

Abstract
A platform was developed to analyze MS/MS spectra from large peptides with low part-per-million mass accuracy, including a commercial-grade software suite. Termed Middle Down Proteomics, this platform identified 7454 peptides from 2–20 kDa (1472 unique) from 555 proteins after 23 LC-MS/MS injections of Lys-C digests of HeLa-S3 nuclear proteins. Along with greatly increased confidence for both peptide identification (expectation values from 10−89 to 10−4) and characterization (up to 18% of peptides were modified in some LC-MS/MS runs), fragmentation data with <2 ppm accuracy enabled error tolerant and routine multiplexed database searching–all clearly demonstrated in this study.
Keywords: Post-Translational Modifications, High Resolution, Mass Accuracy, Mass Spectrometry, Proteomics, Human Nuclei
Introduction
Recent years have seen a large expansion of proteomic work using mass spectrometry (MS) to identify proteins from complex mixtures using two general approaches: “Bottom Up” and “Top Down”.1,2 Bottom Up is far more widespread and refers to using exhaustive, usually tryptic, digestion of proteins either prefractionated by one- or two-dimensional polyacrylamide gel electrophoresis3,4 or proteolyzed in a whole cell extract en masse (“shotgun digestion”).3,5,6 Typically, the resulting peptides are then analyzed via liquid chromatography-tandem mass spectrometry (LC-MS/MS) using ion traps (resolving power ~1000) or time-of-flight instruments (resolving power ~8000), with tandem mass spectral data used for database retrieval of observed peptides. Top Down Proteomics removes the proteolytic digestion step described above, focusing instead on high sequence coverage and complete protein characterization utilizing Fourier-Transform (FT) instruments (resolving power ≫50000) and a variety of techniques for ion dissociation during tandem MS.
Recently, the evolution of higher-performing hybrid instruments (Q-TOF, LTQ-FT, and LTQ-OrbiTrap) has spawned a new generation of data acquisition and data analysis focusing on the use of high-resolution analysis of intact peptides.7–9 The use of precursor FT scans with <10 ppm mass accuracy followed by unit-resolution MS/MS scans (i.e., now the dominant Bottom Up experiment on ion trap-FT hybrids) has been shown to provide more identifications at higher confidence levels than traditional low-resolution experiments.7,9,10 Proteome projects run in this fashion now work well for high-throughput identification of hundreds of proteins from a given sample (<2000 proteins for a whole study)8,10–12 with no information on modifications typically reported.8
Driven in part by improving MS hardware and newer electron-based MS/MS methods,13,14 a fledgling trend in MS-based proteomics involves analysis of larger peptides to improve protein sequence coverage and increase the chance of detecting multiple modifications on the same peptide,15,16 similar in spirit to the information gained by Top Down Proteomics.17–20 While not an altogether new concept,21,22 “Middle Molecule” mass spectrometry can involve simply switching proteolytic agents from trypsin to an alternative protease or chemical digestion procedure to access larger protein pieces. For modern instrumentation, simply acquiring high-resolution MS/MS data would require no change in hardware while providing far more accurate fragmentation data to identify peptides >4 kDa.9,21 Further, confident assignment of modified peptides that are currently missed in most MS/MS data sets would also become routine.23 However, the most popular database searching algorithms do not make good use of ultrahigh accuracy MS/MS data.24 Higher resolution MS/MS data do not produce significantly higher scores, impeding widespread adoption of high-resolution MS/MS experiments even though they are technically feasible.24 Also feasible is the multiplexed fragmentation of many precursors in parallel, up to two for peptides25,26 and between two27 and six28 for intact proteins identified in such a mode with software clearly lagging behind this type of data production.
Here, we have taken an intermediate approach between Bottom Up and Top Down Proteomics. By using the endoprotease Lys-C to generate slightly fewer and larger peptides,9,15,29 acquiring both high-resolution MS and MS/MS scans on a LTQ-FT, and extending ProSight software30,31 including a shotgun annotated peptide database (i.e., a peptide database incorporating all known protein-modifying events9,19,32) to work with peptide fragmentation data with <5 ppm mass accuracy, we have developed the platform envisioned in a recent perspective24 that takes full advantage of the increased capabilities of the current generation of hybrid FT mass spectrometers. The net effect of using high-resolution instead of lower resolution MS/MS data is a slight reduction in the number of proteins identified, a drastic increase in identification confidence, a sharp rise in the number of modifications automatically detected, and a clarified experimental outcome projected for laboratories without expertise in computational proteomics.
Experimental Procedures
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. Solvents were Optima grade from Fisher Chemical (Fair Lawn, NJ). Formic Acid was purchased from Acros Organics.
