

Abstract
Antisense oligonucleotides (AO) or antisense RNA can complementarily bind to a target site in pre-mRNA and regulate gene splicing, either to restore gene function by reprogramming gene splicing or to inhibit gene expression by disrupting splicing. These two applications represent novel therapeutic strategies for several types of diseases such as genetic disorders, cancers and infectious diseases. In this review, the recent developments and applications of antisense-mediated splicing modulation in molecular therapy are discussed, with emphasis on advances in antisense-mediated splice targeting, applications in diseases and systematic delivery.
Keywords: Alternative splicing, antisense oligonucleotide, cancer, genetic disorders, molecular therapy, splicing modulation
Introduction
Pre-mRNA splicing is an essential step in eukaryotic gene expression, and has become a novel target for drug design. Splicing-targeted antisense approaches can lead to potent modulation of disease-related gene expression, either to restore gene function by reprogramming gene splicing or to inhibit gene expression by disrupting splicing. Therefore, antisense-mediated splicing modulation potentially represents a novel therapeutic strategy for several types of diseases such as genetic disorders, cancers and infectious diseases.
Antisense-mediated splicing modulation has generally included the use of antisense oligonucleotides (AOs). An alternative approach to this method uses expression vectors to produce antisense RNA inside the cell. AOs or antisense RNA could complementarily bind to a target site in pre-mRNA, and regulate the splicing process. 2′-O-Methyl (2′-O-Me) and 2′-O-methoxyethyl (2′-MOE) phosphorothioate oligomers are the two most widely used, early-generation, splicing modulation AOs. A newer generation of AOs has been developed, mainly by chemical modifications of the furanose ring of the nucleotide, to further enhance target affinity, biostability and pharmacokinetics [1]. Three of the most promising types of new-generation AOs are: (i) phosphoroamidate morpholino oligomer (PMO); (ii) peptide nucleic acid (PNA); and (iii) locked nucleic acid (LNA) [2–5].
This review discusses the most recent developments and applications of antisense-mediated splicing modulation in molecular therapy in which AOs are most commonly used, with an emphasis on advances in antisense-mediated splicing targeting, applications in diseases, systematic delivery of AOs, as well as the possible future directions for this area of research.
Antisense-mediated splice targeting
AOs or antisense RNA can be used in two contrasting ways to manipulate pre-mRNA splicing: reprogram pre-mRNA splicing or knockdown gene expression (Figure 1).
Figure 1. Schematic demonstration of antisense-mediated splicing modulation.
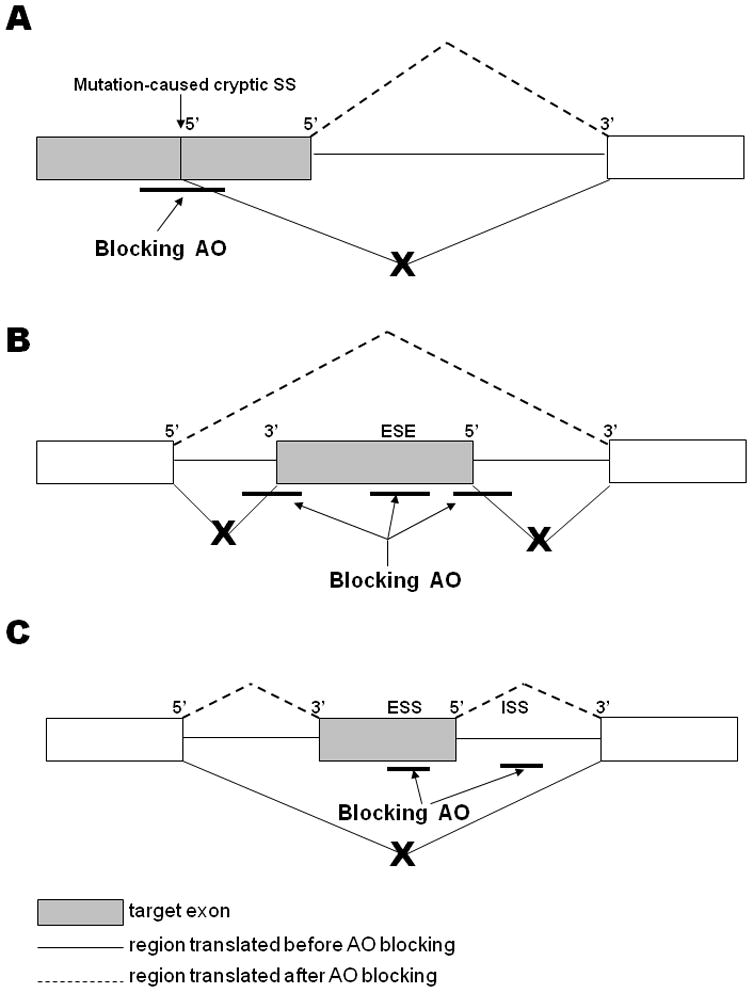
(A) Antisense oligonucleotide (AO) blocking of a mutation-induced cryptic 5′ splicing site (SS). (B) AO blocking of exon-intron junctions and/or exonic splicing enhancer (ESE) site to induce exon skipping. (C) AO blocking of exonic splicing silencer (ESS) or intronic splicing silencer (ISS) sites to enhance exon inclusion.
