

Abstract
In recent years it has become evident that ABC transporters fulfill important barrier functions in normal organs and during disease processes. Most importantly, resistance to drugs in cancer cells led to intense oncological and pharmacological investigations in which researchers were able to highlight important pharmacological interactions of chemotherapeuticals with ABC transporter function. Recently, the development of neurodegenerative diseases and the maintenance of neuronal stem cells have been linked to the activity of ABC transporters. Here, we summarize findings from cell culture experiments, animal models and studies of patients with Alzheimer’s disease. Furthermore, we discuss pharmacological interactions and computational methods for risk assessment.
Keywords: ABC transporter, Alzheimer’s disease, neurodegeneration, MDR1, MRP1, BCRP, systems biology, dementia, Abeta, blood-brain barrier, p-glycoprotein
THE ABC TRANSPORTER SUPERFAMILY
The superfamily of ATP-binding cassette (ABC) transporters regulates drug bioavailability, metabolism, and distribution in cells and the extracellular matrix; the transporters thus play an important role by limiting drug accumulation in tissues, and have been implicated in drug resistance [1, 2]. Some family members are currently being regarded also as markers for neural stem/progenitor cells (NSPC) [3–7]. The superfamily includes several multidrug transporters (see also http://nutrigene.4t.com/humanabc.htm) belonging to one of the seven ABC A to ABC G subfamilies. At the blood-brain barrier (BBB), these subfamilies include the ABCB subfamily corresponding to P-glycoprotein (P-gp) (which mediates multidrug resistance [MDR]), the ABCC subfamily of multidrug resistance-associated proteins (MRP), and the ABCG2 subfamily (breast cancer resistance protein - BRCP) [2]. Multidrug transporters have in common large and partially overlapping substrate specificities for numerous hydrophobic compounds. They are specifically distributed in organs with secretory or barrier functions, including gut, kidney, liver, lung, and, more importantly for neurodegenerative diseases, also at the BBB and the choroid plexus. In endothelia or epithelia, they are sorted either to the apical membrane, facing the luminal surface of vessels, tubuli or canaliculi, or to the basolateral membrane. This functional and polarized arrangement at the cell membrane enables guided transport through these cellular barriers [2]. Thus, ABC transporters fulfill an important function in excreting various compounds and peptides. Especially in diseases with compartmental involvement of organs, this barrier function plays an eminent role in connecting the intra-compartmental cellular and intercellular environment with the extra-compartmental systemic bloodstream. As the brain must be regarded as one of the largest compartments surrounded by a barrier, this function may also play a role in the pathophysiology of neurodegenerative diseases.
ABC TRANSPORTERS AT THE BLOOD-BRAIN BARRIER
The BBB is a physical and metabolic barrier between the brain and the systemic circulation that serves to regulate and protect the microenvironment of the brain. The BBB is composed of a monolayer of brain capillary endothelial cells. The restriction of brain uptake by the BBB is due to the presence of tight junctions (zonulae occludens) between adjacent endothelial cells and a relative paucity of fenestrae and pinocytotic vesicles within the endothelium of cerebral arterioles, capillaries, and venules [8]. The brain capillary endothelial cells are bordered by an extracellular matrix (basal membrane), pericytes, and astrocytic foot processes. Because of the presence of the BBB, circulating molecules gain access to the brain only via lipid-mediated transport of small nonpolar molecules through the BBB by free (passive) diffusion, or, for some substances, by catalyzed transport [8]. The endothelial cells of the BBB contain numerous membrane transporters involved in the influx or efflux of various essential substrates such as electrolytes, nucleosides, amino acids, peptides, and glucose [9]. It was originally believed that membrane carriers localized at the BBB were solely responsible for the transport of such endogenous substances into and out of the brain, and that drug transport across the BBB was largely dependent on the physicochemical characteristics of the drug such as lipophilicity, molecular weight, and ionic state. However, ABC efflux transporters at the BBB limit the brain uptake of a variety of therapeutic agents, including compounds that are relatively lipophilic and would be predicted to permeate the endothelial lining of the brain microvasculature [9].
P-gp (MDR1), MRP (MRP1, 2 and 5) and BCRP play a critical role in the BBB by functioning as active efflux pumps that are able to convey a wide range of structurally diverse compounds out of the CNS. Physiologically, their role is to export endogenous substrates, or ingested or acquired and potentially neurotoxic xenobiotics or waste products from nervous tissue; they thus carry out a critical neuro-protective and detoxifying role for the brain. With regard to neurotoxic waste products, it is of major importance to note that the β-amyloid peptide (Aβ) is also transported by these molecules [10].
Many prescribed therapeutics are substrates for these efflux transporters. The activity of the ABC transporters at the BBB therefore becomes crucial in significantly limiting the entry of a large number of therapeutic drugs into the CNS and the excretion of neurotoxic metabolites. The interactions of drug import and export and the excretion of toxic metabolites play a prominent role in determining the risk for the development of neurological diseases due to hampered excretion function (e.g. Alzheimer’s disease [AD]) and the need for treatment of systemic disorders (e.g. hypertonus, hyperlipidemia). This phenomenon presents a considerable challenge to effective drug delivery and to the investigation of the pathogenesis and the progression of CNS proteopathies, e.g. AD, Parkinson’s disease, progressive supranuclear palsy, and multi systems atrophy [11].
The precise cellular and subcellular distribution of ABC transporters at the BBB remains controversial, mainly due to the difficulty in discriminating their localization in these cells with closely apposed cell membranes. Moreover, several monoclonal antibodies raised against human epitopes do not recognize the murine proteins, whereas polyclonal antibodies made against the corresponding mouse epitopes allow sensitive detection [12]. Thus, numerous investigators have used various experimental approaches such as in situ hybridization, immunohistochemistry and multilabeling immunofluorescence strategies to determine the precise location of transporters in capillary endothelial cells [12–14]. However, there is virtually no information about spatial and temporal expression differences in different brain areas of murine models, human subjects, and more importantly during the development of neurodegenerative diseases.
P-Glycoprotein (P-gp, MDR1, ABCB1)
P-gp was the first ABC transporter that was detected in endothelial cells of the human BBB [15]. Subsequently P-gp was localized in brain capillary endothelial cells of several species, including monkeys, rats, mice, cattle, and pigs, suggesting that P-gp may serve as general defense mechanism in the mammalian BBB, protecting the brain from intoxication by potentially harmful lipophilic compounds that otherwise could penetrate the BBB by simple diffusion [16]. P-gp is encoded by a small group of related genes called multidrug resistance genes (MDR). In rodents three P-gp gene products have been identified - Mdr1a, Mdr1b and Mdr2 [17]. In humans there are two gene products known - MDR1 and MDR2 [18]. Only MDR1 confers multi-drug resistance, since MDR2 is predominantly expressed by hepatocytes at the canalicular side for the secretion of phosphatidylcholine into the bile fluid [19]. P-gp is located at the luminal side of cerebral endothelial cells and transports substrates directly into the bloodstream (Fig. 1). It shuffles a variety of substrates with an immense structural diversity out of the brain (Table 1). With respect to AD, P-gp only recently has been recognized to be involved in Aβ transport [10, 20, 21].
