

Abstract
Cyclophilins belong to the enzyme class of peptidyl prolyl cis/trans isomerases which catalyze the cis/trans isomerization of prolyl bonds in peptides and proteins in different folding states. Cyclophilins have been shown to be involved in a multitude of cellular functions like cell growth, proliferation, and motility. Among the 20 human cyclophilin isoenzymes, the two most abundant members of the cyclophilin family CypA and CypB exhibit specific cellular functions in several inflammatory diseases, cancer development and HCV replication. A small-molecule inhibitor on the basis of aryl 1-indanylketones has now been shown to discriminate between CypA and CypB in vitro. CypA binding of this inhibitor has been characterized by fluorescence anisotropy- and isothermal titration calorimetry-based cyclosporin competition assays. Inhibition of CypA- but not CypB-mediated chemotaxis of mouse CD4+ T cells by the inhibitor provided biological proof of discrimination in vivo.
Keywords: cyclophilin, PPIase, inhibition, chemotaxis
Cyclophilins (Cyps) are folding helper enzymes belonging to the class of peptidyl prolyl cis/trans isomerases (PPIases; EC 5.2.1.8). They catalyze the cis/trans isomerization of peptidyl prolyl bonds in unfolded and native proteins (1). The most abundant member of the cyclophilin family in human tissues is the cytosolic cyclophilin A (CypA, Cyp18), which is the major cellular target for, and mediates the immunosuppressive actions of the cyclic undecapeptide cyclosporin A (CsA) (1, 2). CsA binds to the active site of CypA thereby interfering with its PPIase activity in a very potent manner (1, 2). However, it is believed that the molecular basis of CsA mediated immunosupression is the ternary complex of CypA with CsA and the calcium-calmodulin-activated serine/threonine-specific protein phosphatase calcineurin; formation of this complex inhibits the protein phosphatase activity of calcineurin and thus prevents it from regulating cytokine gene transcription (3).
There are twenty different cyclophilins described in humans at the protein level underlining the importance of selectivity in pharmacological inhibition of a particular family member. The cyclophilin isoforms possess molecular masses ranging from 18 kDa to 352 kDa and all might catalyze prolyl bond cis/trans isomerization utilizing a highly conserved active site (4). Besides the prototypic CypA, seven other cyclophilins (CypB, CypC, PPIL1, CypF, USA-Cyp, CypJ and PPIAL4) consist only of the cyclophilin domain whereas the multidomain cyclophilins contain additional protein domains of different functionality, like RRM, TPR or U-box domains (4). High susceptibility of the cyclophilin isoenzymes to CsA is determined by the presence of a tryptophan residue in the cyclophilin domain. Thus, the W121 residue of CypA was shown to be essential for CsA binding with a complex dissociation constant in the low nanomolar range (5, 6).
Generally, cyclophilins have been shown to be involved in a multitude of cellular functions like cell growth, proliferation, and motility (4, 7). Several of the isoforms are known to exhibit specific cellular functions, participate in specific interactions with other proteins and are related to distinct pathophysiological processes. The mitochondrial CypD represents an important part of the mitochondrial permeability transition pore, which is an essential factor in apoptotic and necrotic cell death (8, 9). Furthermore, interaction of CypD with mitochondrial amyloid-β protein increases mitochondrial, neuronal and synaptic stress and the absence of CypD protects neurons from Aβ- and oxidative stress-induced cell death (10). USA-Cyp and PPIL1 are part of the spliceosomal complex (11, 12).
Based on current knowledge, CypA and CypB are especially interesting members of the cyclophilin family. CypA was found to be overexpressed in many cancer cells (13), including human pancreatic cancer cells, oral squamous cancer cells (14), non–small cell lung cancer (15) and endometrial carcinoma (16). Both CypA and CypB were found to be associated with malignant progression of breast cancer (17, 18). While CypA was shown to interact with the prolactin receptor (19), CypB was found to form a complex with prolactin and to increase prolactin-induced proliferation (20). Besides their intracellular localization, both enzymes are found in the extracellular space and are thought to contribute to intercellular communication inducing signaling responses in target cells (21). The presence of elevated levels of extracellular cyclophilins has been reported in several inflammatory diseases (22) including severe sepsis (23) and rheumatoid arthritis (24). Extracellular CypA and CypB were shown to contribute to inflammatory responses via their chemotactic activity which has been demonstrated for neutrophils, eosinophils, and T lymphocytes (25-27). The matrix metalloproteinase inducer CD147 mediates the signalling and chemotactic activities of both cyclophilins (28, 29).
In addition, it has been shown that CsA suppresses HCV genome replication in HCV replicon cells and human hepatocytes infected with HCV (30, 31). This anti-HCV activity is directly mediated by blocking the PPIase site of cyclophilins activity since the nonimmunosuppressive CsA analogues NIM811 and Debio 025, which do not show calcineurin inhibition via gain of function, attenuate the in vitro replication of HCV subgenomic replicons (32-34). Furthermore, the anti-HCV effect of Debio 025 was confirmed in patient studies suggesting cyclophilin inhibition as a new therapeutic avenue for hepatitis C (35, 36). Among the cyclophilin subfamily, by several lines of evidence, CypA and CypB have been identified as valid drug targets for hepatitis C treatment. Both cyclophilins were found to physically interact with the HCV RNA-dependent RNA polymerase NS5B (37, 38), the essential catalyst for the HCV replication process (39). However, which PPIase, CypA or CypB or both, is involved in virus replication remains unclear. CsA inhibits both CypA and CypB in the lower nanomolar range thus preventing discrimination between the cyclophilins (40). Different RNA interference approaches showed varying results in that either CypB (37) or CypA (38) or both (41) were found to be essential for HCV replication.
The situation with their similar extracellular localization and secretion (21-24) as well as their involvement in HCV replication (37, 38) makes it especially important to clarify their individual contributions in the respective physiological processes. Beside RNAi strategies pharmacological downregulation of enzyme activity by specific inhibitors may be an effective approach in this field. Thus, compounds that interact selectively with either CypA or CypB and inhibit their PPIase activity are urgently required to clarify the function of these cyclophilins in vivo. In addition, those compounds might be applicable to the therapy of human diseases associated with the activity of a distinct cyclophilin. In this case, treatment with isoenzyme specific inhibitors i) would probably elicit less side effects than a global cyclophilin inhibitor like CsA and ii) would not get dispersed into a cellular sink when sequestered by other cyclophilins.