Antisense-mediated splicing reprogramming is primarily used to restore the function of the deficient gene in genetic diseases. The reprogramming blocks cryptic splicing sites, created by an inherited mutation, to redirect splicing back to the correct SSs (Figure 1A). This approach has been successfully used to correct splicing mutations in several genes, such as CFTR (cystic fibrosis transmembrane conductance regulator), HBB (β-globin), tau, LMNA (lamin A/C) and Oa1 (osteoarthritis QTL 1) [6–8]. AOs can also be used to induce exon skipping to delete a region of pre-mRNA that contains a mutation (Figure 1B). For example, this approach has been evaluated for the treatment of Duchenne muscular dystrophy (DMD) [9]. Furthermore, AOs can be used to regulate alternative splicing, such as in Bcl-x, IL-5, SMN2 (survival motor neuron 2), FGFR1 (fibroblast growth factor receptor 1) and MyD88 (myeloid differentiation primary response gene 88) [6,10,11]. In eukaryotes, in addition to the correct recognition of exon-intron junctions, accurate splicing is also dependent on regulatory sequence elements within exons and introns, such as exonic splicing enhancers and silencers (ESEs and ESSs, respectively). Recently, these regulatory elements have been targeted by antisense approaches to modulate splicing outcome (Figure 1C). Moreover, some efforts have been made to improve the efficiency of splicing modification by combining different AOs, or AOs and siRNAs; some recent advances are discussed below. Lastly, AOs or antisense RNA can also be used to knockdown gene expression by interrupting splicing, such as inducing exon skipping (Figure 1B), and several strategies have been developed for this purpose.
Targeting the regulatory elements of splicing
Spinal muscular atrophy (SMA) is caused by the homozygous loss of SMN1. SMN2, an almost identical copy of the SMN1 gene, is present in all patients with SMA, but is nonfunctional due to exon 7 skipping. Exon 7 skipping in SMN2 is mainly caused by a single nucleotide substitution in the affected exon [12]. This exon is included in a small proportion of SMN2 transcripts, thus producing low levels of functional SMN2 protein. In patients with SMA, the low level of full-lengthSMN2 protein is not sufficient to compensate for the loss of SMN1, and thus cannot maintain the viability of α-motor neurons; the loss of α-motor neurons leads to disease development [13]. Theoretically, if exon 7 could be reintroduced, SMN2 would be functional. The reintroduction of exon 7 represents a potential treatment approach for patients with SMA [14,15]. Hua et al identified an essential core sequence in exon 7 of SMN2 that is surrounded by two inhibitory regions containing ESSs [16]. Blocking or masking any part of the central core sequence promoted skipping of exon 7, whereas blocking either of the two ESS-containing regions that flank exon 7 promoted efficient exon 7 inclusion. If combined with blocking of the intronic splicing silencers (ISSs) within intron 7 of SMN2 [17,18], these elements could provide promising molecular targets for treating SMA.
Cystic fibrosis (CF), caused by mutations in the CFTR gene, is a disease model that could be used to study therapeutic approaches targeting ISSs. The approximately 150 nt ISS of CFTR exon 9 promotes the exclusion of exon 9 in the mature mRNA, and exon 9 exclusion is related to the occurrence of monosymptomatic and full forms of CF. Buratti et al successfully mapped the binding sites of serine/arginine-rich protein (SR) trans-acting factors responsible for the exclusion of exon 9 into an approximately 40-nt long region [19]. It would be interesting to determine whether blocking these SR protein binding sites using AOs could restore exon 9 inclusion in the mature CFTR mRNA.
AO-mediated exon skipping to restore a disrupted reading frame caused by mutations has been tested in DMD [9]. AO-blocking of ESEs efficiently induced exon skipping of the dystrophin gene. A collection of AOs targeting putative ESE sites effectively induced the specific skipping of 38 of the 79 different DMD exons [20]. However, this approach is limited to genes with highly repetitive exons, such as DMD.
ESEs could also provide appropriate targets for silencing disease-causing gene expression [21]. Khoo et al used different sequence-targeting AOs, including AOs against ESEs and splice sites, to induce apolipoprotein B (apoB) exon 27 skipping to interrupt the reading frame and silence the expression of apoB [22]. AOs against ESEs induced skipping of exon 27, but much less efficiently than AOs against splice sites [22]. These results suggested that downregulation of apoB100 by exon skipping could be a potential therapeutic approach for lowering circulating LDL and cholesterol levels.
The use of combinations of AOs for modifying splicing
Generally, a well-designed AO can reprogram or silence pre-mRNA splicing. However, efficiency needs to be improved to achieve therapeutic effects in many situations. The use of cocktails or combinations of AOs against different sites in pre-mRNA has been investigated. For example, specific combinations of 2′-O-Me AOs improved exon skipping by targeting two putative splicing regulatory sequences within one exon in DMD. Such double targeting was effective even for the distal ‘unskippable’ exons 47 and 57 [23]. AO cocktails have also been tested with different chemistries, 2′-O-Me and PMOs, in DMD [24]. In the apoB gene, AOs targeting a routine splice site in combination with AOs against a branch-point sequence were the most effective at inducing exon 27 skipping [22].
Combining splicing modification with siRNA-mediated gene knockdown
In patients with β-thalassemia, a mutated β-globin gene causes deficient synthesis of functional β-chain and, consequently, leads to excessive free α-globin chains precipitating at the erythrocyte membrane, resulting in hemolytic anemia. Theoretically, rebalancing the α/β-globin expression ratio could alleviate the disease. Xie et al combined two different strategies to rebalance α/β-globin expression in single-cell zygotes of a β654 mouse model of β-thalassemia: siRNA to knockdown α-globin mRNA, and AOs to correct splicing-deficient β-globin [25]. Three sets of transgenic mice with short hairpin RNA, antisense RNA or combined constructs were analyzed. Significant restoration of β-thalassemia was observed in all transgenic mice. The F1 progeny of these treated mice also demonstrated significant restoration of β-thalassemia, particularly in those receiving combined constructs, implying the utility of the combined strategy for gene therapy.
