

Abstract
Despite the well-documented association between gallstones and the metabolic syndrome1,2, the mechanistic links between these two disorders remain unknown. Here we demonstrate that mice with isolated hepatic insulin resistance created by liver-specific disruption of the insulin receptor (LIRKO mice) are markedly predisposed towards cholesterol gallstone formation due to at least two distinct mechanisms. Disinhibition of the forkhead transcription factor FoxO1 increases expression of the biliary cholesterol transporters Abcg5 and Abcg8, resulting in an increase in biliary cholesterol secretion. Hepatic insulin resistance also decreases expression of the bile acid synthetic enzymes, particularly Cyp7b1, and produces partial resistance to the farnesoid X receptor (FXR), leading to a lithogenic bile salt profile. As a result, after only one week on a lithogenic diet, 36% of LIRKO mice develop gallstones and by 12 weeks, 100% have gallstones. Thus, hepatic insulin resistance provides the critical link between the metabolic syndrome and increased gallstone susceptibility.
Gallstones represent a major health problem in the U.S., affecting more than 20 million people3 and resulting in at least six billion dollars in healthcare costs annually4. There are several predisposing factors to biliary disease, but among the strongest is the association between cholesterol gallstones and obesity, which was first noted over 100 years ago5. We now recognize this association as part of the more complex metabolic syndrome1,2, which includes insulin resistance, central obesity, varying degrees of impaired glucose tolerance, hepatic steatosis, and increased risk of cardiovascular disease. Insulin resistance plays a central role in the pathogenesis of the metabolic syndrome6, but how it is linked to gallstone formation has remained a mystery.
The LIRKO mouse is a novel model for studying the effects of isolated hepatic insulin resistance and its role in gallstone formation. LIRKO mice were generated using the Cre-loxP system to specifically ablate the insulin receptor in hepatocytes7. Prior studies7,8 of the LIRKO mouse showed that hepatic insulin resistance produces many features of the metabolic syndrome, including increased hepatic glucose production, hyperglycemia, hyperinsulinemia, and dyslipidemia.
To determine whether LIRKO mice are also more susceptible to cholesterol gallstone formation, we challenged LIRKO mice and their littermate controls (Lox) with a lithogenic diet containing 0.5 % cholic acid, 1% cholesterol, and 15% dairy fat. After only one week on this diet, LIRKO gallbladders were filled with cholesterol monohydrate crystals, and 36% had acquired gallstones, compared to 0% of the control mice (Fig. 1). After twelve weeks on the lithogenic diet, 100% of LIRKO mice had developed gallstones, whereas 40% of controls still manifested normal appearing gallbladders and no visible stones (Fig. 1d).
Figure 1. LIRKO mice are susceptible to cholesterol gallstone formation.
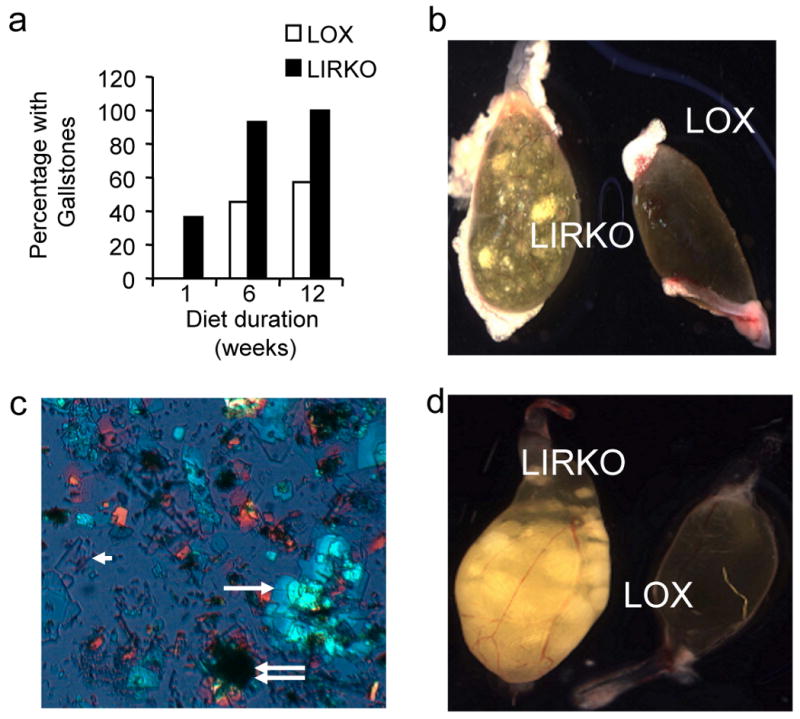
(a) The prevalence of gallstones in male Lox and LIRKO mice after being fed a lithogenic diet for 1, 6 or 12 weeks, and sacrificed at 5-7 months of age (n=10-15 per group). (b) Macroscopic appearance of gallbladders from Lox and LIRKO mice after one week on the lithogenic diet. (c) Polarized light microscopy of bile from a LIRKO mouse fed a lithogenic diet for one week demonstrating solid cholesterol monohydrate crystals (arrowhead) aggregating into sandy stones (single white arrow) and true stones (double arrow) (d) Macroscopic appearance of gallbladders from Lox and LIRKO female mice after 12 weeks of consuming the lithogenic diet. Note that early stones formed after one week on the lithogenic diet developed into numerous large true stones by 12 weeks. Lox mice that did not develop gallstones displayed gallbladders that were normal in appearance and size.