Aryl 1-indanylketones have been reported to be inhibitory to the parvulin type PPIase Pin1 by an interaction based on the twisted amide structure of these compounds resembling the putative transition state of PPIase catalysis (42). Since the spatial arrangements of the active site residues of cyclophilins and Pin1 are similar (57), aryl 1-indanylketones were analysed for their potential to inhibit the PPIase activity of cyclophilins.
In the current studies we describe small-molecule inhibitors on the basis of aryl 1-indanylketones that discriminate between CypA and CypB in vitro. Inhibition of CypA- but not CypB-mediated chemotaxis of mouse CD4+ T cells provided biological proof of discrimination among the isoforms. CypA binding of this inhibitor has been characterized using fluorescence anisotropy- and isothermal titration calorimetry-based cyclosporin competition assays. Hydrophobic as well as polar interactions appear to be the driving forces for the active site binding of the inhibitor. Subtle differences in the spatial arrangement of subsites in the catalytic center of the cyclophilins might form the molecular basis of isoform selectivity.
Experimental Procedures
Chemicals
Buffers were purchased from AppliChem (Darmstadt, Germany) or Merck (Darmstadt, Germany). Yeast extract and Peptone, were purchased from Serva (Heidelberg, Germany). Sodium phosphate was from Merck. All other chemicals were purchased from Sigma (Munich, Germany) and of the highest purity available. Suc-Ala-Ala-Pro-Phe-pNA was purchased from Bachem (Heidelberg, Germany).
Chemical synthesis
Aryl 1-indanylketones and benzofuranones were synthesized as published (42, 43). The side chain carboxyfluorescein (CF)-labeled [Ser]8-CsA derivative [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA was synthesized according to a procedure described by Zhang et al. (44).
Enzymes
Human CypA and CypB were expressed from the vectors pQE70-CypA and pQE60-CypB, respectively in E.coli M15 cells and purified as described (45). To obtain human PPIL1, the gene was PCR-amplified using gene-specific primers from an ORF encoding human PPIL1 (imaGenes, Berlin) and cloned into pET9a. After overexpression in E.coli BL21 cells, purification was performed in 10 mM Tricine, pH 7.0 through a fractogel DEAE 650 column (Merck). The flow-through was passed through a fractogel TSK AF-green matrix (Merck). Bound proteins were eluted with 3 M KCl. The PPIL1 containing fractions were pooled, dialyzed against HEPES buffer (10 mM, pH 6.5), and bound to a fractogel EMD SO3- 650 matrix (Merck). The elution was performed with a linear gradient up to 1 M NaCl. Finally, a size exclusion chromatography step was performed (10 mM HEPES, pH 7.8, 6 mM KCl, 1.5 mM MgCl2, 1 mM DTT). To obtain the expression plasmid of human (His)6-CypD, the gene of the protein without mitochondrial signal sequence was PCR-amplified using gene-specific primers from an ORF encoding human PPIL1 (imaGenes, Berlin) and cloned into pET28a. The expression plasmid of mouse (His)6-CypC (pET26b-mCypC) was obtained from G. Schwartz (Rochester). The expression plasmid of human (His)10-USA-Cyp (pET19-USA-Cyp) was a gift from D. Horowitz (Bethesda). Purifications of His-Tag fusion proteins were performed using affinity chromatography on Ni-NTA resin followed by size exclusion chromatography in 10 mM HEPES, pH 7.8, 6 mM KCl, 1.5 mM MgCl2, 1 mM DTT.
PPIase activity measurements and inhibition experiments
PPIase activity assays were performed at 283 K in quartz cuvettes with a path length of 1 cm under vigorous stirring with a Hewlett-Packard 8453A UV/Vis spectrophotometer in 35 mM HEPES buffer at pH 7.8. PPIases were used at concentrations between 0.7 nM and 30 nM. Enzyme activities towards the substrate Suc-Ala-Ala-Pro-Phe-pNA were measured using the protease free assay according to Janowski et al. (46) in the additional presence of different concentrations of the compounds. A 30 mM stock solution of the substrate in 0.5 M LiCl/TFE (anhydrous) was prepared fresh before the measurement (60 μM final substrate concentration). Prior to every measurement, all components except substrate were incubated for 300 s at 283 K. The measurement was started upon substrate addition and the cis/trans isomerization kinetic of the substrate was followed at 330 nm.
Fluorescence polarization-based ligand displacement
Fluorescence polarization measurements according to (47) were performed on an Envision (PerkinElmer) fluorescence plate reader using black 96 well microtiter plates (PerkinElmer) filled with a total volume of 300 μl. Fluorescence anisotropy values were determined using a filter set for fluorescein polarization measurement (FP480/FP535). The anisotropy values r were calculated from the S and P-polarized fluorescein fluorescence intensity after excitation with a flashlamp according to r = (S-GxP)/(S+Gx2P) (G-factor G = 1) for every well.
All measurements were conducted in 35 mM HEPES buffer (pH 7.8), 10 mM NaCl at room temperature. To prevent adsorption of the CsA derivatives to the walls of the wells 1 μM of CsH was added. CsH is almost inactive in binding CypA and consequently does not compete for the CypA active site. After manual addition of the assay components to the wells the plates were shaken for 30 s prior to measurement. The binding of [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA to CypA was analyzed using a final concentration of 50 nM [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA and different concentrations of CypA between 1 nM and 1 μM. For ligand displacement experiments final concentrations of 50 nM [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA and 50 nM CypA were applied in the presence of various concentration of CsA (10 nM to 10 μM) or the aryl 1-indanylketon compound 1 (Figure 1) (10 nM to 100 μM) respectively. The anisotropy values obtained were fitted according to (eq.1) to yield the dissociation constant KD for the [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA/CypA complex.
Figure 1.

Chemical structure of aryl 1-indanylketones (A, B) and benzofuranones (C)
| (eq. 1) |
In this equation [L]t and [R]t represent the total concentrations of [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA and CypA respectively and r0 is the anisotropy value of [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA in the absence CypA. The proportionality factor c relates the concentration of the complex to the anisotropy value. Ligand displacement experiments were evaluated according to the mathematical model of Wang (48) using Igor (Wavemetrics) for data fitting.