Gene knockdown by antisense-mediated splicing modulation
Pre-mRNA splicing also provides a target for knockdown of disease-related gene expression. Some recent examples of this approach are discussed.
Antisense-induced exon skipping can disrupt reading frames
Disrupting the reading frame of a gene leads to the premature termination of protein synthesis, and could also trigger the degradation of mRNA through a mechanism known as nonsense-mediated decay [26]. Theoretically, disrupting the reading frame provides an alternative method for downregulating gene expression. Antisense-induced exon skipping was used to inhibit HIV-1 multiplication by targeting the HIV-1 regulatory proteins Tat and Rev. Asparuhova et al used antisense U7 RNAs to induce a partial skipping of the Tat and Rev internal exons, and repress the expression of these proteins [27]. HIV-1 multiplication in lymphocytes was inhibited by the most efficient constructs, suggesting a new therapeutic approach in the treatment of HIV.
Inhibition of RNA splicing via branch-point modification
The 2′-OH group of the branch point adenosine is a key moiety that initiates the splicing reaction by attacking the phosphate at the 5′ exon-intron junction [28]. Most eukaryotic box C/D small nucleolar RNAs (snRNAs) guide base-pairing with target RNAs and direct site-specific 2′-O-methylation [29]. Zhao et al designed artificial box C/D snRNAs to target 2′-O-methylate in Saccharomyces cerevisiae pre-mRNAs at the adenosine branch point, resulting in efficient blocking of splicing initiation [30]. This new approach could be applied to target other genes by changing the guide sequence in the C/D snRNAs. However, this system has only been tested in yeast; whether it would be successful in pre-mRNA modification in higher eukaryotic systems remains uncertain.
Antisense U7 snRNAs
To increase the longevity of AOs, an antisense U7 snRNA expression system was originally developed to produce antisense RNA in cells [31]. This method has been investigated in DMD, SMN2 and β-globin [32–34]. Because U7 snRNAs are nuclear, the design ensures the accumulation of antisense RNA in the nucleus; localization to the nucleus is especially useful for splicing modulation. Because the antisense U7 snRNAs are generated continuously by the expression vector within the cell, efficient modulation of splicing is persistent over many cell divisions, as long as the expression vectors are retained.
U7-snRNAs have been successfully used to block mutation-activated cryptic splicing sites in PTCH1, BRCA1 and CYP11A, and restore full-length protein [35]. Bifunctional U7 snRNAs also demonstrated more permanent correction effects than AOs in SMA [36]. When used for gene silencing, U7 antisense snRNA also improved the efficiency of splicing modulation. For example, U7-snRNA-mediated splicing disruption of Tat and Rev efficiently inhibited HIV multiplication [27]. However, this method requires vector expression systems, similar to conventional gene therapy approaches, and the systematic delivery and safety risk of the constructs remain major concerns.
Applications in disease
As alluded to above, splice-targeted antisense approaches have been tested in many genetic diseases, such as DMD, CF, SMA, β-thalassemia, Hutchinson-Gilford progeria syndrome, ocular albinism and ataxia-telangiectasia (A-T). These approaches have also been used in cancers associated with the misregulation of pre-mRNA alternative splicing of Bcl-x [37] and FGFR1 [38]. The applications of this approach in DMD are the most advanced; the first clinical trial of AO therapy for DMD has been completed (see below). In addition, splice-targeted gene knockdown approaches have been developed for treating cancers and infectious diseases.
Genetic disorders for splice-targeted therapy
Ataxia-telangiectasia
A-T is a progressive autosomal recessive disorder resulting from mutations in the ATM (A-T mutated) gene. Approximately half of the unique ATM mutations are splicing mutations [39]. In one study, three types of splicing mutations for correction were selected: a 5′ exonic cryptic splice site variant, a 3′ exonic cryptic splice site variant, and a pseudo-exon inclusion variant. Antisense PMOs corrected ATM splicing, producing up to 20% functional full-length ATM protein that restored the radiosensitive cellular phenotype of A-T cells [40]. If modest increases in functional ATM protein levels could improve the disease phenotype, A-T may provide an exciting model for exploring splice-targeted therapeutic approaches.
Methylmalonic acidemia and propionic academia
In a study similar to the A-T study, Ugarte et al used PMOs to restore normal splicing caused by intronic mutations in the genes for methylmalonic acidemia and propionic acidemia [41]. Three point mutations were located in deep intronic regions of three genes, MUT (methylmalonyl coenzyme A mutase), PCCA (propionyl coenzyme A carboxylase, α polypeptide) and PCCB (propionyl coenzyme A carboxylase, β polypeptide); these mutations caused the introduction of pseudoexons into the transcripts. PMOs were designed to mask the 5′ or 3′ cryptic splice sites of the potential pseudoexonic regions. Restoration of correctly spliced mRNA led to effective protein synthesis, and the activities of the enzymes coded by these genes were consequently restored in the fibroblasts of patients.
Dystrophia myotonica
The neuromuscular disease myotonic dystrophy (DM) is caused by microsatellite repeat expansions in the DMPK (dystrophia myotonica-protein kinase) and ZNF9 (CCHC-type zinc finger, nucleic acid binding protein) genes [42]. These nucleotide repeat expansions are associated with aberrant splicing in DM cells. In DM, an increase in the excitability of skeletal muscle leads to repetitive action potentials, stiffness and delayed relaxation. This constellation of features is associated with abnormal alternative splicing of the muscle-specific chloride channel (ClC-1) and reduced conductance of chloride ions in the sarcolemma [43]. Wheeler et al reported that targeting the 3′ splice site of ClC-1 exon 7a with PMOs could reverse the defect of ClC-1 alternative splicing in two mouse models of DM. This further led to expression of the full-length protein in the surface membrane, normalized muscle ClC-1 current density and deactivation kinetics, and elimination of myotonic discharges [44], indicating the potential of AOs in correcting alternative splicing defects in DM.