To identify the pathophysiological mechanisms by which insulin resistance increases gallstone susceptibility, we studied Lox and LIRKO mice on a normal chow diet. Real-time PCR revealed decreased expression of the major enzymes of the bile acid synthetic pathways Cyp7a1, Cyp7b1, Cyp8b1 and Cyp27a1 by 30-80% (Fig. 2a). Consistent with this, bile acid synthesis, measured by steady state fecal bile acid excretion, was decreased 40% (Fig. 2b). In addition, the LIRKO bile salt profile was more hydrophobic, i.e., more lithogenic9, than bile from control mice (Fig. 2c) with a four-fold decrease in the ratio of muricholates to cholate in LIRKO bile (Fig. 2d).
Figure 2. LIRKO mice show quantitative and qualitative defects in bile acid synthesis on a chow diet.
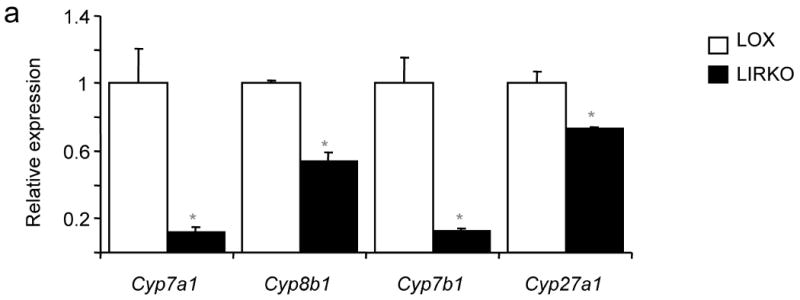
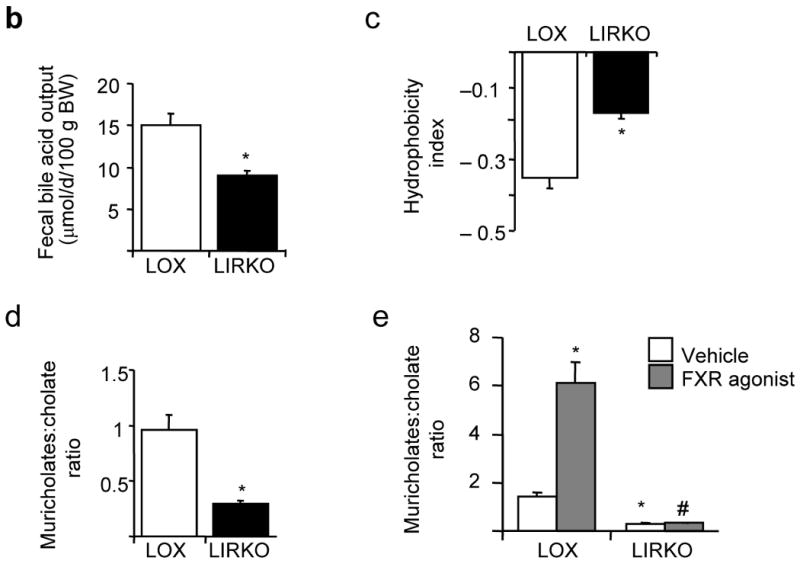
(a) Real-time PCR analysis of cDNA prepared from six-week old mice sacrificed during the light cycle following a 24 hour fast; the expression of each gene was normalized to its value in Lox mice (n=5-8, *p=0.05 versus Lox). (b) Since the synthesis of bile acids is equivalent to fecal excretion in the steady state, we assessed bile acid synthesis by collecting feces from 3 month old Lox and LIRKO mice for 72 hours and measuring total bile acid outputs (n=4, p=0.02). Hepatic bile from 5-6 month old Lox and LIRKO mice was subjected to HPLC analysis as described in Methods (n=4-5). (c) Hydrophobicity indices were calculated as described in Methods (*p=0.002). (d) The ratio of muricholates to cholate was calculated using the molar percentages of tauroα- and tauroβ- muricholate and taurocholate (*p=0.001). (e) Three-month old mice were gavaged with an FXR agonist (GW4064) at 100 mg/kg/d or vehicle for 14 days. Mice were fasted overnight, and sacrificed two hours after the last dose of agonist. Bile was expressed from the gallbladder and ratio of muricholates to cholate was determined as above. *p<0.05 versus Lox untreated; #p<0.05 versus Lox treated.