Isothermal titration (ITC)
ITC experiments were performed using a VP-ITC (MicroCal). Prior to the experiment, all solutions and buffers were filtered through filter membranes with a pore size of 0.2 μm (Whatman) and degassed. Protein solutions were dialyzed against the assay buffer (35 mM HEPES, pH 7.8). 100 mM stock solutions of the inhibitors were prepared in DMSO and diluted into assay buffer. To maintain constant conditions in all samples, DMSO was also added to protein solutions at the appropriate concentration (below 0.1% v/v). In a typical experiment, 300 μl of a 20 μM solution of CypA was titrated in 15 μl-steps to a 2 μM CsA solution at 293 K. Titrations were performed in the absence and presence of 200 μM 1. By adding compound 1 to both the CsA and CypA solutions, a constant concentration of competing ligand was maintained throughout the experiment. The instruments stirring speed was set to 310 RPM and the feedback gain mode was set to “high”. Since the signal from the first injection can usually not be used for data analysis only 2 μl were titrated in this step and the data point was omitted. Measured data were analyzed using the “Origin” software (MicroCal).
According to Zhang and Zhang (49), the thermodynamic parameters for the binding of a low-affinity ligand L2 can be estimated by a competition ITC experiment. First, the protein P is titrated with a high-affinity ligand L1 (ΔH1, K1) which occupies the same binding site as L2 to determine its binding parameters. Next, the titration is performed in the additional presence of the low-affinity ligand L2 to obtain the apparent ITC parameters (ΔHapp, Kapp). The ITC parameters for the low-affinity ligand (ΔH2, K2) can be calculated according to the following equations (eq. 2) and (eq. 3).
| (eq. 2) |
| (eq. 3) |
Chemotaxis assay
Activated CD4+ T cells were generated by overnight stimulation of total mouse spleen cells at 3×106 cells per well with the mitogen Concanavalin A (ConA) at 10 μg/ml in Click's medium containing 5% FCS in a 24-well tissue culture plate at 37°C in the presence of 5% CO2. The following day, the CD4+ T cells were purified from the cultures using a MACS (Miltenyi Biotec Inc., Auburn, CA) negative depletion kit. These purified CD4+ T cells were then tested for chemotaxis in 48-well modified Boyden chambers (Neuroprobe, Gaithersburg, MD), with the two compartments separated by a 5-μm polycarbonate membrane (Neuroprobe). Medium (RPMI-1640 + 1% bovine serum albumin) containing 100 ng/ml recombinant human CypA (Calbiochem, San Diego, CA) or 200 ng/ml recombinant CypB (generated at GWU), or containing 1 ng/ml recombinant mouse RANTES (PeproTech, Rocky Hill, NJ), or containing nothing (medium alone) was placed in wells in the lower compartment of the Boyden chamber. Purified CD4+ T cells in medium (104 per well) were placed in wells in the upper compartment. In some groups, compound 1 was added at a final optimized concentration of 2 μM to matching wells in the top and lower compartments. The loaded chamber was then incubated at 37°C in the presence of 5% CO2 for 50 min. Following incubation, the polycarbonate membrane was removed, non-migrated cells were scraped off, and the membrane was stained with Wright-Giemsa (CAMCO, Fort Lauderdale, FL). A chemotactic index was generated for each well by dividing the number of cells migrating within each test well by the average number of cells migrating to medium alone.
Results
Aryl 1-indanylketone 1 is a specific inhibitor of CypA among cyclophilin isoforms
The chemical structures of aryl 1-indanylketones and benzofuranones used in this work are shown in Figure 1. Among these compounds 1 was found to reversibly inhibit cyclophilin A (CypA, Cyp18) with a KI value of 0.52 ± 0.15 μM (Figure 2A). Notably, CsA as the generic CypA inhibitor with a KI value of 2.9 nM cannot markedly differentiate between six important human cyclophilins (Table 1). CypA is most sensitive to CsA but the level of selectivity for CypB is in the range of 3-fold. In contrast, the aryl 1-indanylketone compound 1 is able to differentiate between the cyclophilins (Table 2). It was found that the PPIase activity of CypB, CypC and USA-Cyp did not respond to compound 1 up to a concentration of 30 μM indicating selectivity of at least 200-fold. CypB, CypC and USA-Cyp share a close homology with CypA (63%, 56% and 55% amino acid identity to CypA in the PPIase domain, respectively). In assays using CypD and PPIL1 compound 1 had a 4.6- and 56-fold selectivity for CypA inhibition, respectively. The KI values were determined using recombinantly produced enzymes in a protease-free assay (46) with Suc-Ala-Ala-Pro-Phe-4-nitroanilide at a substrate concentration [S] ≪ KM. Reversibility of inhibition was thoroughly examined by dilution experiments of the CypA complex formed with 2, which represents the tightest binding compound (Table 2). CypA (0.75 μM) and 2 (0.02 - 200 μM) were incubated for 30 min for gradual inactivation and subsequently diluted. The PPIase activity of CypA completely recovered from the inhibited enzyme when diluted 1:375 in 35 mM HEPES buffer pH 7.8, regardless of whether the incubation time was prolonged or the concentration of 2 was further increased (Figure 2B).
Figure 2.

A) Residual PPIase activity of CypA (closed circles) and CypB (open triangles) by compound 1. B) Reversibility of CypA inhibition by compound 2 analyzed by dilution experiments. CypA (0.75 μM) and 2 (0.02 - 200 μM) were incubated for 30 min. After 1:375 dilution in 35 mM HEPES buffer pH 7.8, the PPIase activity was determined (bars) and compared with the PPIase activity of CypA at the indicated concentrations of compound 2. The PPIase activity of CypA completely recovered from the inhibited enzyme. The PPIase activity was measured in 35 mM HEPES buffer at pH 7.8 and 283 K using Suc-Ala-Ala-Pro-Phe-pNA as substrate. Each data point represents the mean of two measurements with a deviation less than 10 %.
Table 1.