Duchenne muscular dystrophy
Antisense-mediated exon skipping for the treatment of DMD can remove nonsense mutations or frame-shifting mutations from mRNA [9], and thus far presents the most promising therapeutic strategy for DMD. Recently, this strategy has been further validated.
Wilton et al concluded that every exon targeted could be removed from the dystrophin mRNA, not withstanding some variable efficiency for different exons. Interestingly, no single motif was identified as a universal target site of AOs [45]. The length of AOs can affect the efficiency of splicing modulation [46]. Furthermore, AO cocktails have proven to be more efficient than a single AO [23,24]. Gurvich et al identified several novel mutations that cause pseudoexon inclusion in patients with DMD, and concluded that aberrant splicing caused by pseudoexons could be corrected using AOs [47]. In an experimental model, McClorey et al extended the AO-meditated exon-skipping studies from animal models or cultured human muscle cells to human muscle explants, which more closely resemble in vivo conditions; similar results were achieved [48].
Perhaps the most interesting data are those from the first clinical trial of AOs for DMD [49]. A 2-′O-Me AO, PRO-051 (Prosensa Therapeutics BV), was evaulated in patients (n = 4) with DMD. PRO-051 (0.8 mg) efficiently restored dystrophin expression by the introduction of exon 51 skipping in treated muscle fibers of all four patients after a single injection into the tibialis anterior muscle of the leg. In a biopsy taken 4 weeks after the injection, novel dystrophin expression was observed in the majority of muscle fibers (64 to 75%), with protein levels of dystrophin that are expected to be clinically relevant (3 to 12% of the total protein extract of that found in the control specimen and from 17 to 35% of that of the control specimen in the quantitative ratio of dystrophin to laminin α2.). The robust expression of dystrophin protein after a single injection indicates the positive effects of the AO. This is the first clinical trial of an RNA-based therapeutic agent for DMD and, therefore, is an important step towards treatment of this disease. This therapy may represent the beginning of personalized mutation-based molecular medicine [50].
Prior to this clinical trial, only a single case report existed of a patient with DMD treated with an AO to restore the reading frame by skipping exon 19 [51]. The 10-year-old patient with an out-of-frame, exon 20 deletion in the dystrophin gene was treated with a 31-mer phosphorothioate oligonucleotide (0.5 mg/kg iv) against the splicing enhancer sequence of exon 19 for 4 weeks, at 1-week intervals. One week after the final infusion, exon 19-skipped, in-frame mRNA was identified in the muscle. The dystrophin protein was also detected in the sarcolemma of muscle cells after treatment. The AO-induced exon skipping levels in this study were low (≤6% correction in mRNA level) probably due to the low dose and the suboptimal chemistry of the AO.
Targeting diseases using antisense-mediated gene silencing
AO-mediated splice modulation provides a novel method for silencing or knocking down gene expression, and thus extends the clinical potential of AO therapy. As discussed previously, these approaches were developed to knockdown HIV regulatory proteins Tat and Rev [27], as well as to decrease LDL levels by inhibiting apoB expression [22].
Delivery of AOs
One of the major obstacles to the successful application of all RNA-based therapies is that of antisense delivery to target tissues; this obstacle is further complicated by the potential instability of antisense RNA in blood and degradation in cells. The lipidic nature of biological membranes is the major impediment to the intracellular delivery of macromolecules such as AOs. Furthermore, entrapment within endocytic vesicles and degradation in the lysosome also need to be avoided to maintain AO activity. Various carrier systems, such as cell-penetrating peptides (CPPs), liposomes, cationic lipids and polymers, and polymeric nanoparticles, have been developed and evaluated for efficient intracellular AO delivery. Some recent advances are discussed below.
Cell-penetrating peptides
CPPs are a class of small cationic peptides of approximately 10 to 30 amino acids that can be used as transmembrane drug delivery agents through various forms of endocytosis for low-molecular weight compounds, including drugs, imaging agents, oligonucleotides, peptides and proteins [52]. CPPs are also known as ‘protein transduction domains’. Earlier-identified CPPs, such as Tat or penetratin, had been used to deliver neutral AOs, such as PNA and PMO; however, the entrapment of AOs within endocytic vesicles limited delivery efficiency [53]. Substantial progress in developing improved CPPs has been made recently.
Arginine-rich cell-penetrating peptides
Several novel arginine-rich CPPs have been developed for improved delivery of PNAs and PMOs. Morris et al described a peptide, Pep-3, with the potential for in vivo delivery of charged PNA and DNA mimics [54]. This peptide combines a tryptophan/phenylalanine domain with a lysine/arginine-rich hydrophilic motif, and can form stable nano-size complexes with both uncharged and charged PNAs. Pep-3 promoted the efficient delivery of PNAs into several cell lines, without any associated cytotoxicity. Pep-3 also successfully delivered PNA in vivo via intratumoral and intravenous administration in mice. Another arginine-rich peptide, M-918, derived from the tumor suppressor protein p14AR, also improved PNA delivery into several cell types, such as HeLa, human breast cancer cells and CHO [55].