Fig. (1).
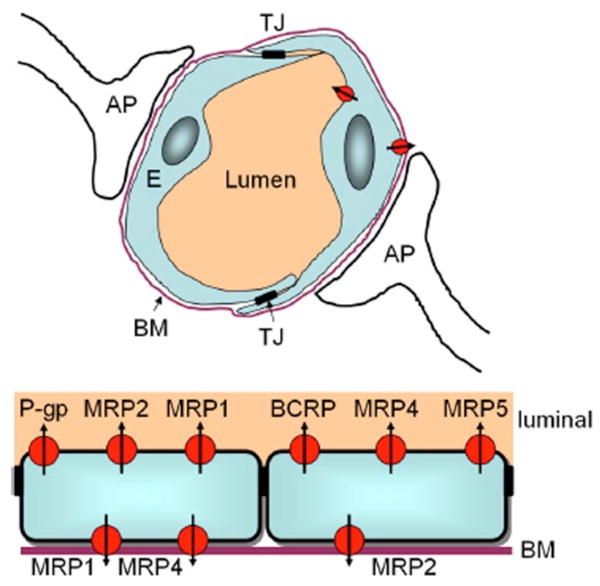
ABC transporters at the blood-brain barrier. Top: Schematic drawing of a brain capillary. The BBB is comprised essentially of the endothelial cells (E), the basal membrane (BM), and astrocytic processes (AP). Pericytes (not shown) also contribute to the cellular barrier. The cerebral endothelial cells are connected via tight junctions (TJ). Bottom: Localization of ABC transporters at the brain capillary endothelial cells. Transporters localized in either the luminal (apical) or basolateral part of the cell membrane restrict uptake (apical) or excretion (basolateral) of drugs and toxic metabolic products. P-gp – P-glycoprotein, MRP – multidrug resistance protein, BCRP – breast cancer resistance protein. Based on data from [2, 9, 12, 59, 96].
Table 1.
Selected Therapeuticals and Chemical Compounds that are Transported by P-gp. Some of these Drugs are Widely Used for the Treatment of Chronic Diseases, e.g. High Blood Pressure (Beta Blockers) and Hyperlipidemia (Statins). A Majority of these Compounds Inhibit the Excretion Function of P-gp at the BBB for Neurotoxic Metabolites from the Brain. Thus, Life-Long Treatment Using these Compounds Might Increase the Intracerebral Content of Metabolites Such as Aβ. In Contrast, the Antibiotic Rifampicin is the Strongest Known Activator of P-gp Function.
|
Ca++ channel blockers Verapamil, Diltiazem, Mibefradil, Nefedipine, Nitrendipine |
HIV protease inhibitors Ritonavir, Saquinavir, Amprenavir |
Antitumor drugs Etoproside, Imatinib, Lonafarmib, Vincaalkaloids, Taxanes |
|
Immunosuppressants Cyclosporine A, Tacrolimus, Sirolimus |
Statins Atorvastatin, Lovastatin, Simvastatin |
Antibiotics Erythromycin, Rifampicin |
|
Opoids Morphine-6-glucuronide Methadone, Loperamide, Fentanyl |
Beta blockers Carvedilol, Celiprolol, Talinolol, Bunitrolol, Carazolol |
Diverse Digoxin, Midazolam, Ivermectin, Octeotride, Ranitidine, Methotrexate, Rhodamine-123, St John’s Wort |
Multidrug Resistance-Associated Proteins (MRP, Mrp)
In contrast to P-gp, data on other ABC transporters at the BBB are much more limited and controversial. Mrp1, Mrp2 and Mrp5 can be detected in brain endothelial cells, with Mrp5 being located at the luminal side, Mrp1 on the abluminal side (basal), and Mrp2 in between. Recent studies of six murine ABC transporters (Mdr1a, Mdr3, Mrp1, Mrp2, Mrp3, and Mrp5) highlighted different cellular and subcellular localizations in different mouse strains [12]. These data emphasize the need for precisely specifying the type of sample, species, and strain analyzed. The authors were able to show endothelial expression of Mrp2 in the brains of C57Bl/6, Swiss, and SvJ mice, but not in FVB mice. It is worth noting that several ABC transporter knockout mice were established in FVB or FVB-related backgrounds [22–25]. Thus, it should be recognized in the experimental setting that ABC transporters might already be expressed differentially in the particular mouse strain employed in the investigations. MRPs comprise a group of closely related gene products that show less than 15% homology to P-gp [26]. The substrate preferences of a number of MRPs and P-gp exhibit overlap, but MRPs show a preference for organic anions and many drugs that are conjugated to glutathione, glucuronate or sulphate due to CNS enzyme activities [27].
Breast Cancer Resistance Protein (BCRP, ABCG2, Bcrp-1)
BCRP appears to be expressed in the luminal membrane of cerebral endothelial cells in a similar manner to P-gp [13]. Interestingly BCRP is essentially a half transporter when compared to P-gp, with only six transmembrane domains and one nucleotide binding site, and it is therefore assumed to form a homodimer in the cell membrane so that it can function as a transporter [28, 29]. P-gp and BCRP seem to share some functional parallels. BCRP mRNA is upregulated 3-fold in P-gp (Mdr1a) knockout mice [30]. This finding highlights the care that must be taken in the analysis of different mouse models in which the lack of the expression of a specific ABC transporter may lead to compensatory changes in gene expression and protein synthesis. BCRP was primarily isolated from human breast tumor cell lines that were negative for P-gp and MRPs, yet displayed a high resistance to mitoxantrone [31].
ABC TRANSPORTER ABLATION ANIMAL MODELS
The majority of multidrug transporter genes have one rodent orthologue. However some of the human genes are encoded in two genes. P-gp, discovered by Juliano and Lin in 1976 [32], is encoded in two genes in mice, Mdr1a and Mdr1b, with ubiquitous, complementary tissue distribution [22, 23]. In recent years, new models with ablation of various BBB ABC transporters have been reported (Table 2). These animal models have been analyzed primarily with respect to alterations in the bioavailability of pharmaceuticals [9, 16, 22, 33–35]. However, the models also provide information about compensatory expression, e.g. triple knockout mice lacking Mdr1a, Mdr1b and Mrp1 show an upregulation of Bcrp-1 during treatment with mitoxantrone, topotecan or doxorubicin [36]. Recently, we established new animal models for the investigation of the involvement of BBB ABC transporters during the initiation of Aβ deposition in the parenchyma and vasculature of AD mouse models. These mice overexpress mutated β-amyloid precursor protein (APP) and presenilin-1 (PS1) [37], or Dutch-type APP [38], or lack the expression of neprilysin [39] and are combined knockouts for one, two, three or four different ABC transporters (Mdr1a & b [23], Mrp1 [24], Bcrp-1 [25]). These different experimental models enable the collection of quantitative data, providing the basis for mathematical models.