The nuclear hormone receptor farnesoid-X-receptor (FXR) plays an important role in regulating the bile salt profile. FXR action is anti-lithogenic10,11, and treatment of Lox mice with an FXR agonist produced a six-fold increase in the ratio of muricholates to cholate (Fig. 2e), decreasing bile lithogenicity. LIRKO mice showed reduced levels of FXR mRNA and protein (Supplementary Fig. 1). Although FXR agonist treatment increased expression of the small heterodimer partner, Shp, and decreased expression of Cyp7a1 in LIRKO mice, as it did in controls, it failed to decrease Cyp8b1, improve the bile salt profile (Fig. 2e) and prevent gallstone formation in LIRKO mice fed the lithogenic diet (Fig. 2e, and Supplementary Fig. 1). Therefore, LIRKO mice are partially resistant to FXR activation.
Further contributing to gallstone formation, biliary cholesterol secretion was increased four-fold in LIRKO mice (Fig. 3a). This resulted in a marked increase in the propensity of LIRKO bile to form cholesterol gallstones, as indicated by equilibrium phase diagrams (Fig. 3b) and cholesterol saturation indices (CSI, Fig. 3c and Supplementary Table 1). Gallbladder volume was also increased three-fold in LIRKO mice (Fig. 3d), consistent with impaired motility and increased biliary cholesterol content 12. Since fractional cholesterol absorption from the gut was similar in Lox and LIRKO mice (Fig. 3e), the absolute amount of cholesterol absorbed from the gut in LIRKO mice would be increased because of their high levels of biliary cholesterol secretion. This was associated with a two-fold increase in hepatic cholesterol (Fig. 3f), and decreased expression of sterol regulatory element binding protein (SREBP)-2 and its targets8.
Figure 3. Biliary cholesterol content is increased in LIRKO mice, promoting supersaturated bile.
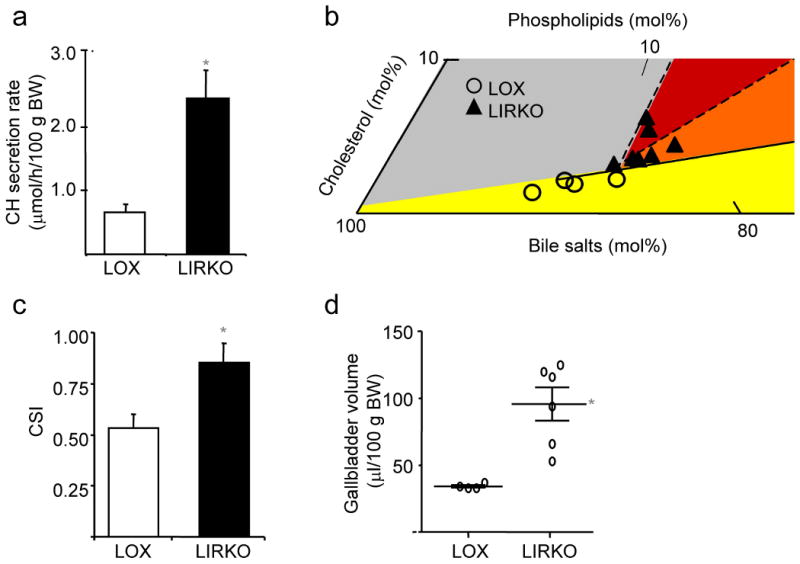
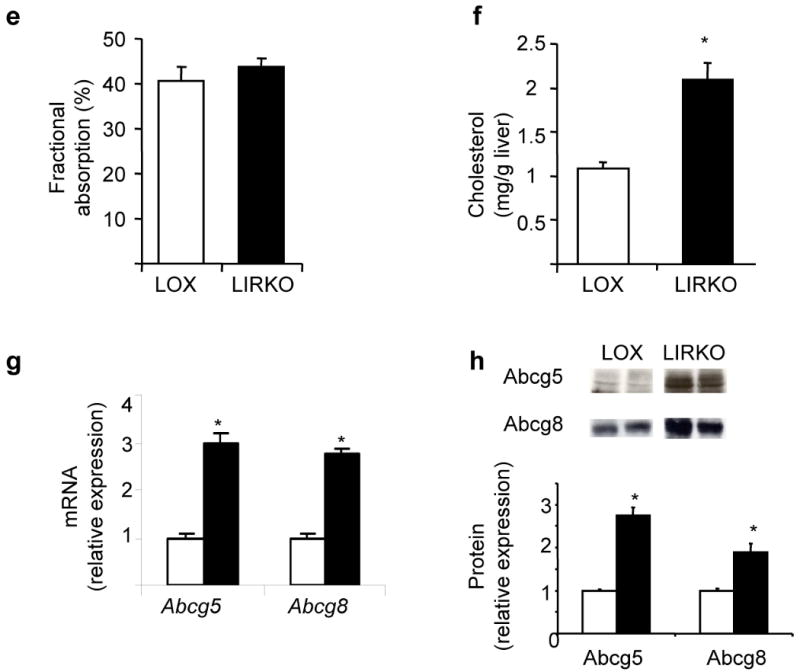
(a) Bile was collected from the common bile duct of 5-6 month old mice. Cholesterol (CH) secretory rates were derived from cholesterol concentrations and normalized to body weight (BW, *p=0.003, n=4-5 per group). (b) Relative lipid compositions (mol per 100 mol) of gallbladder bile obtained from Lox (open circles) and LIRKO (closed triangles) mice fed a chow diet were calculated and plotted on a partial condensed phase diagram (see Methods). The different phases are indicated for equilibrium conditions: micelles only (yellow); micelles and liquid crystals (orange); micelles, liquid crystals and cholesterol monohydrate crystals (red); and micelles and cholesterol monohydrate crystals (gray). Relative lipid compositions of bile from control mice plot in a zone in which only micelles would form at equilibrium, but bile from LIRKO mice plot in zones in which liquid crystals (orange) or both liquid crystals and cholesterol monohydrate crystals (red) in addition to micelles would form at equilibrium. (c) Cholesterol Saturation Indices (CSI) of gallbladder bile obtained from 5-6 month old mice were calculated as described in Methods. (d) Gallbladder volumes from 5-6 month old mice. Each point represents a single mouse, whereas long and short lines represent mean and SEM, respectively (*p=0.004). (e) Cholesterol absorption was measured in 4 month old mice by a fecal dual isotope ratio method. (f) Hepatic lipids were extracted from 2-3 month old Lox and LIRKO mice and total cholesterol content was measured by gas chromatography (n=4-6, *p<0.05). (g) Real-time PCR analysis of RNA isolated from the livers of non-fasted 8-10 week old mice (n=8, *p<1 x 10-5). (h) Membranes purified from 8-10 week old mice were subjected to immunoblotting with antibodies against ABCG5 or ABCG8, and quantified with ImageJ software (*p<0.05, n=3-4).
Biliary cholesterol secretion is regulated by the heterodimeric cholesterol transporters, Abcg5 and Abcg813,14. LIRKO mice displayed a two- to three-fold increase in the expression of these genes at the mRNA (Fig. 3g) and protein levels (Fig. 3h), accounting for the increased biliary cholesterol secretion observed in LIRKO mice. This increase in Abcg5/Abcg8 was a direct effect of insulin resistance. Therefore, in rat hepatoma cells, insulin suppressed expression of Abcg5/Abcg8 mRNA at sub-nanomolar concentrations, in a dose responsive manner that paralleled the suppression of the well-described insulin target, phosphoenolpyruvate carboxykinase (Pck1) (Fig. 4a). Treatment with an FXR agonist had no effect on Abcg5/Abcg8 (Fig. 4b and Supplementary Fig. 1b), but treatment with a liver-X-receptor (LXR) agonist increased expression of these genes and impaired the ability of insulin to suppress them (Fig. 4b), consistent with the known ability of LXR to induce Abcg5/Abcg8 expression in vitro and in vivo 15.
Figure 4. Insulin and LXR regulate Abcg5/ABCG8 expression.
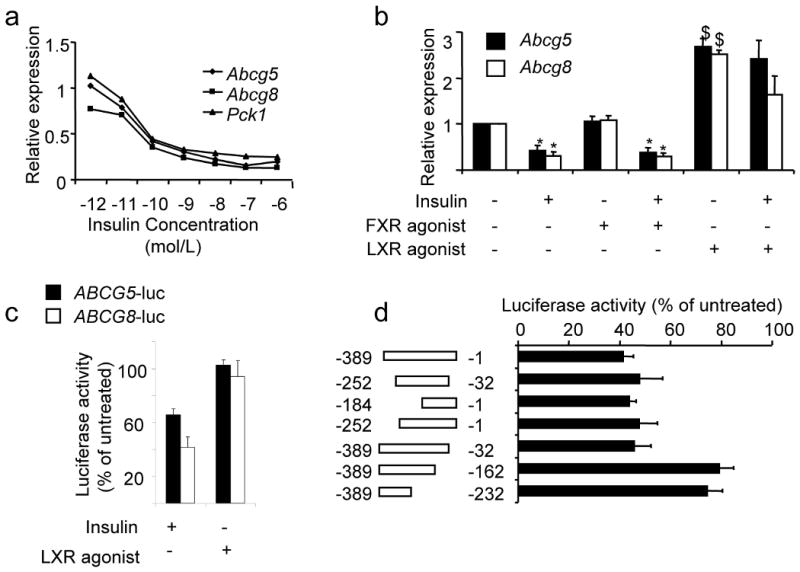
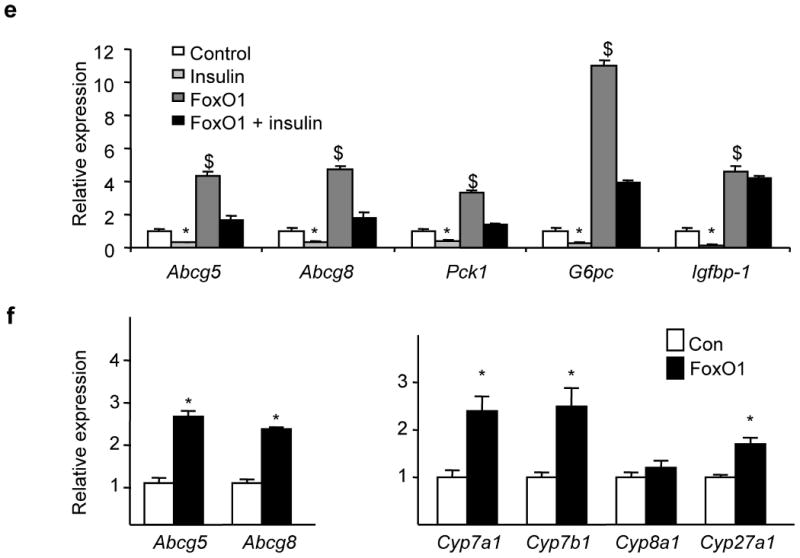
(a) Fao rat hepatoma cells were serum-starved overnight, and then treated with the indicated amounts of insulin for 6-8 hours. We performed real-time PCR and normalized the expression of each gene to its expression level in the absence of insulin (n=3). (b) Cells were cultured overnight in the presence or absence of 100 nM insulin, and either vehicle (DMSO), 50 nM FXR agonist (GW4064) or 5 μM LXR agonist (T090137). Gene expression was measured by real-time PCR analysis (n=3). (c) Cells were transfected with a luciferase reporter fused to the intragenic region of the ABCG5/ABCG8 gene in either the ABCG5 or ABCG8 orientation, and treated overnight with 100 nM insulin