Moreover, Abes et al developed two arginine-rich CPPs, (RXR)4XB and R(6)Pen [56,57], that allowed efficient nuclear delivery of splice-correcting PNAs and PMOs at micromolar concentrations. The (RXR)4XB-PMO conjugate could avoid uptake by lysosomes and access the nuclear compartment and, therefore, more efficiently deliver PMO analogs. This (RXR)4 CPP can deliver PMOs directly into primary murine leukocytes [58]. The in vivo efficacy of (RXR)4-PMO conjugates has been demonstrated in a mouse model of DMD [59]. After intraperitoneal injection, this CPP enhanced cell uptake of the PMO, resulting in widespread dystrophin expression in all muscles examined, except for cardiac muscle.. In vivo disposition of (RXR)4-PMO was also investigated in rats [60]. After intravenous administration, conjugation to CPP increased the uptake of PMO in all tissues, except brain tissue. (RXR)4-PMO did not demonstrate any obvious toxicity at a dose of 15 mg/kg [60].
RVG-9R: The first CPP to cross the blood-brain-barrier
Kumar et al fused a short peptide derived from rabies virus glycoprotein (RVG) to a nine arginine siRNA packaging peptide (RVG-9R); this conjugate enabled the transvascular delivery of siRNA specifically to target neuronal cells expressing the nicotinic ACh receptor (nAChR) in the brain [61]. Intravenous treatment with RVG-9R-bound siRNA against flavivirus induced robust protection against fatal viral encephalitis in mice. This study was the first to demonstrate that the systemic delivery of macromolecular oligonucleotides can traverse the blood-brain barrier and specifically target the brain of adult mice. Considering that the blood-brain barrier precludes the entry of large molecules into the brain, this finding could make treating numerous neurological disorders possible.
Cationic lipids
Cationic lipids and cationic polymers have been widely used for the delivery of charged AO analogs, such as 2′-O-Me or 2′-MOE. However, most of the available commercial formulations are toxic and unstable in the presence of serum proteins, and the delivery efficiency is also limited [62]. Resina et al designed a novel dioctadecyldimethylammonium chloride/dioleoylphosphatidylethanolamine (DOGS/DOPE) liposome formulation, DLS, that mediated the efficient nuclear delivery of negatively charged 2′-O-Me and 2′-MOE in serum-supplemented cell culture [53]. DLS-packed 2′-O-Me or 2′-O-MOE induced more efficient splicing correction than newly-developed R-Ahx-R-conjugated PNAs in vitro.
Nanoparticles
Nanoparticles have been used for the delivery of nucleic acids (DNA, RNA and oligonucleotides) due to their ability to penetrate the cell wall and deliver biomolecules into living systems [63–65]. The majority of these applications were for the delivery of vectors and siRNA; however, nanotechonolgy has also been applied to the deliverly of AOs. The nano-sized dendritic α,ε(-poly(L-lysine)s (DPL) can efficiently deliver DPL-2′-OMe complexes and correct pre-mRNA splicing in cells [66]. A Pep-3-based nanoparticle system can efficiently deliver PNA mimics into living cells and animal tumor models [54]. Seferos et al have described LNA nanoparticle conjugates that form stable duplexes with complementary target sequences, and readily enter cells [67]. Moreover, PMOs were also used to prepare a MORF/streptavidin/tat nanoparticle to improve efficiency of cellular delivery [68]. Taken together, these reports suggest promise for the delivery of the three main AOs (PNA, PMO and LNA) to be delivered as nanoparticles.
Conclusion
Antisense-mediated splicing modification can manipulate gene expression in two ways, either to restore gene function by reprogramming gene splicing or to knockdown gene expression by interfering with normal splicing. These approaches present novel therapeutic strategies for a variety of diseases. Although significant progress has been achieved, the clinical application of antisense-mediated splicing modulation remains limited by the specificity and efficiency of pre-mRNA targeting, systemic delivery, and the largely untested in vivo safety of antisense agents. The novel systemic delivery systems need to be evaluated for in vivo delivery efficiency, especially after long-term administration of AOs. The potential off-target effects caused by unspecific AO targets must also be considered and investigated.
Considering that the majority of human genes undergo alternative splicing and an increasing number of diseases are found to be caused by alternative splicing [69–71], many more potential applications of AOs can be expected. Also, knocking down gene expression by antisense approaches may have potential for treating certain cancers and infectious diseases. Therefore, the fields of targeting strategy, AO chemistry [72–74], and systematic delivery of AOs have a promising future in splice-targeted antisense-mediated therapy.
