Feng W, Huber R, Kushner PJ, Nilsson S, Scanlan T, Fletterick RJ, Schaufele F, Baxter JD. Mechanisms of thyroid hormone action: insights from X-ray crystallographic and functional studies. Recent Prog Horm Res. 1998;53:351–392. discussion 392–354. [PubMed] [Google Scholar]
- 9.Nguyen NH, Apriletti JW, Cunha Lima ST, Webb P, Baxter JD, Scanlan TS. Rational design and synthesis of a novel thyroid hormone antagonist that blocks coactivator recruitment. J Med Chem. 2002;45:3310–3320. doi: 10.1021/jm0201013. [DOI] [PubMed] [Google Scholar]
- 10.Lim W, Nguyen NH, Yang HY, Scanlan TS, Furlow JD. A thyroid hormone antagonist that inhibits thyroid hormone action in vivo. J Biol Chem. 2002;277:35664–35670. doi: 10.1074/jbc.M205608200. [DOI] [PubMed] [Google Scholar]
- 11.Hashimoto A, Shi Y, Drake K, Koh JT. Design and synthesis of complementing ligands for mutant thyroid hormone receptor TRbeta(R320H): a tailor-made approach toward the treatment of resistance to thyroid hormone. Bioorg Med Chem. 2005;13:3627–3639. doi: 10.1016/j.bmc.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 12.Flamant F, Gauthier K, Samarut J. Thyroid hormones signaling is getting more complex: STORMs are coming. Mol Endocrinol. 2007;21:321–333. doi: 10.1210/me.2006-0035. [DOI] [PubMed] [Google Scholar]
- 13.Arnold LA, Estebanez-Perpina E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J Biol Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 14.Arnold LA, Estebanez-Perpina E, Togashi M, Shelat A, Ocasio CA, McReynolds AC, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. A high-throughput screening method to identify small molecule inhibitors of thyroid hormone receptor coactivator binding. Sci STKE. 2006;2006:pl3. doi: 10.1126/stke.3412006pl3. [DOI] [PubMed] [Google Scholar]
- 15.Arnold LA, Kosinski A, Estebanez-Perpina ERJF, Guy RK. Inhibitors of the Interaction of a Thyroid Hormone Receptor and Coactivators: Preliminary Structure-Activity Relationships. J Med Chem. 2007;50:5269–5280. doi: 10.1021/jm070556y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Estebanez-Perpina E, Arnold LA, Jouravel N, Togashi M, Blethrow J, Mar E, Nguyen P, Phillips KJ, Baxter JD, Webb P, Guy RK, Fletterick RJ. Structural Insight into the Mode of Action of a Direct Inhibitor of Coregulator Binding to the Thyroid Hormone Receptor. Mol Endocrinol. 2007;21:2919–2928. doi: 10.1210/me.2007-0174. [DOI] [PubMed] [Google Scholar]
- 17.Bruhova I, Tikhonov DB, Zhorov BS. Access and Binding of Local Anesthetics in the Closed Sodium Channel. Mol Pharmacol. 2008;74:1033–1045. doi: 10.1124/mol.108.049759. [DOI] [PubMed] [Google Scholar]
- 18.Wright SN. Cardiotoxic and antiarrhythmic tertiary amine local anesthetics: sodium channel affinity vs. sodium channel gating. Curr Vasc Pharmacol. 2003;1:239–242. doi: 10.2174/1570161033476628. [DOI] [PubMed] [Google Scholar]
- 19.Mannich C. Synthesis of b-ketonic bases. Arch. Pharm. 1917;255:261–276. [Google Scholar]
- 20.Lehmann F, Pilotti A, Luthman K. Efficient large scale microwave assisted Mannich reactions using substituted acetophenones. Mol Divers. 2003;7:145–152. doi: 10.1023/b:modi.0000006809.48284.ed. [DOI] [PubMed] [Google Scholar]
- 21.Leadbeater NE, Torenius HM, Tye H. Microwave-assisted Mannich-type three-component reactions. Mol Divers. 2003;7:135–144. doi: 10.1023/b:modi.0000006822.51884.e6. [DOI] [PubMed] [Google Scholar]
- 22.Sebille S, Gall D, de Tullio P, Florence X, Lebrun P, Pirotte B. Design, synthesis, and pharmacological evaluation of R/S-3,4-dihydro-2,2-dimethyl- 6-halo-4-(phenylaminocarbonylamino)-2H-1-benzopyrans: toward tissue-selective pancreatic beta-cell KATP channel openers structurally related to (+/−)-cromakalim. J Med Chem. 2006;49:4690–4697. doi: 10.1021/jm060161z. [DOI] [PubMed] [Google Scholar]
- 23.Galinis-Luciani D, Nguyen L, Yazdanian M. Is PAMPA a useful tool for discovery? J Pharm Sci. 2007;96:2886–2892. doi: 10.1002/jps.21071. [DOI] [PubMed] [Google Scholar]
- 24.Obata K, Sugano K, Machida M, Aso Y. Biopharmaceutics classification by high throughput solubility assay and PAMPA. Drug Dev Ind Pharm. 2004;30:181–185. doi: 10.1081/ddc-120028713. [DOI] [PubMed] [Google Scholar]
- 25.Alsenz J, Kansy M. High throughput solubility measurement in drug discovery and development. Adv Drug Deliv Rev. 2007;59:546–567. doi: 10.1016/j.addr.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Johansson T, Weidolf L, Jurva U. Mimicry of phase I drug metabolism--novel methods for metabolite characterization and synthesis. Rapid Commun Mass Spectrom. 2007;21:2323–2331. doi: 10.1002/rcm.3077. [DOI] [PubMed] [Google Scholar]
- 27.Shetty HU, Nelson WL. Chemical aspects of metoprolol metabolism. Asymmetric synthesis and absolute configuration of the 3-[4-(1-hydroxy-2-methoxyethyl)phenoxy]-1-(isopropylamino)-2-propanols , the diastereomeric benzylic hydroxylation metabolites. J Med Chem. 1988;31:55–59. doi: 10.1021/jm00396a009. [DOI] [PubMed] [Google Scholar]
- 28.Engst W, Landsiedel R, Hermersdorfer H, Doehmer J, Glatt H. Benzylic hydroxylation of 1-methylpyrene and 1-ethylpyrene by human and rat cytochromes P450 individually expressed in V79 Chinese hamster cells. Carcinogenesis. 1999;20:1777–1785. doi: 10.1093/carcin/20.9.1777. [DOI] [PubMed] [Google Scholar]
- 29.Hansch CLA, Taft RW. A survey of hammett substituent constants and resonance and field parameters. Chem. Rev. 1991;91:165–195. [Google Scholar]
- 30.Hansch CLA, Hoekman DH. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants. Washington, DC: American Chemical Society; 1995. pp. 219–304. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.