Properties of prototypic cyclophilins
| Enzyme | alias/gene namea | Molecular weight a precursor/processed (Da) |
Identity with CypA (%) |
kcat/KMb (M-1s-1) |
KI CsA (nM) |
|---|---|---|---|---|---|
| CypA | Cyp18/PPIA | 18012 | 100 | 1.4 × 107 | 2.9 |
| CypB | Cyp23/PPIB | 22742/20289 | 63 | 5.6 × 106 | 8.4 |
| CypC | Cyp23a/PPIC | 22763 | 56 | 5.5 × 105 | 7.7 |
| PPIL1 | Cyp18.2a/PPIL1 | 18236 | 46 | 2.0 × 106 | 9.8 |
| CypD | Cyp22/PPIF | 22040/18897 | 75 | 1.3 × 107 | 6.7 |
| USA-Cyp | Cyp19.2/PPIH | 19208 | 55 | 6.9 × 105 | 91 |
Data according to SwissProt.
kcat/KM values were determined with the protease-free PPIase assay according to Janowski et al. (46) in 35 mM HEPES buffer pH 7.8 at 283 K, using Suc-Ala-Ala-Pro-Phe-pNA (60 μM) as substrate
KI values were determined with the protease-free PPIase assay in the presence of increasing concentrations of CsA. SD of the measurements of the kinetic constants were not larger than 10 %.
Table 2.
Inhibition of prototypic cyclophilins by compounds 1 and 2
| Enzyme | KI (μM) | |
|---|---|---|
| 1 | 2 | |
| CypA | 0.52 ± 0.15 | 0.3 ± 0.1 |
| CypB | >100 | 12 ± 5 |
| CypC | >100 | >100 |
| PPIL1 | 29 ± 7 | >100 |
| CypD | 2.42 ± 0.76 | 6.28 ± 0.73 |
| USA-Cyp | >100 | >100 |
KI values were determined with the protease-free PPIase assay according to Janowski et al. (46) in 35 mM HEPES buffer pH 7.8 at 283 K, using Suc-Ala-Ala-Pro-Phe-pNA (60 μM) as substrate and increasing concentrations of aryl 1-indanylketones, respectively.
Beside Pin1 inhibition (42) compound 1 does not influence Par14 and only modestly affects FKBP12 (KI = 38 ± 8μM). The similar inhibitory properties of compound 1 toward Pin1 and CypA prompted us to investigate a series of recently reported Pin1 inhibitors of the aryl 1-indanylketone and spiroannulated benzofuranone type (42, 43) to establish if there is a further enhanced selectivity for CypA over other cyclophilins, especially CypB (Table 3). CypA and CypB can be weakly inhibited by benzofuranones 7 and 9, in a range that was already found for Pin1. Due to its low potency the spirocyclic benzofuranone ring may not be suitable for the design of cyclophilin-directed inhibitors. In the aryl 1-indanylketone series the most active inhibitor against CypA was compound 2 (KI = 0.3 ± 0.1 μM). However, compound 2 inhibited CypB with a KI of 12 ± 5 μM, thereby discriminating between both only by a factor of 40. Thus, compound 1 remained the compound with the highest ability to discriminate between CypA and CypB by a factor of >200.
Table 3.
Inhibition of CypA and CypB by aryl 1-indanylketones and benzofuranones
| compound | KI (μM) | |
|---|---|---|
| CypA | CypB | |
| 1 | 0.52 ± 0.15 | >100 |
| 2 | 0.3 ± 0.1 | 12 ± 5 |
| 3 | 1.7 ± 0.5 | 8.6 ± 0.9 |
| 4 | 1.2 ± 0.4 | 2.1 ± 0.3 |
| 5 | 10 ± 2 | >100 |
| (S)-6 | >100 | >100 |
| (R)-6 | 7.5 ± 1.5 | 40 ± 10 |
| 7 | 21 ± 4 | >100 |
| 8 | >100 | >100 |
| 9 | >100 | 63 ± 12 |
KI values were determined with the protease-free PPIase assay according to Janowski et al. (46) in 35 mM HEPES buffer pH 7.8 at 283 K, using Suc-Ala-Ala-Pro-Phe-pNA (60 μM) as substrate and increasing concentrations of aryl 1-indanylketones.
A considerable difference in the inhibition of CypA and CypB was found, when the two enantiomers of 6 were analyzed. The inhibitory (R)-enantiomer had a 40-fold selectivity for CypA. Notably, (S)-configuration in the 1-methyl position totally compromised the inhibition of both CypA and CypB.
Aryl 1-indanylketone 1 binds to the active site of CypA
Next we analyzed, if compound 1 binds to CypA at the active site, which is composed of residues from four of the eight beta-strands and a loop of aromatic residues forming a hydrophobic pocket at one side of the hydrophobic core of CypA (50). We used a CsA competition assay because this inhibitor was shown to bind to the active site of CypA (47). Displacement of a side chain carboxyfluorescein (CF)-labeled [Ser]8-CsA derivative ([O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA) from the CypA active site was determined at increasing concentrations of compound 1 using fluorescence anisotropy measurements. This compound inhibited the PPIase activity of CypA with a KI value of 2.4 ± 0.3 nM.
The anisotropy change associated with the binding of [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA by increasing concentrations of CypA is shown in Figure 3A. When using various concentrations of CypA at an invariable concentration of the fluorescent derivative (50 nM), we obtained a saturating binding curve with a KD of 4.0 ± 1.5 nM (Figure 3A). According to these data, the fluorescence anisotropy competition assay was set up to use 50 nM of the [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA and 50 nM of CypA. As a reference competition assay CsA was applied at various concentrations to the preformed [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA/CypA complex. As expected, the anisotropy value of the probe decreased with the increasing concentration of CsA providing a KD of 12 ± 2 nM. Considering the different protein concentrations in the assays, this value agrees within the limits of error with the value determined by enzyme inhibition (Figure 3B, Table 1). When various concentrations of compound 1 were applied to compete for CypA of the preformed complex, the anisotropy value of the probe decreased with the increasing concentration of compound 1 and provided a KD of 0.54 ± 0.13 μM (Figure 3B).
Figure 3.

Aryl 1-indanylketone 1 and CsA are able to displace [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA from the complex with CypA. A) Determination of the dissociation constant for the [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA/CsA-complex. 50 nM [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA in 35 mM HEPES buffer pH 7.8, 10 mM NaCl, 1 μM CsH was titrated with increasing concentrations of CypA and fluorescence anisotropy values were recorded. The anisotropy values obtained were fitted according to quadratic equation (eq.1) to yield a dissociation constant (KD) of 4.0 ± 1.5 nM for the CypA/ [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA interaction. B) Titration of the [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA / CypA complex with compound 1 (black circle) or CsA (white triangle). The preformed complex of 50 nM CypA and 50 nM [O-(CF-NH(CH2)2NHC(O)CH2-)d-Ser]8CsA in 35 mM HEPES buffer pH 7.8, 10 mM NaCl, 1 μM CsH, was titrated with increasing amounts of compound 1 or CsA and fluorescence anisotropy values were recorded. Ligand displacement experiments were evaluated according to the mathematical model of Wang (48) resulting in dissociation constants (KD) of 540 ± 130 nM for the CypA/ compound 1 complex and 12 ± 2 nM for the CypA/CsA complex, respectively.