References
•• of outstanding interest
• of special interest
- 1.Chan JH, Lim S, Wong WS. Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol. 2006;33(5–6):533–540. doi: 10.1111/j.1440-1681.2006.04403.x. [DOI] [PubMed] [Google Scholar]
- 2.Morcos PA. Achieving targeted and quantifiable alteration of mRNA splicing with morpholino oligos. Biochem Biophys Res Commun. 2007;358(2):521–527. doi: 10.1016/j.bbrc.2007.04.172. [DOI] [PubMed] [Google Scholar]
- 3.Pande V, Nilsson L. Insights into structure, dynamics and hydration of locked nucleic acid (LNA) strand-based duplexes from molecular dynamics simulations. Nucleic Acids Res. 2008;36(5):1508–1516. doi: 10.1093/nar/gkm1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amantana A, Iversen PL. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr Opin Pharmacol. 2005;5(5):550–555. doi: 10.1016/j.coph.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Lundin KE, Good L, Stromberg R, Graslund A, Smith CI. Biological activity and biotechnological aspects of peptide nucleic acid. Adv Genet. 2006;56:1–51. doi: 10.1016/S0065-2660(06)56001-8. [DOI] [PubMed] [Google Scholar]
- 6.Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest. 2003;112(4):481–486. doi: 10.1172/JCI19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11(4):440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vetrini F, Tammaro R, Bondanza S, Surace EM, Auricchio A, De LM, Ballabio A, Marigo V. Aberrant splicing in the ocular albinism type 1 gene (OA1/GPR143) is corrected in vitro by morpholino antisense oligonucleotides. Hum Mutat. 2006;27(5):420–426. doi: 10.1002/humu.20303. [DOI] [PubMed] [Google Scholar]
- 9.Artsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA. 2007;13(10):1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol. 2004;22(5):535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 11.Vickers TA, Zhang H, Graham MJ, Lemonidis KM, Zhao C, Dean NM. Modification of MyD88 mRNA splicing and inhibition of IL-1β signaling in cell culture and in mice with a 2′-O-methoxyethyl-modified oligonucleotide. J Immunol. 2006;176(6):3652–3661. doi: 10.4049/jimmunol.176.6.3652. [DOI] [PubMed] [Google Scholar]
- 12.Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 13.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48(6):885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Lim SR, Hertel KJ. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J Biol Chem. 2001;276(48):45476–45483. doi: 10.1074/jbc.M107632200. [DOI] [PubMed] [Google Scholar]
- 15.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci USA. 2003;100(7):4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5(4):e73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashima T, Rao N, Manley JL. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci USA. 2007;104(9):3426–3431. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82(4):834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buratti E, Stuani C, De PG, Baralle FE. SR protein-mediated inhibition of CFTR exon 9 inclusion: Molecular characterization of the intronic splicing silencer. Nucleic Acids Res. 2007;35(13):4359–4368. doi: 10.1093/nar/gkm444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Artsma-Rus A, Janson AA, Heemskerk JA, De Winter CL, van Ommen GJ, van Deutekom JC. Therapeutic modulation of DMD splicing by blocking exonic splicing enhancer sites with antisense oligonucleotides. Ann NY Acad Sci. 2006;1082:74–76. doi: 10.1196/annals.1348.058. [DOI] [PubMed] [Google Scholar]
- 21.Goto M, Sawamura D, Nishie W, Sakai K, McMillan JR, Akiyama M, Shimizu H. Targeted skipping of a single exon harboring a premature termination codon mutation: Implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J Invest Dermatol. 2006;126(12):2614–2620. doi: 10.1038/sj.jid.5700435. [DOI] [PubMed] [Google Scholar]
- 22•.Khoo B, Roca X, Chew SL, Krainer AR. Antisense oligonucleotide-induced alternative splicing of the apoB mRNA generates a novel isoform of apoB. BMC Mol Biol. 2007;8:3. doi: 10.1186/1471-2199-8-3. A successful application of antisense-mediated splicing modulation to knockdown gene expression by inducing aberrant splicing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Artsma-Rus A, Kaman WE, Weij R, den Dunnen JT, van Ommen GJ, van Deutekom JC. Exploring the frontiers of therapeutic exon skipping for Duchenne muscular dystrophy by double targeting within one or multiple exons. Mol Ther. 2006;14(3):401–407. doi: 10.1016/j.ymthe.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 24.Adams AM, Harding PL, Iversen PL, Coleman C, Fletcher S, Wilton SD. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: Cocktails and chemistries. BMC Mol Biol. 2007;8:57. doi: 10.1186/1471-2199-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Xie SY, Ren ZR, Zhang JZ, Guo XB, Wang QX, Wang S, Lin D, Gong XL, Li W, Huang SZ, Zeng F, Zeng YT. Restoration of the balanced α/β-globin gene expression in β654-thalassemia mice using combined RNAi and antisense RNA approach. Hum Mol Genet. 2007;16(21):2616–2625. doi: 10.1093/hmg/ddm218. Describes the application of a two technique combined strategy to rebalance protein expression in a animal mode of β-thalassemia: antisense-mediated splicing correction and siRNA-mediated knockdown. [DOI] [PubMed] [Google Scholar]
- 26.Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36(8):801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 27•.Asparuhova MB, Marti G, Liu S, Serhan F, Trono D, Schumperli D. Inhibition of HIV-1 multiplication by a modified U7 snRNA inducing Tat and Rev exon skipping. J Gene Med. 2007;9(5):323–334. doi: 10.1002/jgm.1027. A successful application of antisense-induced exon skipping to inhibit HIV-1 multiplication. [DOI] [PubMed] [Google Scholar]