Nuclei Isolation
Washed HeLa-S3 cell pellets (2 × 108 cells) were suspended in NIB-250: 15 mM Tris-HCl, pH 7.5, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, 1 mM dithiothreitol (DTT), 10 mM sodium butyrate plus 0.3% NP-40 at a 10:1 (v/v) ratio. Cells were lysed by gentle mixing and incubation on ice for 5 min. Nuclei were pelleted at 600g for 5 min at 4 °C and then washed twice with NIB-250 without detergent.
Lysis and In-Solution Digestion
Pelleted nuclei were re-suspended in digestion buffer (50 mM ammonium bicarbonate, 1 mM DTT, 10 mM sodium butyrate, 1 μL DNase, and 10% acetonitrile, pH 8.5) by vortexing and sonicated on ice 6 times for 30 s to lyse. After the protein concentration was determined by Bradford’s method,33 20 μg of freshly prepared Lys-C (100 ng/μL in 50 mM ammonium bicarbonate, pH 8.5, Wako USA, Richmond, VA) was added to the lysate and incubated for 18 h at 37 °C at a substrate to enzyme ratio of roughly 250:1. Lys-C was chosen based on in silico digests of the human proteome and the desire for robust and reproducible digests (Supporting Information and Supplementary Figure 1).
LC-MS/MS of In-Solution Digests
Digested nuclear lysates were aliquoted to give 50–200 nM of protein and were injected through a Gilson 235P autosampler onto a 1 mm × 100 mm polymeric reverse phase column (PLRP-S, Higgins-Analytical, Mountain View, CA) connected to an Agilent 1200 HPLC pump flowing at 100 μL/min following a linear gradient (5% B for 10 min, increasing to 60% B in 90 min, 95% B in 100 min, Buffer A = 10% acetonitrile/water with 0.2% formic acid; Buffer B = 90% acetonitrile/isopropanol with 0.2% formic acid). Eluent flowed through a Advion Triversa NanoMate (Ithaca, NY) with a 300:1 split resulting in ~300 nL/min flow rate at the nanospray source. Data-dependent LC-MS/MS data (5 most intense precursors, 5 m/z isolation window) utilizing dynamic exclusion (2 times, 120 s exclusion, 300 max exclusion list size) were acquired on a 12 T LTQ-FT Ultra (Thermo Fisher Scientific, San Jose, CA) with both the precursor and fragmentation scans analyzed in the ICR cell at a resolution setting of 171 000 at m/z 400. The use of a 5 m/z isolation window was chosen for two reasons. Use of a wider isolation windows improves sensitivity in hybrid LTQ-FTs34 and the rigorous isolation of single species is not required when using accurate mass MS/MS information, with the ProSight multiplexing search option able to routinely handle “chimeric” MS2 spectra (see below).35
Data Analysis
Online MS/MS data sets were run through cRAWler 2.0, a software tool based on the Xtract algorithm (Thermo Fisher Scientific, San Jose, CA). cRAWler is a software application for processing LC-MS/MS data in Thermo Scientific raw files. All algorithms for data processing described here are embedded in a software environment called ProSightPC v2.0, to be distributed by Thermo Scientific starting Fall 2008. The cRAWler application first determines all precursors according to user-specified tolerances on isolation m/z and retention time. The broadband and fragmentation scans corresponding to these precursors are then separately averaged and analyzed by the Xtract algorithm, which provides a list of monoisotopic masses. This information is compiled along with metadata into a series of MS/MS experiments to create a ProSight upload file (.puf) amenable to ProSightHT, a high-throughput version of the Web-based ProSight PTM software30,31 which is now embedded into ProSightPC v2.0. Like MASCOT, ProSight PTM is available over the Internet for performing single searches via a user interface. ProSight PTM was used for scrutinizing single peptides either at the threshold for identification or to localize modifications to a smaller sequence region. During the precursor scan analysis, cRAWler can treat multiple masses within the isolation range as multiple precursors, based on an intensity cutoff (set at 10% here) relative to the base peak of the analysis window. This allows for the case where multiple precursors are fragmented together, although it does also increase database search times roughly linearly with the number of precursors in absolute mass search mode.
Database Searching
Data were searched iteratively using ProSightHT using the embedded binary search tree. First, an absolute mass mode search with ±5Da intact mass window and ±10 ppm fragment tolerance was performed against a shotgun annotated middle down database (see below). Actual MS and MS/MS errors were 2.1 and 4.0 ppm, respectively, at 2σ (data not shown). Those searches returning an expectation value (E-value) below 0.001 were populated to a “good” hit database. Only those searches, which failed, were automatically researched with a ±200 Da intact mass window to account for those unexpected/unknown PTMs not housed within the database. A compiled list of identified peptides was exported into Microsoft Excel (Supplementary Tables 1 and 2 in Supporting Information) by ProsightHT and those with mass shifts were analyzed by hand in ProSightPC to further localize and explain mass discrepancies (Δms). For PTM reporting, residue numbers are based on UniProt annotation, which may or may not account for N-terminal processing housed in the database.