Table 2.
ABC Transporters with Known Function at the Blood Brain Barrier and/or Involvement in AD
| Human transporter | Mouse transporter | BBB expression | Knockout model published by |
|---|---|---|---|
| ABCA2 | ABCA2 | no | [97] |
| ABCB1/P-gp/MDR1-P-gp | Mdr1a & b Mdr1a |
Luminal | [23] |
| ABCC1/MRP1 | Mrp1 | Luminal a/o Basal | [98] |
| ABCC2/MRP2 | Mrp2 | Luminal a/o Basal | [99] |
| ABCC4/MRP4 | Mrp4 | Luminal a/o Basal | [33] |
| ABCC5/MRP5 | Mrp5 | Luminal | [100] |
| ABCG2/BCRP | Bcrp-1 | Luminal | [25] |
PHARMACOLOGICAL INTERACTIONS WITH ABC TRANSPORTER FUNCTION
The role of ABC transporters in neurodegenerative diseases is intriguing, as the activity of these transporters can be modulated by several drugs [40]. Recently, a number of modulators of ABC transporter-mediated drug resistance have been developed, primarily for P-gp activity. Observations made on many of these inhibitors have been largely incidental. Additionally, there are different groups of therapeutics that are currently used for the treatment of cardiovascular diseases, e.g. hypertonus and hyperlipidemia. These drugs also coincidentally interfere with ABC transporter function at the BBB. Some of the drugs do not interfere with just a single ABC transporter, and moreover, the vast majority of the drugs change the transport kinetics of a variety of related transport proteins. These interactions have already been recognized, since the multi-drug treatment of various disorders can raise plasma and tissue drug levels to toxic amounts. Numerous problems may be associated with general inhibition of ABC transporter activity. An impressive example is congestive heart failure caused by a combined therapy of verapamil and doxorubicin at sufficient doses to inhibit P-gp [41]. Inhibitors of efflux transporters, when applied coincidently for general treatment or to enhance CNS uptake of therapeutics, are better suited to acute treatment regimes where the object is to achieve short maximal concentrations, for example in the treatment of brain tumors, than for chronic administration where long-term inhibition of ABC transporter-mediated efflux will disturb normal homeostasis. With respect to the cerebral proteopathies, this means that extended treatment with ABC transporter inhibitors will lead to raised intracerebral concentrations of a range of neurotoxic substances that enter the brain from the bloodstream or are being produced within the brain. These changes of the transport kinetics and the accumulation of neurotoxic peptides or peptide fragments must also be taken into account when evaluating the pathogenesis of neurodegenerative diseases such as AD, Lewy body disease, and Parkinson’s disease (PD). Most importantly, the extent of transport kinetics changes that take place during long-term treatment, in relation to the intracerebral concentration of neurotoxic peptide moieties, has yet to be investigated intensively in vivo.
ABC TRANSPORTERS ARE MARKERS FOR NEURAL STEM/PROGENITOR CELLS
Stem cells can be purified based on the efflux of fluorescent dyes such as rhodamine 123 and Hoechst 33342. This method is widely used to extract hematopoietic stem cells [42, 43]. However, these dyes also can be used to evaluate ABC transporter function [44]. Just recently, it has become clear that ABC transporters are expressed in a variety of stem/progenitor cells. Various groups have demonstrated expression of P-gp, BCRP, and ABCA2 in human and murine NPSCs [3–5]. The expression of different ABC transporters in NSPCs of adult mouse and rat brains appears to be region-dependent. For example, it was found that ABCA2 expression is limited to the oligodendrocytic lineage (unambiguously distinguished from astrocytes), and to a subset of cortical GABAergic interneurons and pyramidal glutamatergic neurons where it could be localized to lysosome-related organelles [4]. Considering that the latter are targets of Alzheimer’s lesions, these data provide a new rationale to explore the neuropathological consequences of ABC transporter functional dysregulation. ABCA2 also has been suggested to be a marker of neural progenitors, as it is expressed in the subventricular zone and the dentate gyrus, sites of ongoing neurogenesis in the adult brain. Moreover, it was found by another research group that all nestin-positive cells within neurospheres simultaneously express P-gp [4]. Using fluorescence-activated cell sorting (FACS) with an antibody against P-gp, it was possible to extract even more human NSPCs than the nestin-positive population [7]. In an additional investigation, the same researchers showed that both BCRP and P-gp are expressed functionally at the plasma membrane of NSPCs. Furthermore, these experiments highlighted that the expression of P-gp, BCRP and nestin was reduced during differentiation, while expression of the astrocytic marker glial fibrillary acid protein (GFAP) was upregulated [3]. Thus, ABC transporters may have a functional role in maintaining the undifferentiated status of NSPCs, as the downregulation of nestin and upregulation of GFAP are considered to be indicators of NSPC differentiation or maturation. ABC transporters have emerged as an important new field of investigation in the regulation of stem cell biology and the development of neurodegenerative diseases. It will be of major interest for the AD research community to highlight the interactions and pathways involved therein – regulation of compound excretion (including Aβ efflux) and stem cell maintenance function. Altered neurogenesis in AD patients has been recognized by various groups [45, 46]. Suggestive as a key player is the Wnt signaling pathway and its key enzyme glycogen synthase kinase-3 beta (GSK3β), as this enzyme is known to be involved in pathways that contribute to the progression of AD [47–51].
INVOLVEMENT OF ABC TRANSPORTERS IN ALZHEIMER’S DISEASE
The development of Aβ plaques in the extracellular compartment of the brain parenchyma is a hallmark of AD, and biochemical and genetic findings underscore the importance of the Aβ peptide in the pathogenesis of AD [52]. Amyloid plaques consist predominantly of Aβ peptides, mainly Aβ40 and Aβ42. The Aβ peptides are produced continuously in cells of the nervous system and systemic tissues by the cleavage of the amyloid precursor protein (APP). With age, soluble Aβ accumulates over time in the brain extracellular space, multimerizes into oligomers, protofibrils and insoluble fibrils, and eventually forms diffuse and dense-cored amyloid plaques [53–55]. Studies in AD patients have detected increased levels of peripherally circulating Aβ [56, 57]. It was suggested that Aβ in blood plasma and cerebrospinal fluid exists in equilibrium (which is controlled by an unknown mechanism) that shifts the concentration toward the brain during plaque development [58]. Normally, the brain endothelium, a key element of the BBB in vivo, does not allow free exchange of polar solutes such as Aβ between brain and blood, or blood and brain, due to the presence of a continuous cellular monolayer of tightly junctioned endothelial cells [59]. Thus, specialized transporters for Aβ must exist in brain endothelium to expel the peptide from the CNS into the bloodstream, and/or to shuttle circulating Aβ into the CNS. It has been proposed that the Aβ equilibrium between plasma and CSF is regulated at the BBB by an influx receptor (RAGE, receptor for advanced glycation end products) and an efflux receptor (LRP1, low-density lipoprotein receptor-related protein) [60]. Apolipoprotein E is considered to be involved in the RAGE/LRP1 shuffling mechanism [61], and polymorphisms in apolipoprotein E (ApoE) are known to markedly influence the risk of late-onset AD [53, 62–68].