or 5 μM LXR agonist, or left untreated (n=3-5). (d) As described in Methods, we made constructs containing deletions in the ABCG5/ABCG8 intragenic region (in the ABCG8 orientation) and tested their ability to respond to insulin. For each construct, data are presented as luciferase activity in the presence of insulin divided by luciferase activity in the absence of insulin x 100% (n=3-8). (e) Cells were infected with control adenovirus or adenovirus encoding constitutively active FOXO1. Two days following infection, cells were washed, and incubated overnight in the presence or absence of 100 nM insulin, before being harvested for real-time PCR analysis (n=3). (f, g) Real-time PCR was used to measure gene expression in 8-week old male mice overexpressing constitutively active FOXO1 under the α1-antitrypsin promoter and their controls, after being fed a high carbohydrate diet for six hours after a 24 hour fast, as described previously 18 (*p<0.05 versus value in the absence of insulin; $p<0.05 versus value in the absence of agonist or constitutively active FOXO1)
In humans, the ABCG5 and ABCG8 genes are arranged head-to-head on chromosome 2, separated by 374 base pairs16. This region lacks a canonical LXR response element16, and failed to respond to an LXR agonist in a reporter assay (Fig. 4c). Nonetheless, it responded to insulin in both the ABCG5 and ABCG8 orientations. These data indicate the presence of an element in the intragenic region of the ABCG5/ABCG8 gene that responds to insulin independently of LXR.
To better define this element, we studied the activity of a series of ABCG8 deletion constructs (Fig. 4d). Insulin suppressed the full length construct by approximately 60%, whereas constructs lacking the −32 to −162 region were suppressed by only 20%, indicating at least one insulin responsive element in this region. Examination of this region revealed the sequence TGTTT, which is part of a well characterized insulin response element that is present in Pck1, glucose-6-phosphatase (G6pc), and insulin-like growth factor binding protein 1(Igfbp-1) 17. This motif binds the winged helix/forkhead transcription factors, such as FoxO1, and can mediate the suppressive effect of insulin on these genes17.
Indeed, overexpression of a constitutively active form of FOXO1 18 in rat hepatoma cells produced a four-fold increase in Abcg5/Abcg8 expression (Fig. 4e), and significantly blunted the ability of insulin to suppress these genes. Similar effects were seen in Pck1, G6pc, and Igfbp-1, consistent with prior reports 17. The fact that insulin still retained some ability to suppress Abcg5/Abcg8 expression in the presence of constitutively active FOXO1 suggests that insulin regulates these genes through both FOXO1-dependent and independent mechanisms.