Database Construction
The Middle Down database was created with a Top Down data query mindset; all known modifications (~68 000 “mod res” entries in the UniProt database) were shotgun annotated32 onto theoretical peptides from the human proteome. To create this database, the human protein database from UniProt (2007–11–02) was in silico digested with a custom script that allowed for any protein annotation to be carried onto the appropriate amino acid in the resulting peptide with a maximum of 14 features (e.g., PTMs, N-terminal processing, etc.) per peptide sequence. Peptides containing up to four missed cleavages and masses between 1–50 kDa were included. In addition, N-terminal methionine on/off and N-terminal acetylation were shotgun annotated into the database to cover all combinations of N-terminal processing. After merging of duplicate entries, the database was batch uploaded into ProSight PC resulting in 3 378 894 basic sequences and 6 051 898 peptide forms populating the database (2.5 GB).
In-Gel Digestion with Capillary LC-MS/MS
Isolated nuclei from ~5 × 107 HeLa-S3 cells were resuspended in 1 mL of 50 mM ammonium bicarbonate (pH 8.5) with 0.1% SDS and 1 μL of DNase and sonicated 6 times for 30 s. The lysate was then clarified at 14 000 rpm for 20 min before mixing with SDS-PAGE gel loading buffer and boiling for 5 min at 95 °C. Ten micrograms of protein lystate was separated using a 4–20% SDS-PAGE gel (Bio-Rad, Hercules, CA) at a constant 125 V and then stained with Coomassie blue for 30 min. After destaining, the gel was cut into 16 sections and an in-gel digestion was performed (Supplementary Figure 2 in Supporting Information). Briefly, sections were diced into 1 mm3 pieces, washed with 100 mM ammonium bicarbonate buffer (pH 8.5), and reduced and alkylated using 10 mM DTT and 50 mM iodoacetamide. Gel pieces were dehydrated and then rehydrated in a minimal volume of 100 mM ammonium bicarbonate buffer containing 50 ng/μL Lys-C and incubated overnight at 37 °C. Peptides were extracted into a solution of 50% acetonitrile containing 5% formic acid, followed by further extraction with 100% acetonitrile. Extracted peptides were concentrated by vacuum centrifugation and resuspended in 20 μL of 0.1% acetic acid. The peptides were then bomb loaded onto homemade 75 μm × 10 cm nanocapillary columns packed with C18 media (YMC Co., Ltd. Kyoto, Japan). An Agilent 1200 was spilt ~300:1 to give ~300 nL/min flow rates with high-resolution LC-MS/MS data acquired and piped through the workflow described above.
Results and Discussion
Shotgun Lys-C Digest of Human Nuclei
In a single high-resolution LC-MS/MS run with multiplexed27,36 data searching enabled, 578 peptides (222 unique) were identified. Without manual interpretation, 21 N-terminal acetylations, 2 phosphorylations, 1 methylation, and 1 acetylation were automatically detected. Peptide masses ranged from 1.2–15.5 kDa (Supplementary Figure 3A in Supporting Information) with 20% of these identified above 3 kDa. The median expectation value was 10−21 (Supplementary Figure 3B in Supporting Information). Without multiplexing enabled during data reduction and database searching, the number of peptides identified decreased to 303 (197 unique) and the median expectation value decreased to 10−23.
Figure 1 highlights a search result using multiplexed tandem MS. In panel A, the 5 m/z region around m/z 1053.3 is shown with two overlapping isotopic distributions from an FT precursor scan. In automated fashion, cRAWler assigned the appropriate charge state to each distribution and determined a monoisotopic mass for each one. These masses were passed into ProSightHT and searched independently using the entire fragment ion list derived from the Figure 1B MS/MS spectrum. Figure 1C shows Lys-C peptides from histone H4 (P62805) and lamin A/C (Q5TCI9) were identified with the mass accurate fragment ions (matching 10 b-ions, 9 y-ions and 4 b-ions, 5 y-ions, respectively) allowing for conclusive identification of two peptides from one tandem MS experiment. For the direct LC-MS/MS sampling of digested nuclear extracts, multiplexing almost doubled the absolute number of peptides identified and led to a 10% increase in the number of unique peptides identified.
Figure 1.