In 2001, Lam and colleagues [10] investigated ABC transporter-mediated Aβ efflux in vitro in transfected HEK293 cells. By applying the P-gp inhibitors RU486 and RU49953, they showed that P-gp serves as an Aβ efflux pump. Additionally, fluorescence quenching binding affinity determinations and transport competition experiments supported their data. The results were confirmed in in vitro cell culture experiments using MDR1-transfected polarized kidney epithelial cells (LLC-PK1) [44]. In patients, differences in cerebrovascular P-gp expression were shown to influence Aβ deposition rates, although the determination of three known polymorphisms failed to be of significance [69]. Interestingly, the inverse correlation between Aβ deposition and P-gp expression was most striking with regard to Aβ40 positivity. Investigations of the correlation between cerebrovascular β-amyloid angiopathy (CAA) and P-gp function indicated a loss of P-gp in vessels with abundant cerebrovascular Aβ in non-demented elderly humans [20]. However, in that study, it was unclear whether the correlation was due to a primary loss of P-gp expression or a diminution of P-gp expression secondary to Aβ deposition. Kinetic studies of Aβ influx/efflux in wild type and double transgenic AD mice [70] using 125I-labeled Aβ40 yielded conflicting results [71, 72]. Interestingly, Kandimalla and colleagues demonstrated that higher Aβ40 levels in the peripheral circulation in AD mice compared to wild-type mice are not due to differences in the plasma, hepatic, or renal metabolism of the peptide.
ABC transporters, in particular P-gp, may also play a role in the intracerebral/intracellular distribution of Aβ, as they have been detected in different intracerebral (neurons, astrocytes, microglia) and intracellular compartments [73–79]. These findings are supported by our own observation of early intracellular accumulations of Aβ in small vesicles in Mdr1a/b double knockout mice expressing the Dutch-type variant of APP F (Fig. 2). New APP-overexpressing AD mouse models (APPPS1 [37]) that were depleted of P-gp (Mdr1a and Mdr1b [23]) show that BCRP function partially compensates for P-gp-mediated Aβ clearance. BCRP functions as a clearance transporter with limited capacity relative to P-gp. BCRP compensates for rising soluble Aβ concentrations up to an intracerebral concentration of 200ng/mg brain protein, thus leading to double sigmoidal deposition kinetics in these murine models.
Fig. (2).

Accumulation Aβ-immunoreactive material (antibody clone 4G8) in a 175 day-old Mdr1a/b-deficient mouse (B) expressing the Dutch-type variant of APP. Large pyramidal neurons in layer 5 (D – cortex & F – layer 5) and neurons in CA1-3 of the hippocampal formation (E) show small, Aβ-positive endolysosomes. Control DTAPP-type mice (A), which are not deficient for Mdr1a/b, do not show intracellular deposits (C – CA1 of hippocampal formation). Scale bars: A, B and D: 500μm; E: 100μm; C and F: 50μm.
Large genetic studies using 326 microsatellite markers in 466 late-onset AD families have demonstrated various new chromosomal regions with linkage to AD [80]. Matching ABC transporter locations with these new regions, a number of transporters can be found in close proximity to genetic hotspots, for example ABCA2 located on chromosome 9 (marker D9S1818). The most exciting finding was on chromosome 9, where the nonparametric affected sibpair maximum LOD score (MLS) in the combined sample was 2.97 [77]. The results for chromosome 9 were enhanced when only the autopsy-confirmed subset with an MLS of 4.31 was analyzed. This was the highest LOD score yet seen in a genomic screen for AD, even larger than the score for D19S246, the closest screening marker to apolipoprotein E in that study. Later, an in vitro cell culture investigation using neuroblastoma cells demonstrated a close relationship of APP and ABCA2 in endolysosomal compartments [81]. Analyzing brain tissue samples, Chen and colleagues demonstrated that ABCA2 levels were highest in temporal and frontal regions of the AD brain, with lower but significant levels in parietal, occipital, and cerebellar regions. However, similar ABCA2 protein expression patterns were observed in normal and schizophrenia brain samples. Thus, it will be important to define precisely whether there is indeed a role of ABCA2 in the pathogenesis of AD.
Unlike ABCA2, ABCA1 and ABCG2 have been suggested to play a significant role in the regulation of neuronal cholesterol efflux to apoE discs, and in the regulation of APP processing to generate Aβ peptides [82]. ABCA1 serves as a major regulator of cholesterol efflux and high density lipoprotein metabolism. It was demonstrated that targeted disruption of ABCA1 increases Aβ deposition in APP23 mice [83]. The lack of ABCA1 also considerably increased levels of CAA and exacerbated CAA-related hemorrhages in these mice. Remarkably, the augmentation of parenchymal and vascular Aβ was accompanied by a dramatic decrease in the level of soluble brain apoE, whereas insoluble apoE was not changed. Just recently, genetic analysis of polymorphisms in the promoter and coding region of ABCA1 indicated that the development of AD might be influenced by either a qualitative change of the ABCA1 protein caused by coding region variants (219K, 883I, and 1587R), or by a quantitative change in ABCA1 expression caused by promoter region variant (−14T) in concert with the APOE epsilon4 allele [84].
LONG-TERM INHIBITION OF ABC TRANSPORTER FUNCTION – WHERE DO WE GO?
Various treatment regimes for cardiovascular diseases include therapeutics that interfere with ABC transporter function. As described earlier, ABC transporters can be found at the BBB, in membranes of intracellular compartments, and in the choroid plexus. This complex system of membrane-bound “locks and elevators” is highly vulnerable to interference. The large number of patients with obesity and high blood pressure is not limited to the elderly, but more importantly begins to rise even in the younger population of <40 years of age. These patients will be treated with beta-blockers, anti-hyperlipidemic drugs and calcium antagonists for many years before they eventually reach the age at which AD and other proteopathies become increasingly common. There are several questions arising from this scenario, for example: 1) Does a reduction of ABC transporter function over a long period of time put the patients at risk for developing neurodegenerative diseases? 2) If so, how much reduction in transporter function is required to significantly raise the risk − 10%, 5%, 0.5%, or less? 3) What types of mechanisms play a role therein? 4) What types of toxic agents harm cerebral homeostasis – primarily intracerebral or primarily extracerebral (exogenous)? 5) How can we assess the risk of certain therapeutic drugs? and 6) Is stem cell maintenance altered by agents that affect transporters?