Thermodynamic parameters of the interaction
Isothermal titration calorimetry was used to determine the thermodynamic parameters of the association reaction of the inhibitor compound 1 with CypA (Table 4). Initially, the titration of 50 μM compound 1 with 200 μM CypA and the titration of 10 μM CypA with 100 μM compound 1 at 293 K were performed. These direct titrations did not allow reliable determination of the thermodynamic parameter. Therefore, we have used a competitive method to obtain the thermodynamic parameters and association constant for the binding of compound 1 (49). This method is based on the thermodynamic coupling of two ligands that bind to the same site of a protein. The thermodynamic parameters for the low-affinity ligand can be obtained by conducting ITC experiments with a high-affinity ligand in the absence and presence of the low-affinity ligand. In this experiment, compound 1 constituted the low-affinity ligand and CsA was used as the high-affinity ligand. First, the titration of 2 μM CsA with 20 μM CypA at 293 K was performed (Figure 4A). It revealed a binding enthalpy ΔHITC of -13.2 ± 0.1 kcal mol-1, a TΔSITC of -3.0 kcal mol-1 and a ΔGITC of -10.2 kcal mol-1 which results in an association constant KA of (4.05 ± 0.94) 107 M-1 (Table 4). These values agree well with the values already described (45). In the presence of 200 μM compound 1, the apparent binding constant for CsA was (4.12 ± 1.59) 106 M-1, which is about 10 fold smaller than that determined in the absence of compound 1 (Figure 4B). The association constant and the ΔH for the binding of compound 1 to CypA were obtained by using equations (eq. 2) and (eq. 3), respectively. Thus, using the competition ITC approach, compound 1 revealed a binding enthalpy ΔH of -2.8 kcal mol-1, a TΔS of 3.5 kcal mol-1, a ΔG of -6.3 kcal mol-1, and an association constant KA of 4.4 × 104 M-1. The positive TΔS showed that the association of CypA with compound 1 was entropically driven. The positive entropic contribution hints to a burial of solvent-accessible surface area on binding, since the release of ordered water molecules often contributes extensively and positively to the entropy of an interaction (51). The enthalpic contribution suggests that the enzyme/inhibitor complex is stabilized to some extent by hydrogen bonds or van der Waals interactions (51).
Table 4.
Thermodynamic parameters for the interactions of CypA with CsA and compound 1
| Syringe | Cell | ΔH (kcal mol-1) |
TΔS (kcal mol-1) |
ΔG (kcal mol-1) |
KA (M-1) |
N |
|---|---|---|---|---|---|---|
| CypA | CsA | -13.2 ± 0.1 | -3.0 | -10.2 | (4.05 ± 0.94)×107 | 1.3 |
| CypA, 1 | CsA, 1 | -10.8 ± 0.1 | -1.9 | -8.9 | (4.12 ± 1.59)×106 | 1.6 |
| 1 | CsA | n.s. | n.s. | n.s. | n.s. | n.s. |
A solution of 2 μM CsA (initial conc.) in 35 mM HEPES buffer pH 7.8 was titrated at 293 K with 20 μM CypA (initial conc.) in the absence and presence of 200 μM compound 1. The error of each parameter represents the error of fitting. n.s. no signal of heat formation.
Figure 4.
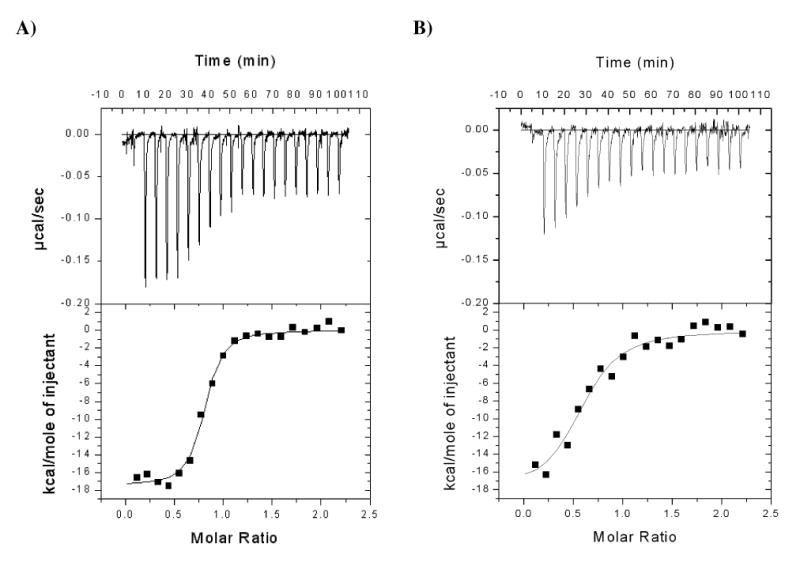
Isothermal titration of CsA with CypA in the absence (A) and presence (B) of compound 1. A solution of 2 μM CsA (initial conc.) in 35 mM HEPES buffer pH 7.8 was titrated at 293 K with 20 μM CypA in the absence and presence of 200 μM compound 1. By adding compound 1 to both, the CsA and CypA solutions, a constant concentration of the competing ligand was maintained throughout the experiment.
Remarkably, the dissociation constant KD of 22.6 μM as determined by the ITC probe of CsA/compound 1 competition is much higher than the KI value determined by the fluorescence anisotropy competition assay using a CsA derivative. This may be caused by differences in the experimental setup of the respective measurements. In the ITC, the final CypA concentration was ≫103 fold higher than the protein concentration applied in the other binding assays.
Aryl 1-indanylketone 1 inhibits the chemotactic property of CypA but not of CypB
Next we asked whether the application of the CypA specific inhibitor compound 1 would elicit an influence on a cellular function already described to be associated with inhibition of the PPIase activity of cyclophilins by CsA. CypA as well as CypB have been shown to function as chemotactic agents for leukocytes (26, 28, 29). This function is based on the activity of CD147 as a signalling receptor for extracellular cyclophilins (21). The chemotactic activity of these cyclophilins can be inhibited by CsA (22).