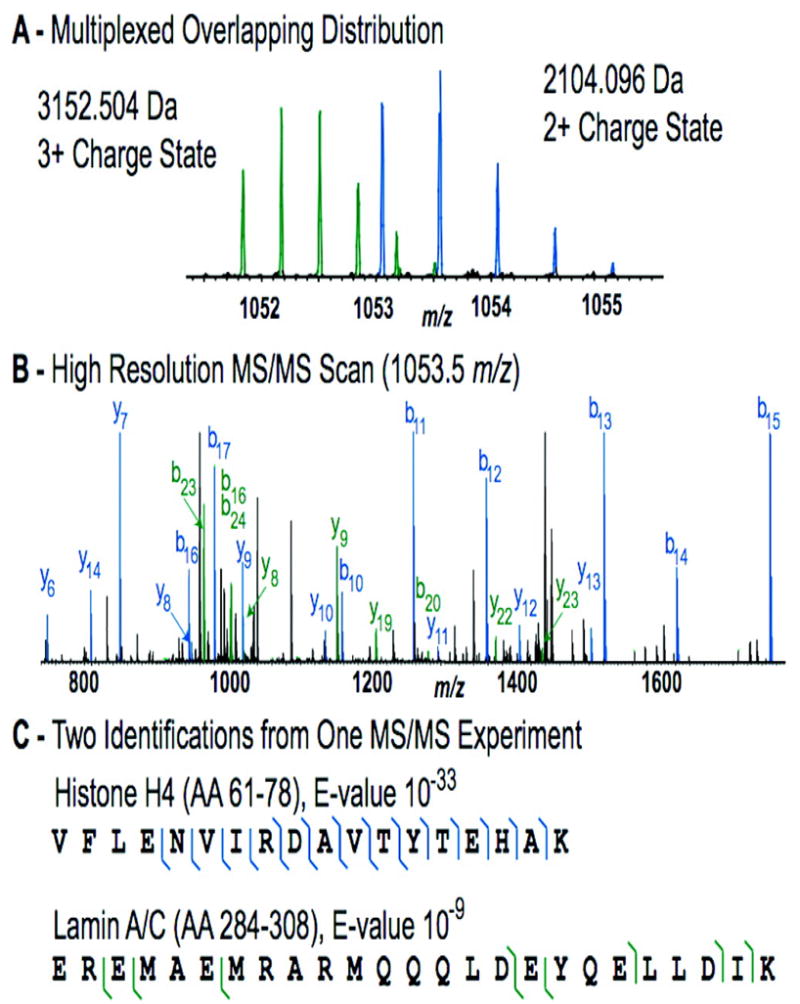
Multiplexed peptide identification using high-resolution MS/MS and tailored software. In a single MS/MS experiment, multiple precursor peptide ions were identified by high-resolution fragment ions. Panel A shows overlapping isotopic distributions from a histone H4 (P62805) peptide (AA 61–78) with 10 b-ion and 9 y-ions identifying it and a lamin A/C (Q5TCI9) peptide (AA 284–308) with 4 b-ions and 5 y-ions matching (B) and (C).
Three peptide identifications exemplify the utility of a platform based on high-resolution MS/MS and highly annotated databases to automatically detect and localize diverse biological features, without extensive variable modification searches. The N-terminal peptide from histone H1.4 (P10412) was automatically identified (10−28 E-value) and precisely characterized as N-terminally acetylated (no start methionine), with a phosphothreonine also localized to residue 16 by 5 b-ions and 16 y-ions. In addition to demonstrating this system’s ability to automatically characterize diverse modifications simultaneously without manual interpretation (Figure 2A), the platform was able to identify H1.4 specifically, from the rest of the histone H1 family. This example illustrates the power and promise of Middle Down Proteomics using accurate mass MS/MS data and housing known PTM information for primary searching (i.e., Shotgun Annotation32). Another peptide example from γ/β-actin (P63261, P60709) was characterized with a methyl-histidine at residue 73, which was cleanly distinguished from a γ-actin variant form with a glycine to alanine switch at residue 74 by virtue of a complimentary pair of fragment ions between residues 73 and 74 (Figure 2B). Since the peptides are isomeric, the fragmentation data definitively confirm that the modified form is present, which was only predicted by similarity (bovine and murine) in the human UniProt database. With all fragment ions measured with low part-per-million mass accuracy, peptides that contain modifications that are not known and annotated in the UniProt database are also confidently identified. In a third example, the N-terminal peptide of RNA binding motif protein 25 (Q2TA72, Q9H6A1), whose existence was only known at the transcript level, was identified solely by y-ions encompassing the C-terminus with a nominal +60 Da or +28 Da mass shift depending on whether the N-terminus was considered acetylated (E-value 10−15). Upon closer inspection in the ProSightPC, the peptide was putatively characterized as N-terminally acetylated and arginine 8 dimethylated, with 8 additional b-ions matching (Figure 2C). Confirmation of this assignment was provided by accurate mass measurements. The Δm between the N-terminal acetylated form and the nominal +28 species is exactly 28.0328 Da, within 1.5 mDa of a dimethylation, and the positive mass defect eliminates formylation (27.9999 Da) or any other PTMs from consideration.23
Figure 2.