Environmental and genetic factors have been discussed as causes for late-onset AD. Genetic disposition and diet also influence ABC transporter function. A large number of polymorphisms in coding and non-coding regions of ABC transporter genes have been described that affect drug clearance [85–87].
AD RISK ASSESSMENT USING SYSTEMS BIOLOGY COMPUTATIONAL MODELING
A basic problem is that the many risk factors for complex diseases are poorly understood, let alone the way in which they interact [88]. The very fact that there are multiple drivers for these conditions suggests that a reductionist approach focusing on individual entities in isolation is no longer appropriate, and may even be misleading. Investigating the possible combinations by trial and error in humans is onerous, but feasible, when only two components are considered. However, it becomes extremely complicated with three components, and well nigh impossible with four or more. Such circumstances will require a systems approach and the use of sophisticated and progressively refined models. Systems Biology, therefore, promises to assist in the development of more specific compounds and in the identification of optimal drug targets on the basis of their importance as key ‘nodes’ within an overall network, rather than on the basis of their properties as isolated components.
Only very recently have systems biology approaches been applied towards elucidating the dynamics underlying complex diseases such as AD. The systems biology approach includes mathematical modeling of cellular biochemical reaction networks and the investigation of their properties through computer simulations [89, 90]. This strategy allows us to explore the structure and function of networks using methods from dynamical systems theory.
A number of different factors or presumed ‘drivers’ that are involved in AD pathophysiology can be included in these analyses: Aβ, tau, apoE, inflammation, oxidative stress, protein degradation, and protein transport systems. Since aging is the most important risk factor for AD, and aging is associated with multiple changes that might influence the probability of developing AD, a systems biological approach is particularly valuable in this instance. Computer simulations enable fast and large-scale quantitative predictions of networks dynamics. An important application is to understand disease mechanisms and to support the development of therapies. Mathematical modeling of AD has, so far, focused on the kinetics of Aβ aggregation, a downstream consequence of the decision between the amyloidogenic and non-amyloidogenic pathways. Up to now, systems biology has been one-directional, aiming at the mathematical modeling of biochemical reactions. Only recently has feedback to the wet-lab, in the form of quantitative predictions of pathway dynamics, been possible. A systems biology approach supports not only the formulation and testing of hypotheses, but could also form the basis for a risk assessment of ABC transporter involvement in AD mouse models. The phenomena described here are of a dynamic nature, most likely involving nonlinear feedback mechanisms. In the long run, once the relevant cellular components are identified and characterized at the molecular level, it will be necessary to investigate dynamic features for an understanding of transport mechanisms. A systems biology approach does, however, require quantitative time-series data, which for many omics-type technologies remains a challenge to this day.
Clearance kinetics and toxicity evaluation of various compounds, pharmaceuticals and peptides can be assessed in cell culture experiments and animal models. These data help to evaluate the risk to prevent side effects or other diseases. Risk assessment in AD research can be supported with systems biology methods. Systems biology aims to unravel the complex dynamics of cellular biochemical reaction networks by mathematical modeling and computer simulations. These simulations can help to analyze interaction networks with respect to internal critical events or to the impact of external influences. Aβ aggregation and fibrillization have been extensively analyzed in silico and in vivo with respect to different APP mutations [91, 92]. Computational models beyond the scope of Aβ aggregation are rare.
Simplified interaction schemes (Fig. 3) and quantitative data from different ABC transporter knockout mouse models can be used to describe the chemical kinetics of Aβ aggregation and its clearance through the BBB in mathematical models. The resulting differential equations describe the interaction between Aβ moieties and different ABC transporters, and allow predictions of common regulation mechanisms. Knowledge and quantification of toxic aggregates, e.g. Aβ*56, can be included and used to assess critical constellations [55]. The application of computer simulations helps to predict the influence of drug interactions within the network. Due to the fact that processes that underlie AD are of a nonlinear dynamic nature, different strategies to those from genomics and bioinformatics are required. Systems biology has emerged as a paradigm to address such phenomena, but relies on accurate, quantitative, sufficiently rich time series. The systems approach investigates the dynamic behavior of a system through systematic perturbations. Given that an appropriate set of stimulus-response data is available, methods from dynamical systems theory allow the identification of mathematical models from time-course experiments. These computational techniques support the formulation of hypotheses about alternative network structures and through simulation studies, metabolic control analysis, sensitivity and bifurcation analysis, key elements of a network can be investigated with respect to their role within the overall network [93, 94]. Systematic perturbation experiments are necessary if the underlying processes involved feedback mechanisms to realize a robust or adaptive system. Apart from the study of intracellular data, a mathematical approach could also be useful for phenomenological data, capturing relationships between conditions and helping extract rules for AD risk assessment. This area of research is in its infancy, but given the increasing recognition and success of such approaches in the study of metabolic and signal transduction pathways, the time has come to strengthen interdisciplinary links between the medical and clinical domain with systems biology. The establishment of interaction maps and models of AD-related pathology with a correlation to the inhibition of ABC transporter function, especially those transporters that exhibit BBB excretion function (e.g. P-gp), will help us to evaluate clinical information of different drug therapies.
Fig. (3).

Schematic model for the mathematical evaluation of clearance, degradation, and aggregation of Aβ involving ABC transporters at the BBB. Aβ aggregates into oligomers, protofibrils and fibrils. The kinetics of aggregation and disaggregation vary, thus the timepoints of plaque development and the onset of neurotoxic effects are highly dependent on these factors. The aggregates and monomers can be degraded by different proteases. Additionally, Aβ monomers and small Aβ multimers are transported by BBB transporters. The excretion capacity and its velocity are dependent on the charge, size and conformation of the Aβ peptides. Charge changes as known from Dutch-Aβ (E22Q) lead to hampered transport. After several months, the restricted clearance of Dutch-Aβ results in deposition at the BBB. Intracerebral toxic levels of metabolites and peptide oligomers impair the normal function of neurons and disturb the maintenance of NSPCs. Importantly, long-term inhibition of the ABC transporter excretion function at the BBB by therapeuticals (e.g. statins and beta-blockers) could put the patients at higher risk for developing neurodegenerative diseases. The knowledge of clearance, degradation, aggregation and excretion kinetics as well as the amount of a defined Aβ moiety needed to exert toxicity for specific neuronal or stem cell populations and its clinical outcome, can be used to calculate individual risk profiles.