To investigate the influence of compound 1 on cyclophilin-induced chemotaxis, we used CypA and CypB to induce the migration of mouse CD4+ T cells (Figure 5). Groups of CD4+ T cells were set up in Boyden chambers in the presence of a previously optimized dose of recombinant CypA or CypB. The chemotactic index represents the ratio of cells migrating within each test well to the average number of cells migrating to medium alone. Based on published studies and previous experience, a chemotactic index of >1.2 was considered as significant chemotaxis (52). As already described in the literature (26, 28, 29), CypA as well as CypB induced the migration of the T cells (Figure 5). Migration was also observed in response to a positive controlchemokine, RANTES, which we had previously established was not sensitive to the indanylketone (data not shown). The addition of compound 1 inhibited the leukocyte chemotaxis induced by CypA almost completely, indicating the presence of an active site mediating the chemotactic properties of CypA. As expected from the inability of compound 1 to inhibit the PPIase activity of CypB, CypB-induced chemotaxis was barely impacted by addition of compound 1. The diluent had no significant effect on the migration induced by either CypA or CypB. Taken together, these findings indicate that the ability of compound 1 to selectively inhibit CypA in vitro is mirrored by a distinct response of a relevant biological system.
Figure 5.

Aryl 1-indanylketone 1 inhibits the chemotactic activity of CypA but not CypB. Activated CD4+ T cells were purified using MACS separation after overnight stimulation of spleen mononuclear cells with ConA. Chemotaxis assays were conducted using a 48-well modified Boyden chamber. RANTES was used as a positive control for cell migration. CypA and CypB were used at 100 ng/ml and 200 ng/ml, respectively. Compound 1 was used at 2 μM. Data show mean (± SE) chemotactic indices for each group with n=6 wells per group. A chemotactic index was generated for each well by dividing the number of cells migrating within each well by the average number of cells migrating to medium alone.
Discussion
Here we report aryl 1-indanylketones as the first PPIase inhibitors found to exhibit a potent inhibitory effect on CypA along with a considerable degree of selectivity among isoenzymes of the human family of cyclophilins. This structural template gave rise to the phenolic biphenyle ketone 1 which not only enabled a clear discrimination between CypA and CypB activities but also left FKBP12 and Par14 activities nearly unchanged at concentrations inhibitory to CypA. Previous biochemical data for compound 1 reported a comparable inhibitory action on Pin1 (42). The proposed model for the interaction of aryl 1-indanylketones with Pin1 is based on the twisted amide structure of compound 1 which resembles the putative transition state of PPIase catalysis (42). The torsion angle between the aromatic-carbonyl and the indanyl moiety of 92° shown in the X-ray structure of an aryl 1-indanylketone corresponds with the features of a twisted amide supposed to form the transition state (42). However, the features which make the ring substitution pattern of compound 1 especially favorable for Pin1 inhibition remained open. Obviously, similar stereochemical inhibition characteristics for CypA and Pin1 as revealed by the relative inertness of (S)-6 enantiomer (Table 3) support the hypothesis of a transition state inhibitor, since stereochemical selectivity forms an integral part of transition state binding. Other structure-activity relationships are not evident from the limited set of compounds for both selectivity and potency (Table 3). The tight binding of CsA to cyclophilins might indicate its ability to mimic the conformation of the transition state of the reaction. However, structural comparison of the CypA/CsA complex with unligated CypA and the CypA/Ala-Pro complex suggested that CsA is unlikely to be a transition state analog of the substrate (66), in contrast with an original proposal that CsA mimics a transition state of the PPIase reaction (67). Designed inhibitory compounds developed for CypA have been ground state analogues such as alkene isosteres of prolyl bonds or bicyclic cis-Pro mimics or transition state analogues such as phosphoamides (53-56).
By demonstrating that the active site of CypA but not of CypB resembles Pin1 in the affinity for compound 1 we have revealed previously unidentified similarities and differences in the catalytic mechanisms of PPIases. Interestingly, despite the lack of a common global fold in cyclophilins and Pin1, the spatial arrangement of their active site residues is similar (57). The CypA active site residues superimposed with the active site of Pin1 show four CypA residues having identical counterparts in the human Pin1 and two residues show mirror symmetry (57).
Elements contributing to the differential inhibition of CypA and CypB might become visible in X-ray structures of the respective Cyp/CsA complexes. However, the active sites of CypA and CypB are nearly identical (58). There are no obvious differences in sequence or structure in the X-ray structures of both proteins in complex with CsA for any residues of the binding pocket. Even the water-mediated contacts in these complexes were found to be essentially conserved (58). In contrast, comparison of structural parameters of the Cyp/CsA complexes in solution showed a small difference in the NMR chemical shifts of one of the NOE peaks involving the MeVal11 residue of CsA, which occupies the prolyl binding pocket of the cyclophilins. Despite the strong similarities in the X-ray structures of the active sites of CypA and CypB, the catalytic efficiency of CypB is about 2.5-fold less than that of CypA for the same tetrapeptide model substrate (59, Table 1). This suggests that for both cyclophilins the active sites in the vicinity of the prolyl binding sites might exhibit functionally significant differences, probably in the transition state of catalysis (60). It follows that the discriminative power of compound 1 can result from the fine tuning of the contacts at the P1-S1 subsite. Prolyl cis/trans isomerizations undergo two dramatic steric changes due to the loss of the pseudo-double bond character of the peptide bond in the transition state: (a) adopting the perpendicular arrangement of the carbonyl group and the proline ring and, (b) distortion of the proline nitrogen atom hybridization in direction of sp3. Obviously, the degree of the steric changes of the cyclophilin-catalyzed reaction depends on the transition state structure and any slight change in these parameters might alter the transition state binding of substrates or inhibitors. We hypothesize that the state of hybridization of the carbon 1 atom of the indanyl residue of compound 1, which resembles the hybridization pattern of the proline nitrogen in the transition state, is an especially sensitive parameter of transition state binding, and thus inhibitory power and selectivity factors of aryl 1-indanylketones. By examining Pin1 inhibitor constants using a panel of substrate-like compounds a six-membered pipecolinyl residue was found to be far superior than the five-membered prolyl residue (64). Notably, the X-ray structures of two Pin1/pipecolinyl inhibitor complexes support the role of nitrogen hybridization for inhibition power (65).