Automated detection and localization of diverse types of protein variation. Panel A shows the fragmentation map of a histone H1.4 (P10412) peptide (AA 1–20) with 5 b-ions and 11 y-ions characterizing the peptide as N-terminally acetylated and threonine 17 phosphorylated. Panel B compares the fragmentation maps of β/γ-actin (P60709, P63261) to a γ-actin variant (AA 69–84). A complementary pair of fragment ions clearly distinguishes the modified form of β/γ-actin from the γ-actin variant. Panel C shows the fragmentation map of a RBM25 (Q2TA72) peptide (AA 1–62) with 5 b-ions partially characterizing a putative dimethylation on arginine 8 with an N-terminal acetylation.
Several studies have shown the effect of replicate injections on peptide count, verifying that peptide-driven proteomics is a stochastic data acquisition process37,38 (i.e., the instrument cannot keep up with the number of peptides presented to it per unit time). To investigate this effect using high-resolution MS/MS (~10-fold lower data acquisition rate for FT/FT vs FT/ion trap acquisition), the same Lys-C digest analyzed above was injected 7 times and the count of unique peptides was plotted (Figure 3). This plot shows the same basic shape of a curve published in ref 38, with the larger number of injections required to reach a plateau consistent with the slower MS/MS spectral acquisition rate for FT versus unit resolution data. However, this slower rate of data acquisition is partially compensated by the ability to routinely analyze multiplexed MS/MS data and the confident assignment of “one-hit wonders” (i.e., proteins identified from the MS/MS spectrum of a single peptide). With high-resolution scans for MS and MS/MS, one can confidently identify >1 peptide per MS/MS spectrum. Also noteworthy is that between 30–40% of the MS/MS experiments attempted here led to a successful database search results; this compares quite favorably to the 5–15% estimates for common data acquisition approaches used in Bottom Up today.39 Overall, 2701 total peptides identifications were made, with 679 of these unique. This led to a total of 280 identified proteins from a one-dimensional analysis of the shotgun digested human nuclear lysate. In total, 80 modifications were detected (64 N-terminal acetylations, 9 phosphorylations, 2 methylation, 1 dimethylation, 1 trimethylation, and 3 internal acetylations) with an average peptide mass of 2.5 kDa and a median E-value of 10−19.
Figure 3.
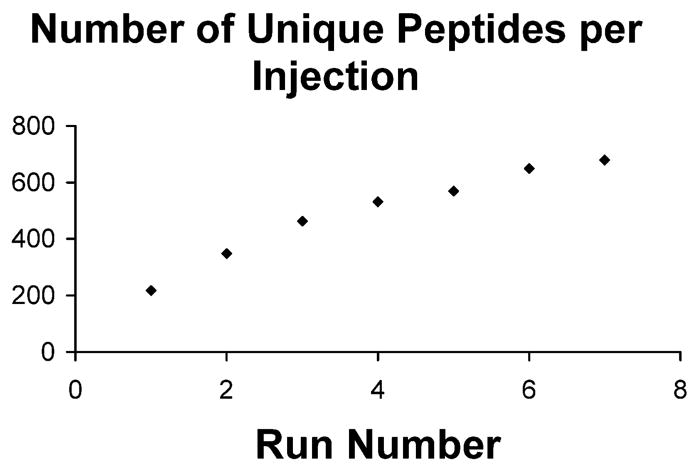
Effect of multiple injections on the number of new unique identifications. Because of the stochastic nature of data-dependent acquisition of MS/MS spectra, the number of unique identifications can be increased by replicate LC-MS/MS injections.
In Gel Digestion
In a single GeLC experiment (16 nanoLC-MS/MS injections) on HeLa nuclei, 4470 peptides were identified with E-values <0.0001 (989 were unique) from 502 distinct proteins. The peptides’ masses ranged from 1.1–7.3 kDa with a median expectation value of 10−15 (Figure 4). Without manual interpretation, 57 N-terminal acetylations, 8 phosphorylations, 1 methylation, and 1 dimethylation were detected in these data.
Figure 4.

Summary of peptide masses identified and their expectation values from a GeLC-MS/MS analysis of the human nuclear proteome. Panel A shows the number of peptides identified in each 1000 Da mass bin, emphasizing the ability of high-resolution MS/MS ability to identify peptides greater than 2 kDa (26% of the peptides detected). Panel B shows the distribution of the E-values from each of the peptides identified. E-values below 10−4 are confidently identified and do not require manual validation.
Of the 989 unique peptide mass values, 802 were identified within experimental error. The remaining 177 identifications had intact mass shifts greater than ±0.3 Da, with 30 of these arising from difficulties in determining the monoisotopic peak from resolved isotopic distributions. Manual interpretation of those unique species with nominal +16 Da mass shifts led to the characterization of 19 peptides with oxidized methionines where the unoxidized form was not identified. Moreover, five unique peptides were identified where the expected carbamidomethyl (+57 Da) at a cysteine residue was substituted for acrylamine (+71 Da), resulting in a +14 Da mass shift that was localized exclusively to cysteine in each case. Presumably, the free cysteines reacted with unpolymerized acrylamide during electrophroresis. Further, six examples of iodoacetamide reacting with serine or threonine hydroxyls were unambiguously detected (data not shown). Since common in-gel protocols call for alkylation post 1-D or 2-D electrophoresis, the results here are likely general and missed by common database searches where detection of unexpected Δms is limited.