CONCLUSIONS
Given that most idiopathic AD, which accounts for the vast majority of AD cases, cannot be explained simply by increased Aβ production [52], it is important to elucidate fully the mechanisms whereby the concentrations of AD-related proteins are regulated in the brain. To this end, a detailed understanding of the molecular and genetic basis of ABC transporter function for clearance mechanisms at the BBB and neural stem cell maintenance holds at least a part of the key to understanding the pathogenesis of AD. Using model organisms [95], the kinetics of Aβ transport/exchange between blood and brain can be computed and matched for healthy and diseased persons. Furthermore, these systems-biology computational models can be used to integrate known interactions of currently used therapeutics with the excretion function at the BBB. It is of emerging importance to investigate the effect of long-term pharmacological treatment of systemic diseases on the removal of APP-associated peptides, since some of these cleavage products have been noted for their neuronal toxicity in vitro and in vivo [55, 91, 92]. ABC transporters may also play a crucial role in NSPC self-renewal by preventing cellular differentiation, and hence could be vital in future stem cell-based therapies for neurodegenerative disorders such as AD and PD.
Acknowledgments
The work was funded by intramural grants of the University of Rostock (FORUN 2007 889023 and 889047 to J.P.) and by NIH grant RR-00165 (L.C.W.).
References
- 1.Linton KJ. Structure and function of ABC transporters. Physiology (Bethesda) 2007;22:122–30. doi: 10.1152/physiol.00046.2006. [DOI] [PubMed] [Google Scholar]
- 2.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 3.Islam MO, Kanemura Y, Tajria J, et al. Functional expression of ABCG2 transporter in human neural stem/progenitor cells. Neurosci Res. 2005;52:75–82. doi: 10.1016/j.neures.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 4.Broccardo C, Nieoullon V, Amin R, et al. ABCA2 is a marker of neural progenitors and neuronal subsets in the adult rodent brain. J Neurochem. 2006;97:345–55. doi: 10.1111/j.1471-4159.2006.03714.x. [DOI] [PubMed] [Google Scholar]
- 5.Islam MO, Kanemura Y, Tajria J, et al. Characterization of ABC transporter ABCB1 expressed in human neural stem/progenitor cells. FEBS Lett. 2005;579:3473–80. doi: 10.1016/j.febslet.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 6.Mohan A, Kandalam M, Ramkumar HL, Gopal L, Krishnakumar S. Stem cell markers: ABCG2 and MCM2 expression in retinoblastoma. Br J Ophthalmol. 2006;90:889–93. doi: 10.1136/bjo.2005.089219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin T, Islam O, Heese K. ABC transporters, neural stem cells and neurogenesis--a different perspective. Cell Res. 2006;16:857–71. doi: 10.1038/sj.cr.7310107. [DOI] [PubMed] [Google Scholar]
- 8.Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol. 1999;5:556–69. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
- 9.Loscher W, Potschka H. Blood-Brain Barrier Active Efflux Transporters: ATP-Binding Cassette Gene Family. Neurorx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lam FC, Liu R, Lu P, et al. beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem. 2001;76:1121–8. doi: 10.1046/j.1471-4159.2001.00113.x. [DOI] [PubMed] [Google Scholar]
- 11.Bartels AL, Willemsen AT, Kortekaas R, et al. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm. 2008 doi: 10.1007/s00702-008-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soontornmalai A, Vlaming ML, Fritschy JM. Differential, strain-specific cellular and subcellular distribution of multidrug transporters in murine choroid plexus and blood-brain barrier. Neuroscience. 2006;138:159–69. doi: 10.1016/j.neuroscience.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Cooray HC, Blackmore CG, Maskell L, Barrand MA. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport. 2002;13:2059–63. doi: 10.1097/00001756-200211150-00014. [DOI] [PubMed] [Google Scholar]
- 14.Demeule M, Regina A, Jodoin J, et al. Drug transport to the brain: key roles for the efflux pump P-glycoprotein in the blood-brain barrier. Vascul Pharmacol. 2002;38:339–48. doi: 10.1016/s1537-1891(02)00201-x. [DOI] [PubMed] [Google Scholar]
- 15.Cordon-Cardo C, O’Brien JP, Casals D, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A. 1989;86:695–8. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schinkel AH. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv Drug Deliv Rev. 1999;36:179–194. doi: 10.1016/s0169-409x(98)00085-4. [DOI] [PubMed] [Google Scholar]
- 17.Hsu SI, Cohen D, Kirschner LS, Lothstein L, Hartstein M, Horwitz SB. Structural analysis of the mouse mdr1a (P-glycoprotein) promoter reveals the basis for differential transcript heterogeneity in multidrug-resistant J774.2 cells. Mol Cell Biol. 1990;10:3596–606. doi: 10.1128/mcb.10.7.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chin JE, Soffir R, Noonan KE, Choi K, Roninson IB. Structure and expression of the human MDR (P-glycoprotein) gene family. Mol Cell Biol. 1989;9:3808–20. doi: 10.1128/mcb.9.9.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–62. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 20.Vogelgesang S, Warzok R, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, Siegmund W, Walker LC, Pahnke J. The role of P-glycoprotein in Cerebral Amyloid Angiopathy - Implications for the early pathogenesis of AD. Current Alzheimer Research. 2004;1:121–126. doi: 10.2174/1567205043332225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cirrito JR, Deane R, Fagan AM, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–90. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 23.Schinkel AH, Mayer U, Wagenaar E, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drugtransporting) P-glycoproteins. Proc Natl Acad Sci U S A. 1997;94:4028–33. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wijnholds J, Evers R, van Leusden MR, et al. Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat Med. 1997;3:1275–9. doi: 10.1038/nm1197-1275. [DOI] [PubMed] [Google Scholar]
- 25.Jonker JW, Buitelaar M, Wagenaar E, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci U S A. 2002;99:15649–54. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Han H, Elmquist WF, Miller DW. Expression of various multidrug resistance-associated protein (MRP) homologues in brain microvessel endothelial cells. Brain Res. 2000;876:148–53. doi: 10.1016/s0006-8993(00)02628-7. [DOI] [PubMed] [Google Scholar]
- 27.Dallas S, Miller DS, Bendayan R. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58:140–61. doi: 10.1124/pr.58.2.3. [DOI] [PubMed] [Google Scholar]
- 28.Litman T, Brangi M, Hudson E, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J Cell Sci. 2000;113 (Pt 11):2011–21. doi: 10.1242/jcs.113.11.2011. [DOI] [PubMed] [Google Scholar]