Binding of compound 1 to the active site of CypA when measured by direct ITC of the components did not allow reliable determination of the thermodynamic parameters. Calculation of the thermodynamic parameter of this interaction using an ITC-based competition assay according to Zhang and Zhang (49) revealed enthalpic as well as entropic contributions to the free energy of ligand binding. The positive TΔS calculated for the binding of compound 1 to the CypA active site indicates entropically favored binding. Favorable entropy changes are generally attributed to hydrophobic interactions, to an increase in solvent entropy from burial of hydrophobic groups, and to release of water upon binding (51). The favourable enthalpic term indicates that hydrogen bond formation or van der Waals interactions take place. Interestingly, polar substituents of the biaryl group were necessary to mediate the specific nanomolar interaction with CypA. Since hydrophobic interactions are generally considered to be inherently non-specific, selectivity may be attained by shape complementarity of spatially separated polar groups (61).
The presence of specific polar substituents in compound 1 could also induce transition state complementary of the extended hydrophobic binding patches of the aromatic rings via indirect effects on the indanyl carbon 1 atom hybridization. In contrast, the less selective but potent binding of CsA to cyclophilins is mainly mediated through conserved hydrogen bonds within the cyclophilin family for which W121 of CypA for example, provides a directing template. Direct hydrophobic interactions contribute to a smaller degree to the total binding energy (45). Thus, as revealed by ITC, CypA/CsA complex formation is completely enthalpically driven and CsA binding is associated with unfavorable entropy. The finding that different ligands can bind by enthalpy- or entropy-driven mechanisms has been already described in the case of other protein-ligand interactions. For example, the binding of biotin to streptavidin is entirely enthalpy driven whereas the binding of azobenzene ligands to the same protein is entropically favored (62). Similarly, the development of HIV-1 protease inhibitors had demonstrated either enthalpically or entropically driven ligand binding against the same binding cavity (63).
Both extracellular CypA and CypB can function as chemotactic agents for leukocytes (26, 28, 29), with CD147 being the principal signalling receptor for this function(28, 29). Elevated levels of extracellular cyclophilins have been observed in many inflammatory diseases including severe sepsis, rheumatoid arthritis, and vascular smooth muscle cell disease (22-24). The application of compound 1 specifically inhibited the chemotaxis induced by CypA, whereas the CypB-induced chemotaxis was not influenced by the compound (Figure 5). The fact that cell migration is not impacted in the absence of exogenously provided CypA (for example when using RANTES) demonstrates that only CypA is a target for compound 1 in these assays. These findings also suggest that inhibition of endogenous Pin1 by compound 1 has no effect on the chemotaxis of activated CD4+ T cells. Alternatively, the inhibitor cannot reach the particular fraction of Pin1 required for mediating an effect.
In conclusion, we have shown that the aryl 1-indanylketone 1 selectively blocks the catalytic activity of CypA without interfering with the PPIase activity of CypB. This inhibition inhibits the chemotactic function of CypA for CD4+ T cells. These results emphasize the specificity of compound 1 for CypA in a physiological context and support the use of this compound as a biological probe to analyze and dissect CypA- and CypB-dependent pathways.
Acknowledgments
SLC was funded by NIH grant AI-067254. MB and CSF were funded by Deutsche Forschungsgemeinschaft grants Br 604/14, Br 604/17 (to M. B.) and Graduiertenkolleg 1026 “Conformational transitions in macromolecular interactions” (to C.S.-F). We thank M. Malesevic for synthesis of the fluorescently labeled CsA derivative and M. Frost for providing PPIL1. We are grateful to I. Kunze, S. Ross and B. Korge for technical assistance, A. Schierhorn for protein analyses and G. Fischer for helpful discussions and critical reading of the manuscript.
Abbreviations
- ITC
isothermal titration calorimetry
- pNA
4-nitroanilide
- PPIase
peptidyl prolyl cis/trans isomerase
- wt
wild type
- CsA
cyclosporin A
- CypA
cyclophilin A
- CypB
cyclophilin B
- CF
carboxyfluorescein
References
- 1.Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi N, Hayano T, Suzuki M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature. 1989;337:473–475. doi: 10.1038/337473a0. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 4.Fischer G, Aumüller T. Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev Physiol Biochem Pharmacol. 2003;148:105–150. doi: 10.1007/s10254-003-0011-3. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Chen CM, Walsh CT. Human and Escherichia coli cyclophilins: sensitivity to inhibition by the immunosuppressant cyclosporin A correlates with a specific tryptophan residue. Biochemistry. 1991;30:2306–2310. doi: 10.1021/bi00223a003. [DOI] [PubMed] [Google Scholar]
- 6.Fejzo J, Etzkorn FA, Clubb RT, Shi Y, Walsh CT, Wagner G. The mutant Escherichia coli F112W cyclophilin binds cyclosporin A in nearly identical conformation as human cyclophilin. Biochemistry. 1994;33:5711–5720. doi: 10.1021/bi00185a007. [DOI] [PubMed] [Google Scholar]
- 7.Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin DT, Lechleiter JD. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J Biol Chem. 2002;277:31134–31141. doi: 10.1074/jbc.M112035200. [DOI] [PubMed] [Google Scholar]
- 9.Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horowitz DS, Lee EJ, Mabon SA, Misteli T. A cyclophilin functions in pre-mRNA splicing. EMBO J. 2002;21:470–480. doi: 10.1093/emboj/21.3.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Folk P, Puta F, Skruzny M. Transcriptional coregulator SNW/SKIP: the concealed tie of dissimilar pathways. Cell Mol Life Sci. 2004;61:629–640. doi: 10.1007/s00018-003-3215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao Q, Li M, Yang H, Chai H, Fisher W, Chen C. Roles of cyclophilins in cancers and other organ systems. World J Surg. 2005;29:276–280. doi: 10.1007/s00268-004-7812-7. [DOI] [PubMed] [Google Scholar]