Gene family members and peptides with high sequence identity also contribute to a number of peptides with mass discrepancies and is a result of increasing complexity in higher eukaryotes. For example, in one tandem MS experiment, a Lys-C peptide from a variant form of ribosomal protein L14 (Q53G20, AA 148–163) and CAG-ISL 7 (Q45RF0, AA-148–160) were both confidently identified (10−32 and 10−27, respectively), with large intact mass shifts (−142.074 Da) and (+71.037 Da). Inspection of the sequences show they differ by three alanines (GTAAAAAAAAAAAAAK vs GTAAAAAAAAAAK), and their intact mass shifts reflect multiple alanine mass differences (3 × 71.0371 Da). A blastp search against the presumptive sequence GTAAAAAAAAAAAK identified two sequences related to the ribosomal L14 family of proteins (NCBI Gene Accession: BAF83363 and AH00606) that were not yet annotated in UniProt. The high-resolution MS (0.3 ppm mass difference) and MS/MS data (9 b-ions and 9 y-ions) verify this assignment.
In cases where the exact peptide form is not annotated in the database, multiple identifications from highly similar peptides can complicate the database retrieval output (vide supra). However, this is a problem with precise characterization of the peptides like these, and does not make their identifications less valid. Such complexity is usually blurred or lost in typical Bottom Up experiments, even with manual interrogation or de novo approaches to probe for unexpected mass shifts. The new approach of spectral alignment alleviates some of this,40 but for laboratories without expertise in computational proteomics, accurate mass MS/MS data yields a simple and reliable list of identifications with known modifications automatically placed in the output file. For expert laboratories, accurate mass MS/MS data provide the opportunity to characterize peptides with mass discrepancies to detect unexpected artifactual or unknown biological events that change the mass of protein molecules (e.g., cSNPs, alternative splice events, or unknown PTMs).23
Comparison with Existing Literature Benchmarks
Using SEQUEST and RawExtract for unit resolution MS/MS data from an LTQ-OrbiTrap, Lu et al. reported ~1300 proteins identified with a ~5% false positive rate in a single MudPIT run for yeast.8 This goes to ~900 proteins if >2 peptides are required, or a total of 1119 yeast proteins with replicate runs and an estimated 0.4% false positive rate. With a single GeLC run here (16 nano-LC/MS injections), we identified 502 proteins with a ≪0.1% false positive rate, even using a conservative 0.0001 E-value cutoff to allow for an order-of-magnitude leeway in the Poisson-based probability model used here.27 With the use of three separate types of proteome fractionation approaches followed by GeLC-MS, a report using solely ion trap MS and Bottom Up gave 1174 proteins detected from 780 530 MS/MS spectra from human nuclei at a projected false positive rate of <1%.41 Our approach on the same sample yielded approximately half the number of protein identifications with only 1/25 the number of MS/MS scans. This means the apparent efficiency of high resolution MS/MS with multiplexing is almost an order-of-magnitude higher for identifying peptides (25% vs 2%) and/or proteins (2% vs 0.2%). In neither of the two reports noted above were modifications reported, though in Lu et al. six standard modifications were included in database searches. In the future, head-to-head comparisons of the high-resolution MS/MS approach taken here with the more widespread unit-resolution MS/MS methodology will produce a clarified picture of the pros and cons of running in each mode.
Future Development of Middle Down MS
With the current implementation, Middle Down Proteomics16 using high-resolution MS/MS spends much of its “high value” data acquisition time fragmenting different charge states of the same species and implementation of accurate mass-based rather than m/z-based exclusion and inclusion lists is ongoing. Given that only a 5 m/z precursor isolation window was used here, the potential of fragmenting several peptides at once could be explored more aggressively by increasing the size of this window. Moreover, the system design is readily extensible to digestion techniques which generate a fewer number of larger peptides. In fact, there is a strong need in “Middle Molecule” MS21 for protein digestions that can create >3 kDa peptides selectively. The chromatography, MS instrumentation, and software are now ready to process 3–20 kDa peptides in a high throughput fashion.
Conclusions
Utilizing the high-resolution, mass accurate MS/MS data historically associated with Top Down Proteomics to instead analyze intermediate-sized peptides brings a new level of clarity for the simple act of creating a list of identified peptides measured in mass spectral analysis. This Middle Down approach provides some information on unexpected or multiple modifications (like Top Down), while competing reasonably well with the proteome coverage that can be achieved by Bottom Up. By adapting software designed from the ground up for ultrahigh resolution MS/MS data, we have shown very high confidence in peptide identification with simple probability-based scoring, automatic characterization of modifications, detection of unexpected mass shifts, database searching of more than one peptide in a given search, and simultaneous localization of multiple modifications of diverse types on the same peptide. We therefore project continued application of the high-resolution tandem MS platform described here to compete favorably with the established Bottom Up MS workflows.