- 29.Rocchi E, Khodjakov A, Volk EL, et al. The product of the ABC half-transporter gene ABCG2 (BCRP/MXR/ABCP) is expressed in the plasma membrane. Biochem Biophys Res Commun. 2000;271:42–6. doi: 10.1006/bbrc.2000.2590. [DOI] [PubMed] [Google Scholar]
- 30.Cisternino S, Mercier C, Bourasset F, Roux F, Scherrmann JM. Expression, up-regulation, and transport activity of the multidrug-resistance protein Abcg2 at the mouse blood-brain barrier. Cancer Res. 2004;64:3296–301. doi: 10.1158/0008-5472.can-03-2033. [DOI] [PubMed] [Google Scholar]
- 31.Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–62. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 33.Leggas M, Adachi M, Scheffer GL, et al. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24:7612–21. doi: 10.1128/MCB.24.17.7612-7621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schinkel AH. The roles of P-glycoprotein and MRP1 in the blood-brain and blood-cerebrospinal fluid barriers. Adv Exp Med Biol. 2001;500:365–72. doi: 10.1007/978-1-4615-0667-6_60. [DOI] [PubMed] [Google Scholar]
- 35.Sun H, Dai H, Shaik N, Elmquist WF. Drug efflux transporters in the CNS. Adv Drug Deliv Rev. 2003;55:83–105. doi: 10.1016/s0169-409x(02)00172-2. [DOI] [PubMed] [Google Scholar]
- 36.Allen JD, Brinkhuis RF, Wijnholds J, Schinkel AH. The mouse Bcrp1/Mxr/Abcp gene: amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res. 1999;59:4237–41. [PubMed] [Google Scholar]
- 37.Radde R, Bolmont T, Kaeser SA, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–6. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herzig MC, Winkler DT, Burgermeister P, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–60. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 39.Iwata N, Tsubuki S, Takaki Y, et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–2. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 40.Tan B, Piwnica-Worms D, Ratner L. Multidrug resistance transporters and modulation. Curr Opin Oncol. 2000;12:450–8. doi: 10.1097/00001622-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 41.Ozols RF, Cunnion RE, Klecker RW, et al. Verapamil and adriamycin in the treatment of drug-resistant ovarian cancer patients. J Clin Oncol. 1987;5:641–7. doi: 10.1200/JCO.1987.5.4.641. [DOI] [PubMed] [Google Scholar]
- 42.Spangrude GJ, Johnson GR. Resting and activated subsets of mouse multipotent hematopoietic stem cells. Proc Natl Acad Sci U S A. 1990;87:7433–7. doi: 10.1073/pnas.87.19.7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolf NS, Kone A, Priestley GV, Bartelmez SH. In vivo and in vitro characterization of long-term repopulating primitive hematopoietic cells isolated by sequential Hoechst 33342-rhodamine 123 FACS selection. Exp Hematol. 1993;21:614–22. [PubMed] [Google Scholar]
- 44.Kuhnke D, Jedlitschky G, Grube M, et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007;17:347–53. doi: 10.1111/j.1750-3639.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ziabreva I, Perry E, Perry R, et al. Altered neurogenesis in Alzheimer’s disease. J Psychosom Res. 2006;61:311–6. doi: 10.1016/j.jpsychores.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 46.Jin K, Peel AL, Mao XO, et al. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:343–7. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inestrosa N, De Ferrari GV, Garrido JL, et al. Wnt signaling involvement in beta-amyloid-dependent neurodegeneration. Neurochem Int. 2002;41:341–4. doi: 10.1016/s0197-0186(02)00056-6. [DOI] [PubMed] [Google Scholar]
- 48.De Ferrari GV, Inestrosa NC. Wnt signaling function in Alzheimer’s disease. Brain Res Brain Res Rev. 2000;33:1–12. doi: 10.1016/s0165-0173(00)00021-7. [DOI] [PubMed] [Google Scholar]
- 49.Caricasole A, Bakker A, Copani A, Nicoletti F, Gaviraghi G, Terstappen GC. Two sides of the same coin: Wnt signaling in neurodegeneration and neurooncology. Biosci Rep. 2005;25:309–27. doi: 10.1007/s10540-005-2893-6. [DOI] [PubMed] [Google Scholar]
- 50.Terstappen GC, Gaviraghi G, Caricasole A. The Wnt signaling pathway as a target for the treatment of neurodegenerative disorders. IDrugs. 2006;9:35–8. [PubMed] [Google Scholar]
- 51.Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 53.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 54.Cirrito JR, May PC, O’Dell MA, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–53. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lesné S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 56.Kuo YM, Emmerling MR, Lampert HC, et al. High levels of circulating Abeta42 are sequestered by plasma proteins in Alzheimer’s disease. Biochem Biophys Res Commun. 1999;257:787–91. doi: 10.1006/bbrc.1999.0552. [DOI] [PubMed] [Google Scholar]
- 57.Matsubara E, Ghiso J, Frangione B, et al. Lipoprotein-free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer’s disease and Down’s syndrome. Ann Neurol. 1999;45:537–41. [PubMed] [Google Scholar]
- 58.DeMattos RB, Bales KR, Parsadanian M, et al. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem. 2002;81:229–36. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- 59.Begley DJ. ABC transporters and the blood-brain barrier. Curr Pharm Des. 2004;10:1295–312. doi: 10.2174/1381612043384844. [DOI] [PubMed] [Google Scholar]
- 60.Zlokovic BV. Clearing amyloid through the blood-brain barrier. J Neurochem. 2004;89:807–11. doi: 10.1111/j.1471-4159.2004.02385.x. [DOI] [PubMed] [Google Scholar]
- 61.Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–99. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warzok RW, Kessler C, Apel G, et al. Apolipoprotein E4 promotes incipient Alzheimer pathology in the elderly. Alzheimer Dis Assoc Disord. 1998;12:33–9. doi: 10.1097/00002093-199803000-00005. [DOI] [PubMed] [Google Scholar]
- 63.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strittmatter WJ, Weisgraber KH, Huang DY, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:8098–102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 66.Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker LC, Pahnke J, Madauss M, et al. Apolipoprotein E4 promotes the early deposition of Abeta42 and then Abeta40 in the elderly. Acta Neuropathol (Berl) 2000;100:36–42. doi: 10.1007/s004010051190. [DOI] [PubMed] [Google Scholar]
- 68.Pahnke J, Walker LC, Schroeder E, et al. Cerebral beta-amyloid deposition is augmented by the −491AA promoter polymorphism in non-demented elderly individuals bearing the apolipoprotein E epsilon4 allele. Acta Neuropathol (Berl) 2003;105:25–9. doi: 10.1007/s00401-002-0602-0. [DOI] [PubMed] [Google Scholar]
- 69.Vogelgesang S, Cascorbi I, Schroeder E, et al. Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics. 2002;12:535–41. doi: 10.1097/00008571-200210000-00005. [DOI] [PubMed] [Google Scholar]