- 28.Query CC, Moore MJ, Sharp PA. Branch nucleophile selection in pre-mRNA splicing: evidence for the bulged duplex model. Genes Dev. 1994;8(5):587–597. doi: 10.1101/gad.8.5.587. [DOI] [PubMed] [Google Scholar]
- 29.Cavaille J, Nicoloso M, Bachellerie JP. Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature. 1996;383(6602):732–735. doi: 10.1038/383732a0. [DOI] [PubMed] [Google Scholar]
- 30•.Zhao X, Yu YT. Targeted pre-mRNA modification for gene silencing and regulation. Nat Methods. 2008;5(1):95–100. doi: 10.1038/nmeth1142. Describes a novel approach to inhibit splicing process by branch-point modification. [DOI] [PubMed] [Google Scholar]
- 31.Gorman L, Suter D, Emerick V, Schumperli D, Kole R. Stable alteration of pre-mRNA splicing patterns by modified U7 small nuclear RNAs. Proc Natl Acad Sci USA. 1998;95(9):4929–34. doi: 10.1073/pnas.95.9.4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ. Correction of SMN2 pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12(6):1013–1022. doi: 10.1016/j.ymthe.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 33.Gorman L, Suter D, Emerick V, Schumperli D, Kole R. Stable alteration of pre-mRNA splicing patterns by modified U7 small nuclear RNAs. Proc Natl Acad Sci USA. 1998;95(9):4929–4934. doi: 10.1073/pnas.95.9.4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denti MA, Rosa A, D’Antona G, Sthandier O, De Angelis FG, Nicoletti C, Allocca M, Pansarasa O, Parente V, Musaro A, Auricchio A, Bottinelli R, Bozzoni I. Chimeric adeno-associated virus/antisense U1 small nuclear RNA effectively rescues dystrophin synthesis and muscle function by local treatment of mdx mice. Hum Gene Ther. 2006;17(5):565–574. doi: 10.1089/hum.2006.17.565. [DOI] [PubMed] [Google Scholar]
- 35.Uchikawa H, Fujii K, Kohno Y, Katsumata N, Nagao K, Yamada M, Miyashita T. U7 snRNA-mediated correction of aberrant splicing caused by activation of cryptic splice sites. J Hum Genet. 2007;52(11):891–897. doi: 10.1007/s10038-007-0192-8. [DOI] [PubMed] [Google Scholar]
- 36.Marquis J, Meyer K, Angehrn L, Kampfer SS, Rothen-Rutishauser B, Schumperli D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol Ther. 2007;15(8):1479–1486. doi: 10.1038/sj.mt.6300200. [DOI] [PubMed] [Google Scholar]
- 37.Taylor JK, Zhang QQ, Wyatt JR, Dean NM. Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat Biotechnol. 1999;17(11):1097–1100. doi: 10.1038/15079. [DOI] [PubMed] [Google Scholar]
- 38.Bruno IG, Jin W, Cote GJ. Correction of aberrant FGFR1 alternative RNA splicing through targeting of intronic regulatory elements. Hum Mol Genet. 2004;13(20):2409–2420. doi: 10.1093/hmg/ddh272. [DOI] [PubMed] [Google Scholar]
- 39.Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengüt S, Tolun A, Chessa L, Sanal O, Bernatowska E, Gatti RA, Concannon P. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64(6):1617–1631. doi: 10.1086/302418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Du L, Pollard JM, Gatti RA. Correction of prototypic ATM splicing mutations and aberrant ATM function with antisense morpholino oligonucleotides. Proc Natl Acad Sci USA. 2007;104(14):6007–6012. doi: 10.1073/pnas.0608616104. Describes a genetic disease model (A-T) for exploring splice-targeted approaches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ugarte M, Aguado C, Desviat LR, Sanchez-Alcudia R, Rincon A, Perez B. Propionic and methylmalonic acidemia: Antisense therapeutics for intronic variations causing aberrantly spliced messenger RNA. Am J Hum Genet. 2007;81(6):1262–1270. doi: 10.1086/522376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooper TA. A reversal of misfortune for myotonic dystrophy? N Engl J Med. 2006;355(17):1825–1827. doi: 10.1056/NEJMcibr064708. [DOI] [PubMed] [Google Scholar]
- 43.Lueck JD, Mankodi A, Swanson MS, Thornton CA, Dirksen RT. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J Gen Physiol. 2007;129(1):79–94. doi: 10.1085/jgp.200609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117(12):3952–3957. doi: 10.1172/JCI33355. Describes that PMOs could reverse the defect of ClC-1 alternative splicing in mouse models of DM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilton SD, Fall AM, Harding PL, McClorey G, Coleman C, Fletcher S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol Ther. 2007;15(7):1288–1296. doi: 10.1038/sj.mt.6300095. [DOI] [PubMed] [Google Scholar]
- 46.Harding PL, Fall AM, Honeyman K, Fletcher S, Wilton SD. The influence of antisense oligonucleotide length on dystrophin exon skipping. Mol Ther. 2007;15(1):157–166. doi: 10.1038/sj.mt.6300006. [DOI] [PubMed] [Google Scholar]
- 47.Gurvich OL, Tuohy TM, Howard MT, Finkel RS, Medne L, Anderson CB, Weiss RB, Wilton SD, Flanigan KM. DMD pseudoexon mutations: Splicing efficiency, phenotype, and potential therapy. Ann Neurol. 2008;63(1):81–89. doi: 10.1002/ana.21290. [DOI] [PubMed] [Google Scholar]
- 48.McClorey G, Fall AM, Moulton HM, Iversen PL, Rasko JE, Ryan M, Fletcher S, Wilton SD. Induced dystrophin exon skipping in human muscle explants. Neuromuscul Disord. 2006;16(9–10):583–590. doi: 10.1016/j.nmd.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 49••.van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, den Dunnen JT, Koop K, van der Kooi AJ, Goemans NM, de Kimpe SJ, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357(26):2677–2686. doi: 10.1056/NEJMoa073108. Describes the first clinical trial of a therapeutic AO for DMD. [DOI] [PubMed] [Google Scholar]
- 50.Hoffman EP. Skipping toward personalized molecular medicine. N Engl J Med. 2007;357(26):2719–2722. doi: 10.1056/NEJMe0707795. [DOI] [PubMed] [Google Scholar]