Supplementary Material
Acknowledgments
The authors thank the National Institutes of Health (Grant GM 067193-06 to N.L.K.) and the Packard Foundation for support. We also thank Kelleher group members, Michael Senko, and particularly those on the ProSight development team, including Rich LeDuc and Bryan Early.
Footnotes
Supporting Information Available: In silico digestions, digestion options for Middle Down, comparison of available protein digestion reagents, HeLa nuclear lysate separated by one-dimensional SDS-PAGE, summary of peptide masses identified and their expectation values from 1D LC-MS/MS analysis of the human nuclear proteome. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kelleher NL. Top Down proteomics. Anal Chem. 2004;76(11):197A–203A. [PubMed] [Google Scholar]
- 2.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. Top Down versus Bottom Up protein characterization by tandem high-resolution mass spectrometry. J Am Chem Soc. 1999;121(4):806–812. [Google Scholar]
- 3.McCormack AL, Schieltz DM, Goode B, Yang S, Barnes G, Drubin D, Yates JR. Direct analysis and identification of proteins in mixtures by LC/MS/MS and database searching at the low-femtomole level. Anal Chem. 1997;69(4):767–776. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 4.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 5.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimension protein identification technology. Nat Biotechnol. 2001;19(3):242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 6.Wolters DA, Washburn MP, Yates JR. An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73(23):5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 7.Bakalarski CE, Haas W, Dephoure NE, Gygi SP. The effects of mass accuracy, data acquisition speed, and search algorithm choice on peptide identification rates in phosphoproteomics. Anal Bioanal Chem. 2007;389:1409–1419. doi: 10.1007/s00216-007-1563-x. [DOI] [PubMed] [Google Scholar]
- 8.Lu B, Motoyama A, Ruse C, Venable J, Yates JR. Improving protein indentification sensitivity by combining MS and MS/MS information for shotgun proteomics using LTQ-Orbitrap high mass accuracy data. Anal Chem. 2008;80(6):2018–2025. doi: 10.1021/ac701697w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu SL, Kim J, Hancock WS, Karger B. Extended Range Proteomic Analysis (ERPA): A new and sensitive LC-MS platform for high sequence coverage of complex proteins with extensive post-translational modifications-comprehensive analysis of beta-casein and epidermal growth factor receptor (EGFR) J Proteome Res. 2005;4(4):1155–1170. doi: 10.1021/pr050113n. [DOI] [PubMed] [Google Scholar]
- 10.Everley PA, Bakalarski CE, Elias JE, Waghorne CG, Beausoleil SA, Gerber SA, Faherty BK, Zetter BR, Gygi SP. Enhanced analysis of metastatic prostate cancer using stable isotopes and high mass accuracy instrumentation. J Proteome Res. 2006;5(5):1224–1231. doi: 10.1021/pr0504891. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Gerber SA, Rudner AD, Beausoleil SA, Haas W, Villen J, Elias JE, Gygi SP. Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J Proteome Res. 2007;6(3):1190–1197. doi: 10.1021/pr060559j. [DOI] [PubMed] [Google Scholar]
- 12.Adachi J, Kumar C, Zhang Y, Olsen JV, Mann M. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol. 2006;7(9):R80.1–R80.16. doi: 10.1186/gb-2006-7-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. 2004;101(26):9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. a nonergodic process. J Am Chem Soc. 1998;120(13):3265–3266. [Google Scholar]
- 15.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JEP, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci USA. 2007;104(7):2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat Methods. 2007;4(6):487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 17.Meng F, Forbes AJ, Miller LM, Kelleher NL. Detection and localization of protein modifications by high resolution tandem mass spectrometry. Mass Spectrom Rev. 2005;24(2):126–134. doi: 10.1002/mas.20009. [DOI] [PubMed] [Google Scholar]
- 18.Parks BA, Jiang L, Thomas PM, Wenger CD, Roth MJ, Boyne MT, Burke PV, Kwast KE, Kelleher NL. Top Down Proteomics on a chromatographic time scale using linear ion trap Fourier transform hybrid mass spectrometers. Anal Chem. 2007;79(21):7984–7991. doi: 10.1021/ac070553t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roth MJ, Forbes AJ, Boyne MT, II, Kim Y-B, Robinson DE, Kelleher NL. Precise and parallel characterization of coding polymorphisms, alternative splicing, and modifications in human proteins by mass spectrometry. Mol Cell Proteomics. 2005;4(7):1002–1008. doi: 10.1074/mcp.M500064-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth MJ, Parks BA, Ferguson JT, Boyne MT, Kelleher NL. “Proteotyping”: population proteomics of human leukocytes using Top Down mass spectrometry. Anal Chem. 2008;80(8):2857–2866. doi: 10.1021/ac800141g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yergey JA, Cotter RJ, Heller D, Fenselau C. Resolution requirements for middle molecule mass spectrometry. Anal Chem. 1984;56(12):2262–2263. [Google Scholar]