- 70.Holcomb L, Gordon MN, McGowan E, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 71.Kandimalla KK, Curran GL, Holasek SS, Gilles EJ, Wengenack TM, Poduslo JF. Pharmacokinetic analysis of the blood-brain barrier transport of 125I-amyloid beta protein 40 in wild-type and Alzheimer’s disease transgenic mice (APP,PS1) and its implications for amyloid plaque formation. J Pharmacol Exp Ther. 2005;313:1370–8. doi: 10.1124/jpet.104.081901. [DOI] [PubMed] [Google Scholar]
- 72.Poduslo JF, Curran GL, Wengenack TM, Malester B, Duff K. Permeability of proteins at the blood-brain barrier in the normal adult mouse and double transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2001;8:555–67. doi: 10.1006/nbdi.2001.0402. [DOI] [PubMed] [Google Scholar]
- 73.Rajagopal A, Simon SM. Subcellular localization and activity of multidrug resistance proteins. Mol Biol Cell. 2003;14:3389–99. doi: 10.1091/mbc.E02-11-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Volk H, Potschka H, Loscher W. Immunohistochemical localization of P-glycoprotein in rat brain and detection of its increased expression by seizures are sensitive to fixation and staining variables. J Histochem Cytochem. 2005;53:517–31. doi: 10.1369/jhc.4A6451.2005. [DOI] [PubMed] [Google Scholar]
- 75.Volk HA, Burkhardt K, Potschka H, Chen J, Becker A, Loscher W. Neuronal expression of the drug efflux transporter P-glycoprotein in the rat hippocampus after limbic seizures. Neuroscience. 2004;123:751–9. doi: 10.1016/j.neuroscience.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 76.Bendayan R, Lee G, Bendayan M. Functional expression and localization of P-glycoprotein at the blood brain barrier. Microsc Res Tech. 2002;57:365–80. doi: 10.1002/jemt.10090. [DOI] [PubMed] [Google Scholar]
- 77.Schlachetzki F, Pardridge WM. P-glycoprotein and caveolin-1alpha in endothelium and astrocytes of primate brain. Neuroreport. 2003;14:2041–6. doi: 10.1097/00001756-200311140-00007. [DOI] [PubMed] [Google Scholar]
- 78.Lee G, Schlichter L, Bendayan M, Bendayan R. Functional expression of P-glycoprotein in rat brain microglia. J Pharmacol Exp Ther. 2001;299:204–12. [PubMed] [Google Scholar]
- 79.Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J Comp Neurol. 2006;495:70–83. doi: 10.1002/cne.20840. [DOI] [PubMed] [Google Scholar]
- 80.Pericak-Vance MA, Grubber J, Bailey LR, et al. Identification of novel genes in late-onset Alzheimer’s disease. Exp Gerontol. 2000;35:1343–52. doi: 10.1016/s0531-5565(00)00196-0. [DOI] [PubMed] [Google Scholar]
- 81.Chen ZJ, Vulevic B, Ile KE, et al. Association of ABCA2 expression with determinants of Alzheimer’s disease. Faseb J. 2004;18:1129–31. doi: 10.1096/fj.03-1490fje. [DOI] [PubMed] [Google Scholar]
- 82.Kim WS, Rahmanto AS, Kamili A, et al. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J Biol Chem. 2007;282:2851–61. doi: 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- 83.Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280:43224–35. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- 84.Rodriguez-Rodriguez E, Mateo I, Llorca J, et al. Association of genetic variants of ABCA1 with Alzheimer’s disease risk. Am J Med Genet B Neuropsychiatr Genet. 2007;144:964–8. doi: 10.1002/ajmg.b.30552. [DOI] [PubMed] [Google Scholar]
- 85.Marzolini C, Paus E, Buclin T, Kim RB. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75:13–33. doi: 10.1016/j.clpt.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 86.Schwab M, Eichelbaum M, Fromm MF. Genetic polymorphisms of the human MDR1 drug transporter. Annu Rev Pharmacol Toxicol. 2003;43:285–307. doi: 10.1146/annurev.pharmtox.43.100901.140233. [DOI] [PubMed] [Google Scholar]
- 87.Hoffmeyer S, Burk O, von Richter O, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A. 2000;97:3473–8. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Walker LC, Rosen RF, Levine H., III Diversity of Abeta deposits in the aged brain: a window on molecular heterogeneity? Rom J Morphol Embryol. 2008;49:5–11. [PubMed] [Google Scholar]
- 89.Wolkenhauer O. Defining Systems Biology: An Engineering Perspective. IET Systems Biology. 2007;1:204–206. doi: 10.1049/iet-syb:20079017. [DOI] [PubMed] [Google Scholar]
- 90.Wolkenhauer O, Mesarovic M. Feedback Dynamics and Cell Function: Why Systems Biology is called Systems Biology. Molecular BioSystems. 2005;1:14–16. doi: 10.1039/b502088n. [DOI] [PubMed] [Google Scholar]
- 91.Luheshi LM, Tartaglia GG, Brorsson AC, et al. Systematic in vivo analysis of the intrinsic determinants of amyloid Beta pathogenicity. PLoS Biol. 2007;5:e290. doi: 10.1371/journal.pbio.0050290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheon M, Chang I, Mohanty S, Luheshi LM, Dobson CM, Vendruscolo M, Favrin G. Structural reorganisation and potential toxicity of oligomeric species formed during the assembly of amyloid fibrils. PLoS Comput Biol. 2007;3:1727–38. doi: 10.1371/journal.pcbi.0030173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tyson J, Chen K, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Current Opinion in Cell Biology. 2003;15:221–223. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- 94.Aldridge BB, Burke JM, Lauffenburger DA, Sorger PK. Physicochemical modelling of cell signalling pathways. Nat Cell Biol. 2006;8:1195–203. doi: 10.1038/ncb1497. [DOI] [PubMed] [Google Scholar]
- 95.Radde R, Duma C, Goedert M, Jucker M. The value of incomplete mouse models of Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2008 doi: 10.1007/s00259-007-0704-y. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Y, Schuetz JD, Elmquist WF, Miller DW. Plasma membrane localization of multidrug resistance-associated protein homologs in brain capillary endothelial cells. J Pharmacol Exp Ther. 2004;311:449–55. doi: 10.1124/jpet.104.068528. [DOI] [PubMed] [Google Scholar]
- 97.Sakai H, Tanaka Y, Tanaka M, et al. ABCA2 deficiency results in abnormal sphingolipid metabolism in mouse brain. J Biol Chem. 2007 doi: 10.1074/jbc.M611056200. [DOI] [PubMed] [Google Scholar]
- 98.Wijnholds J, deLange EC, Scheffer GL, et al. Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J Clin Invest. 2000;105:279–85. doi: 10.1172/JCI8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vlaming ML, Mohrmann K, Wagenaar E, et al. Carcinogen and anticancer drug transport by Mrp2 in vivo: studies using Mrp2 (Abcc2) knockout mice. J Pharmacol Exp Ther. 2006;318:319–27. doi: 10.1124/jpet.106.101774. [DOI] [PubMed] [Google Scholar]
- 100.Wijnholds J, Mol CA, van Deemter L, et al. Multidrug-resistance protein 5 is a multispecific organic anion transporter able to transport nucleotide analogs. Proc Natl Acad Sci U S A. 2000;97:7476–81. doi: 10.1073/pnas.120159197. [DOI] [PMC free article] [PubMed] [Google Scholar]