Finally, we examined transgenic mice overexpressing constitutively active human FOXO1 in liver under the α1-antitrypsin promoter18. These mice manifested several features of the metabolic syndrome, including hyperglycemia and hyperinsulinemia18. Abcg5 and Abcg8 were increased two-to three-fold at the mRNA level, showing that FOXO1 activation is sufficient to drive expression of Abcg5/Abcg8 in vivo, as in vitro (Fig. 4f) 18. However, overexpression of activated FOXO1 also increased expression of the bile acid synthetic enzymes, Cyp7a1, Cyp7b1, and Cyp27a1 (Fig. 4g), indicating that other insulin-dependent pathways are more important in the regulation of these enzymes in the LIRKO mouse.
Although FOXO1 clearly activates transcription of Abcg5 and Abcg8 expression in vitro and in vivo, whether this is mediated by direct or indirect mechanisms, such as co-activation of the xenobiotic receptors, CAR and PXR 19, is not known. In either case, this appears to be a general mechanism by which insulin resistance promotes cholesterol gallstone formation, as increased expression of ABCG5/ABCG8 is associated with increased gallstone formation in both humans 20 and mice 21. Moreover, increased expression of Abcg5 and Abcg8 is observed in mice with diet-induced obesity (Supplementary Table 3), and increased biliary cholesterol secretion has been documented in mice with insulin deficiency22 as well as humans with the metabolic syndrome23,24.
Insulin resistance also plays a role in gallstone formation through regulation of bile acid synthesis, but this is more complicated. Cyp7a1 is increased in the insulin deficient mouse25, but decreased in the LIRKO mouse, the high fat fed obese mouse, and the leptin deficient ob/ob mouse, whereas Cyp8b1 is decreased in LIRKO mice, but increased or unchanged in the other models (Supplementary Tables 2, 3). In contrast, Cyp7b1 is consistently decreased by 40-70% in all of these models, indicating that insulin is the major regulator of this gene.
Cyp7b1 is the first enzyme specific to the acidic pathway of bile acid synthesis, which predominantly produces muricholates26 that are protective against cholesterol gallstone formation. Consistent with this, LIRKO and insulin deficient diabetic mice display a marked decrease in the muricholate to cholate ratio and increased gallstone susceptibility 22. Furthermore, this effect of insulin involves FXR and/or its downstream targets, as FXR agonist treatment of LIRKO mice fails to improve the bile salt profile or reduce gallstone formation. In humans, the end product of the acidic pathway of bile acid synthesis is chenodeoxycholate (CDCA). CDCA is more hydrophobic than the muricholates, but when given in pharmacological doses, desaturates human gallbladder bile27. Therefore, it is not clear how a defect in the acidic pathway of bile acid synthesis would affect the lithogenicity of human bile, and studies in humans will be necessary to resolve this issue.
In summary, hepatic insulin resistance is sufficient to increase biliary cholesterol secretion and promote cholesterol gallstone formation, both features of the human metabolic syndrome 23,24,28. These effects are due to disinhibition of FOXO1, which drives expression of Abcg5/Abcg8 in addition to the enzymes of gluconeogenesis, as well as other factors, including resistance to FXR action. Therefore, hepatic insulin resistance links the hyperglycemia7, dyslipidemia8 and gallstone formation of the metabolic syndrome. These findings also suggest that therapies that improve hepatic insulin resistance and decrease FOXO1 activity might prevent formation of cholesterol gallstones, in addition to having beneficial effects on glucose homeostasis.
Methods
Mice and Diets
Generation and genotyping of LIRKO (cre+/-; IR lox/lox) mice, their littermate Lox controls (cre-/-; IRlox/lox) 7, and constitutively active FOXO1 transgenic mice 18 were performed as previously described. LIRKO mice were on a mixed genetic background, including 129/sv, C57BL/6, FVB and DBA, and inbred for more than ten generations. The FOXO1 transgenics were on an FVB background. We fed mice ad lib with a standard chow diet (Mouse Diet 9F; PMI Nutrition International) or a lithogenic diet (0.5 % cholic acid, 1% cholesterol, and 15% dairy fat; Jackson Laboratories, Bar Harbor, ME). Unless otherwise indicated, mice studied were male, chow-fed, and sacrificed in the non-fasted state.
The FXR agonist, GW4064, was administered at 100 mg/kg/day in PEG400: Tween 80, 4:1 (vol/vol) as described10. We sacrificed mice fed the lithogenic diet for three months after an overnight fast, during which they had free access to water. All animal studies followed the US National Institutes of Health guidelines and were approved by the institutional animal care and use committees at the Joslin Diabetes Center or University of Illinois at Chicago College of Medicine and the Jesse Brown Veterans Affairs Medical Center, or the Harvard University Medical Area Standing Committee on Animals.