- 22.Forbes AJ, Mazur MT, Patel HM, Walsh CT, Kelleher NL. Toward efficient analysis of >70 kDa proteins with 100% sequence coverage. Proteomics. 2001;1(8):927–933. doi: 10.1002/1615-9861(200108)1:8<927::AID-PROT927>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 23.Savitski MM, Nielsen ML, Zubarev R. ModifiComb, a new proteomic tool for mapping subtoichiometric post-translational modification, finding novel types of modifications, and finger-printing complex protein mixtures. Mol Cell Proteomics. 2006;5(5):935–948. doi: 10.1074/mcp.T500034-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Zubarev R, Mann M. On the proper use of mass accuracy in proteomics. Mol Cell Proteomics. 2007;6(3):377–381. doi: 10.1074/mcp.M600380-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Purvine S, Eppel JT, Yi EC, Goodlett DR. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3(6):847–850. doi: 10.1002/pmic.200300362. [DOI] [PubMed] [Google Scholar]
- 26.Williams JD, Flanagan M, Lopez L, Fischer S, Miller LAD. Using accurate mass electrosprapy ionization-time-of-flight mass spectrometry with in-source collision-induced dissociation to sequence peptide mixtures. J Chromatogr, A. 2003;1020:11–26. doi: 10.1016/j.chroma.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 27.Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat Biotechnol. 2001;19(10):952–957. doi: 10.1038/nbt1001-952. [DOI] [PubMed] [Google Scholar]
- 28.Johnson JR, Meng F, Forbes AJ, Cargile BJ, Kelleher NL. Fourier-transform mass spectrometry for automated fragmentation and identification of 5–20 kDa proteins in mixtures. Electrophoresis. 2002;23(18):3217–3223. doi: 10.1002/1522-2683(200209)23:18<3217::AID-ELPS3217>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 29.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance characteristics of electron transfer dissociation mass spectrometry. Mol Cell Proteomics. 2007;6(11):1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Leduc RD, Kelleher NL. Using ProSight PTM and related tools for targeted protein identification and characterization with high mass accuracy tandem MS data. Curr Protoc Bioinform. 2007;19:13.6.1–13.6.28. doi: 10.1002/0471250953.bi1306s19. [DOI] [PubMed] [Google Scholar]
- 31.Taylor GK, Kim YB, Forbes AJ, Meng F, McCarthy R, Kelleher NL. Web and database software for identification of intact proteins using ”Top Down” mass spectrometry. Anal Chem. 2003;75(16):4081–4086. doi: 10.1021/ac0341721. [DOI] [PubMed] [Google Scholar]
- 32.Pesavento JJ, Kim YB, Taylor GK, Kelleher NL. Shotgun annotation of histone modifications: a new approach for streamlined characterization of proteins by Top Down mass spectrometry. J Am Chem Soc. 2004;126(11):3386–3387. doi: 10.1021/ja039748i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 34.Scherl A, Shaffer SA, Taylor GK, Hernadez P, Appel RD, Binz P, Goodlett DR. On the benefits of acquiring peptide fragment ions at high measured mass accuracy. J Am Soc Mass Spectrom. 2008;19(6):891–901. doi: 10.1016/j.jasms.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wenger CD, Boyne MT, II, Ferguson JT, Robinson DE, Kelleher NL. Versatile online-offline engine for automated acquisition of high-resolution tandem mass spectra. Anal Chem. 2008;80(21):8055–8063. doi: 10.1021/ac8010704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patrie SM, Robinson DE, Meng F, Du Y, Kelleher NL. Strategies for automating top-down protein analysis with Q-FTICR MS. Int J Mass Spectrom. 2004;234:175–184. [Google Scholar]
- 37.Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods. 2005;2(9):667–675. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
- 38.Liu H, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 39.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 40.Frank AM, Savitski MM, Nielsen ML, Zubarev RA, Pevzner PA. De novo peptide sequencing and identification with precision mass spectrometry. J Proteome Res. 2007;6(1):114–123. doi: 10.1021/pr060271u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwang S, Lundgren DH, Mayya V, Rezaul K, Cowan AE, Eng JK, Han DK. Systematic characterization of nuclear proteome during apoptosis. Mol Cell Proteomics. 2006;5:1131–1145. doi: 10.1074/mcp.M500162-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Cagney G, Amiri S, Premawaradena T, Lindo M, Emili A. In silico proteome analysis to facilitate proteomics experiments using mass spectrometry. Protein Sci. 2003;1(1):5. doi: 10.1186/1477-5956-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.