- 51•.Takeshima Y, Yagi M, Wada H, Ishibashi K, Nishiyama A, Kakumoto M, Sakaeda T, Saura R, Okumura K, Matsuo M. Intravenous infusion of an antisense oligonucleotide results in exon skipping in muscle dystrophin mRNA of Duchenne muscular dystrophy. Pediatr Res. 2006;59(5):690–694. doi: 10.1203/01.pdr.0000215047.51278.7c. A case report on a DMD patient treated with an AO to restore the reading frame by skipping exon 19. [DOI] [PubMed] [Google Scholar]
- 52.Foged C, Nielsen HM. Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin Drug Deliv. 2008;5(1):105–117. doi: 10.1517/17425247.5.1.105. [DOI] [PubMed] [Google Scholar]
- 53.Resina S, Abes S, Turner JJ, Prevot P, Travo A, Clair P, Gait MJ, Thierry AR, Lebleu B. Lipoplex and peptide-based strategies for the delivery of steric-block oligonucleotides. Int J Pharm. 2007;344(1–2):96–102. doi: 10.1016/j.ijpharm.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 54•.Morris MC, Gros E, Aldrian-Herrada G, Choob M, Archdeacon J, Heitz F, Divita G. A non-covalent peptide-based carrier for in vivo delivery of DNA mimics. Nucleic Acids Res. 2007;35(7):e49. doi: 10.1093/nar/gkm053. Describes a new peptide, Pep-3, with potential for in vivo delivery of charged PNA and DNA mimics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El-Andaloussi S, Johansson HJ, Holm T, Langel U. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol Ther. 2007;15(10):1820–1826. doi: 10.1038/sj.mt.6300255. [DOI] [PubMed] [Google Scholar]
- 56•.Abes S, Moulton HM, Clair P, Prevot P, Youngblood DS, Wu RP, Iversen PL, Lebleu B. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release. 2006;116(3):304–313. doi: 10.1016/j.jconrel.2006.09.011. Describes a novel (RXR)4XB peptide with potential for efficient delivery of PMOs in vivo. [DOI] [PubMed] [Google Scholar]
- 57.Abes S, Turner JJ, Ivanova GD, Owen D, Williams D, Arzumanov A, Clair P, Gait MJ, Lebleu B. Efficient splicing correction by PNA conjugation to an R6-penetratin delivery peptide. Nucleic Acids Res. 2007;35(13):4495–4502. doi: 10.1093/nar/gkm418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marshall NB, Oda SK, London CA, Moulton HM, Iversen PL, Kerkvliet NI, Mourich DV. Arginine-rich cell-penetrating peptides facilitate delivery of antisense oligomers into murine leukocytes and alter pre-mRNA splicing. J Immunol Methods. 2007;325(1–2):114–126. doi: 10.1016/j.jim.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 59.Fletcher S, Honeyman K, Fall AM, Harding PL, Johnsen RD, Steinhaus JP, Moulton HM, Iversen PL, Wilton SD. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol Ther. 2007;15(9):1587–1592. doi: 10.1038/sj.mt.6300245. [DOI] [PubMed] [Google Scholar]
- 60.Amantana A, Moulton HM, Cate ML, Reddy MT, Whitehead T, Hassinger JN, Youngblood DS, Iversen PL. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem. 2007;18(4):1325–1331. doi: 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- 61••.Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448(7149):39–43. doi: 10.1038/nature05901. Describes a novel rabies-derived peptide, RVG-9R, with the ability to cross the blood-brain-barrier and deliver siRNAs specifically to the brain. [DOI] [PubMed] [Google Scholar]
- 62.Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114(1):100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 63.Sokolova V, Epple M. Inorganic nanoparticles as carriers of nucleic acids into cells. Angew Chem Int Ed Engl. 2008;47(8):1382–1395. doi: 10.1002/anie.200703039. [DOI] [PubMed] [Google Scholar]
- 64.Fenske DB, Cullis PR. Liposomal nanomedicines. Expert Opin Drug Deliv. 2008;5(1):25–44. doi: 10.1517/17425247.5.1.25. [DOI] [PubMed] [Google Scholar]
- 65.Pirollo KF, Rait A, Zhou Q, Hwang SH, Dagata JA, Zon G, Hogrefe RI, Palchik G, Chang EH. Materializing the potential of small interfering RNA via a tumor-targeting nanodelivery system. Cancer Res. 2007;67(7):2938–2943. doi: 10.1158/0008-5472.CAN-06-4535. [DOI] [PubMed] [Google Scholar]
- 66.Eom KD, Park SM, Tran HD, Kim MS, Yu RN, Yoo H. Dendritic α,ε-poly(L-lysine)s as delivery agents for antisense oligonucleotides. Pharm Res. 2007;24(8):1581–1589. doi: 10.1007/s11095-006-9231-y. [DOI] [PubMed] [Google Scholar]
- 67.Seferos DS, Giljohann DA, Rosi NL, Mirkin CA. Locked nucleic acid-nanoparticle conjugates. Chembiochem. 2007;8(11):1230–1232. doi: 10.1002/cbic.200700262. [DOI] [PubMed] [Google Scholar]
- 68.Wang Y, Nakamura K, Liu X, Kitamura N, Kubo A, Hnatowich DJ. Simplified preparation via streptavidin of antisense oligomers/carriers nanoparticles showing improved cellular delivery in culture. Bioconjug Chem. 2007;18(4):1338–1343. doi: 10.1021/bc070032c. [DOI] [PubMed] [Google Scholar]
- 69.Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30(1):13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 70.Caceres JF, Kornblihtt AR. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18(4):186–193. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 71.Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci. 2006;119(Pt 13):2635–2641. doi: 10.1242/jcs.03053. [DOI] [PubMed] [Google Scholar]
- 72.Wojciechowski F, Hudson RH. Nucleobase modifications in peptide nucleic acids. Curr Top Med Chem. 2007;7(7):667–679. doi: 10.2174/156802607780487795. [DOI] [PubMed] [Google Scholar]
- 73.Pensato S, Saviano M, Romanelli A. New peptide nucleic acid analogues: Synthesis and applications. Expert Opin Biol Ther. 2007;7(8):1219–1232. doi: 10.1517/14712598.7.8.1219. [DOI] [PubMed] [Google Scholar]
- 74.Shiraishi T, Hamzavi R, Nielsen PE. Subnanomolar antisense activity of phosphonate-peptide nucleic acid (PNA) conjugates delivered by cationic lipids to HeLa cells. Nucleic Acids Res. 2008;36(13):4424–4432. doi: 10.1093/nar/gkn401. [DOI] [PMC free article] [PubMed] [Google Scholar]