Biliary and Hepatic Lipid Studies and Cholesterol Absorption
These are detailed in the Supplementary Methods section. Briefly, we obtained hepatic bile by cannulating the common bile duct, and gallbladder bile by digitally expressing the contents of the gallbladder. We measured bile salt species using high performance liquid chromatography (HPLC), comparing retention times with bile salt standards. Gallbladder bile obtained from mice treated with an FXR agonist showed some peaks attributable to the agonist itself which represented less than 10% of the total bile acids. Biliary phospholipid concentrations were determined by the inorganic phosphorus method; biliary cholesterol was measured by HPLC after extraction with hexane; total bile salts were measured by a spectrophotometric assay based on 3α-hydroxysteroid dehydrogenase. We measured hepatic cholesterol by gas chromatography or using an enzymatic assay (Wako Chemicals). Cholesterol absorption was measured by the fecal dual isotope ratio method.
Real-time-PCR analysis
Quantitative real-time PCR analysis of FOXO1 transgenic livers was performed as described previously 18 and expression levels normalized to L32 ribosomal protein. In all other experiments, RNA (RNeasy, Qiagen) was isolated from mouse liver and used to direct cDNA synthesis (RT for PCR, Clontech). Quantitative real-time PCR was performed in a fluorescent temperature cycler (ABI) with either SYBR Green Master Mix (Roche, Mannheim, Germany) or the Taqman Universal master mix (ABI; primers purchased through Assay on Demand) 29. We normalized expression values to the level of input RNA.
Western Blot Analysis
Preparation of liver and nuclear extract samples is detailed in the Supplementary Methods section. Antibodies against ABCG530 and ABCG813 have been described previously. Antibodies against FXR were obtained commercially (Santa Cruz). Quantitation was performed with ImageJ software.
Gallbladder Volumes and Microscopy
Following cholecystectomy, gallbladder contents were expressed manually and weighed. Volume was calculated by assuming a density of 1 g/mL. For prevalence studies, gallbladders were placed on a glass slide, incised to release their contents, visualized by direct and polarized light microscopy, and scored for the presence of stones and cholesterol crystals.
Plasmids, Transfections and Luciferase Assays
Luciferase constructs, encoding −1 to −374 base pairs of the human ABCG5/ABCG8 intragenic region have been described16. Deletions were made by PCR amplification of the pABCG8-luc construct and ligated into PGL3 basic (Promega), using KpnI and BglII sites introduced by the PCR primers. For transfection experiments, we cultured cells in 24-well plates and transfected them at 50-80% confluence with 800 μg of the reporter plasmid and Lipofectamine 2000, per the manufacturers instructions. For each plasmid, the same transfection mixture was used for all conditions. Luciferase activity was measured using the Dual Assay Luciferase kit (Promega) and normalized to activity in the absence of insulin or agonist. We performed at least three independent experiments, each in triplicate, with each plasmid. Data are presented as the means of three or more experiments.
Cell Culture Studies
Fao rat hepatoma cells were grown in RPMI supplemented with 10% serum, washed three times in RPMI, and cultured in RPMI with or without insulin, 5 μM TO901317 (a synthetic LXR agonist, Sigma) or 50 nM GW4064 (a synthetic FXR agonist). For adenoviral studies, we treated cells with equivalent amounts of control adenovirus (encoding LacZ) or adenovirus encoding constitutively active FOXO1, in which the three sites phosphorylated in response to insulin (Thr-24, Ser 256, Ser-319) were mutated to alanines 18. After 48 hours, cells were treated with insulin and assayed as above. Each experiment was performed at least twice, in triplicate, and the results of a representative experiment are shown.
Statistical Analyses
Statistical significance was calculated by an unpaired Student's t-test. All data are expressed as the mean ± SEM.
Supplementary Material
Acknowledgments
We thank C. Rask-Madsen for photography, and M. Leonard and D.E. Cohen for helpful discussions. We also thank H. Hobbs (University of Texas Southwestern Medical Center at Dallas) for antibodies against ABCG5 and ABCG8, T. Willson (GlaxoSmithKline) for supplying GW4064, and J. Sakai (University of Tokyo) for the ABCG5 and ABCG8 luciferase reporter constructs. This work was funded in part by grants from the US National Institutes of Health, including DK063696-05 (SBB), DK31036 and DK45935 (CRK), DK036588 and DK073687 (MCC), the Joslin Diabetes and Endocrine Research Center grant DK036836-20, and the Veterans Affairs Merit Review Program (TGU).
Reference List
- 1.Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Cirulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 2.Diehl AK. Cholelithiasis and the insulin resistance syndrome. Hepatology. 2000;31:528–530. doi: 10.1002/hep.510310238. [DOI] [PubMed] [Google Scholar]
- 3.Everhart JE, Khare M, Hill M, Maurer KR. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology. 1999;117:632–639. doi: 10.1016/s0016-5085(99)70456-7. [DOI] [PubMed] [Google Scholar]
- 4.Sandler RS, et al. The burden of selected digestive diseases in the United States. Gastroenterology. 2002;122:1500–1511. doi: 10.1053/gast.2002.32978. [DOI] [PubMed] [Google Scholar]
- 5.Osler W. The Principles and Practice of Medicine. D. Appleton and Company; New York: 1892. p. 432. [Google Scholar]
- 6.Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- 7.Michael MD, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Molecular Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 8.Biddinger SB, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heuman DM. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J Lipid Res. 1989;30:719–730. [PubMed] [Google Scholar]
- 10.Moschetta A, Bookout AL, Mangelsdorf DJ. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat Med. 2004;10:1352–1358. doi: 10.1038/nm1138. [DOI] [PubMed] [Google Scholar]
- 11.Kok T, et al. Enterohepatic circulation of bile salts in farnesoid X receptor-deficient mice: efficient intestinal bile salt absorption in the absence of ileal bile acid-binding protein. J Biol Chem. 2003;278:41930–41937. doi: 10.1074/jbc.M306309200. [DOI] [PubMed] [Google Scholar]
- 12.Chen Q, Amaral J, Biancani P, Behar J. Excess membrane cholesterol alters human gallbladder muscle contractility and membrane fluidity. Gastroenterology. 1999;116:678–685. doi: 10.1016/s0016-5085(99)70190-3. [DOI] [PubMed] [Google Scholar]
- 13.Graf GA, et al. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem. 2003;278:48275–48282. doi: 10.1074/jbc.M310223200. [DOI] [PubMed] [Google Scholar]
- 14.Yu L, et al. Expression of ABCG5 and ABCG8 is required for regulation of biliary cholesterol secretion. J Biol Chem. 2005;280:8742–8747. doi: 10.1074/jbc.M411080200. [DOI] [PubMed] [Google Scholar]
- 15.Repa JJ, et al. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J Biol Chem. 2002;277:18793–18800. doi: 10.1074/jbc.M109927200. [DOI] [PubMed] [Google Scholar]
- 16.Remaley AT, et al. Comparative genome analysis of potential regulatory elements in the ABCG5-ABCG8 gene cluster. Biochem Biophys Res Commun. 2002;295:276–282. doi: 10.1016/s0006-291x(02)00652-6. [DOI] [PubMed] [Google Scholar]
- 17.Foufelle F, Ferre P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J. 2002;366:377–391. doi: 10.1042/BJ20020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281:10105–10117. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 19.Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24:7931–7940. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang ZY, et al. Increased expression of LXRalpha, ABCG5, ABCG8, and SR-BI in the liver from normolipidemic, nonobese Chinese gallstone patients. J Lipid Res. 2008;49:464–472. doi: 10.1194/jlr.M700295-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Uppal H, et al. Activation of liver X receptor sensitizes mice to gallbladder cholesterol crystallization. Hepatology. 2008;47:1331–1342. doi: 10.1002/hep.22175. [DOI] [PubMed] [Google Scholar]
- 22.Akiyoshi T, Uchida K, Takase H, Nomura Y, Takeuchi N. Cholesterol gallstones in alloxan-diabetic mice. J Lipid Res. 1986;27:915–924. [PubMed] [Google Scholar]
- 23.Shaffer EA, Small DM. Biliary lipid secretion in cholesterol gallstone disease. The effect of cholecystectomy and obesity. J Clin Invest. 1977;59:828–840. doi: 10.1172/JCI108705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennion LJ, Grundy SM. Effects of obesity and caloric intake on biliary lipid metabolism in man. J Clin Invest. 1975;56:996–1011. doi: 10.1172/JCI108180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishida H, Yamashita C, Kuruta Y, Yoshida Y, Noshiro M. Insulin is a dominant suppressor of sterol 12 alpha-hydroxylase P450 (CYP8B) expression in rat liver: possible role of insulin in circadian rhythm of CYP8B. J Biochem (Tokyo) 2000;127:57–64. doi: 10.1093/oxfordjournals.jbchem.a022584. [DOI] [PubMed] [Google Scholar]
- 26.Swell L, et al. An in vivo evaluation of the quantitative significance of several potential pathways to cholic and chenodeoxycholic acids from cholesterol in man. J Lipid Res. 1980;21:455–466. [PubMed] [Google Scholar]
- 27.Grundy SM, Lan SP, Lachin J. The effects of chenodiol on biliary lipids and their association with gallstone dissolution in the National Cooperative Gallstone Study (NCGS) J Clin Invest. 1984;73:1156–1166. doi: 10.1172/JCI111301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Attili AF, et al. Factors associated with gallstone disease in the MICOL experience. Multicenter Italian Study on Epidemiology of Cholelithiasis. Hepatology. 1997;26:809–818. doi: 10.1002/hep.510260401. [DOI] [PubMed] [Google Scholar]
- 29.Biddinger SB, et al. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes. 2005;54:1314–1323. doi: 10.2337/diabetes.54.5.1314. [DOI] [PubMed] [Google Scholar]
- 30.Yu L, et al. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A. 2002;99:16237–16242. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.