- 14.Shen J, Person MD, Zhu J, Abbruzzese JL, Li D. Protein expression profiles in pancreatic adenocarcinoma compared with normal pancreatic tissue and tissue affected by pancreatitis as detected by two-dimensional gel electrophoresis and mass spectrometry. Cancer Res. 2004;64:9018–9026. doi: 10.1158/0008-5472.CAN-04-3262. [DOI] [PubMed] [Google Scholar]
- 15.Howard BA, Zheng Z, Campa MJ, Wang MZ, Sharma A, Haura E, Herndon JE, 2nd, Fitzgerald MC, Bepler G, Patz EF., Jr Translating biomarkers into clinical practice: prognostic implications of cyclophilin A and macrophage migratory inhibitory factor identified from protein expression profiles in non–small cell lung cancer. Lung Cancer. 2004;46:313–323. doi: 10.1016/j.lungcan.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Zhao X, Bai S, Wang Z, Chen L, Wei Y, Huang C. Proteomics identification of cyclophilin a as a potential prognostic factor and therapeutic target in endometrial carcinoma. Mol Cell Proteomics. 2008;7:1810–1823. doi: 10.1074/mcp.M700544-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng J, Koblinski JE, Dutson LV, Feeney YB, Clevenger CV. Prolyl isomerase cyclophilin A regulation of Janus-activated kinase 2 and the progression of human breast cancer. Cancer Res. 2008;68:7769–7778. doi: 10.1158/0008-5472.CAN-08-0639. [DOI] [PubMed] [Google Scholar]
- 18.Fang F, Flegler AJ, Du P, Lin S, Clevenger CV. Expression of cyclophilin B is associated with malignant progression and regulation of genes implicated in the pathogenesis of breast cancer. Am J Pathol. 2009;174:297–308. doi: 10.2353/ajpath.2009.080753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Syed F, Rycyzyn MA, Westgate L, Clevenger CV. A novel and functional interaction between cyclophilin A and prolactin receptor. Endocrine. 2003;20:83–90. doi: 10.1385/ENDO:20:1-2:83. [DOI] [PubMed] [Google Scholar]
- 20.Rycyzyn MA, Reilly SC, O'Malley K, Clevenger CV. Role of cyclophilin B in prolactin signal transduction and nuclear retrotranslocation. Mol Endocrinol. 2000;14:1175–1186. doi: 10.1210/mend.14.8.0508. [DOI] [PubMed] [Google Scholar]
- 21.Bukrinsky MI. Cyclophilins: unexpected messengers in intercellular communications. Trends Immunol. 2002;23:323–325. doi: 10.1016/s1471-4906(02)02237-8. [DOI] [PubMed] [Google Scholar]
- 22.Arora K, Gwinn WM, Bower MA, Watson A, Okwumabua I, MacDonald HR, Bukrinsky MI, Constant SL. Extracellular cyclophilins contribute to the regulation of inflammatory responses. J Immunol. 2005;175:517–522. doi: 10.4049/jimmunol.175.1.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tegeder I, Schumacher A, John S, Geiger H, Geisslinger G, Bang H, Brune K. Elevated serum cyclophilin levels in patients with severe sepsis. J Clin Immunol. 1997;17:380–386. doi: 10.1023/a:1027364207544. [DOI] [PubMed] [Google Scholar]
- 24.Billich A, Winkler G, Aschauer H, Rot A, Peichl P. Presence of cyclophilin A in synovial fluids of patients with rheumatoid arthritis. J Exp Med. 1997;185:975–980. doi: 10.1084/jem.185.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherry B, Yarlett N, Strupp A, Cerami A. Identification of cyclophilin as a proinflammatory secretory product of lipopolysaccharide-activated macrophages. Proc Natl Acad Sci U S A. 1992;89:3511–3515. doi: 10.1073/pnas.89.8.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Q, Leiva MC, Fischkoff SA, Handschumacher RE, Lyttle CR. Leukocyte chemotactic activity of cyclophilin. J Biol Chem. 1992;267:11968–11971. [PubMed] [Google Scholar]
- 27.Allain F, Vanpouille C, Carpentier M, Slomianny MC, Durieux S, Spik G. Interaction with glycosaminoglycans is required for cyclophilin B to trigger integrin-mediated adhesion of peripheral blood T lymphocytes to extracellular matrix. Proc Natl Acad Sci U S A. 2002;99:2714–2719. doi: 10.1073/pnas.052284899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yurchenko V, O'Connor M, Dai WW, Guo H, Toole B, Sherry B, Bukrinsky M. CD147 is a signaling receptor for cyclophilin B. Biochem Biophys Res Commun. 2001;288:786–788. doi: 10.1006/bbrc.2001.5847. [DOI] [PubMed] [Google Scholar]
- 29.Yurchenko V, Zybarth G, O'Connor M, Dai WW, Franchin G, Hao T, Guo H, Hung HC, Toole B, Gallay P, Sherry B, Bukrinsky M. Active site residues of cyclophilin A are crucial for its signaling activity via CD147. J Biol Chem. 2002;277:22959–22965. doi: 10.1074/jbc.M201593200. [DOI] [PubMed] [Google Scholar]
- 30.Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282–1288. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- 31.Nakagawa M, Sakamoto N, Enomoto N, Tanabe Y, Kanazawa N, Koyama T, Kurosaki M, Maekawa S, Yamashiro T, Chen CH, Itsui Y, Kakinuma S, Watanabe M. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem Biophys Res Commun. 2004;313:42–47. doi: 10.1016/j.bbrc.2003.11.080. [DOI] [PubMed] [Google Scholar]
- 32.Goto K, Watashi K, Murata T, Hishiki T, Hijikata M, Shimotohno K. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem Biophys Res Commun. 2006;343:879–884. doi: 10.1016/j.bbrc.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 33.Ma S, Boerner JE, TiongYip C, Weidmann B, Ryder NS, Cooreman MP, Lin K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob Agents Chemother. 2006;50:2976–2982. doi: 10.1128/AAC.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paeshuyse J, Kaul A, De Clercq E, Rosenwirth B, Dumont JM, Scalfaro P, Bartenschlager R, Neyts J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43:761–770. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- 35.Flisiak R, Horban A, Gallay P, Bobardt M, Selvarajah S, Wiercinska-Drapalo A, Siwak E, Cielniak I, Higersberger J, Kierkus J, Aeschlimann C, Grosgurin P, Nicolas-Métral V, Dumont JM, Porchet H, Crabbé R, Scalfaro P. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008;47:817–826. doi: 10.1002/hep.22131. [DOI] [PubMed] [Google Scholar]
- 36.Crabbé R, Vuagniaux G, Dumont JM, Nicolas-Métral V, Marfurt J, Novaroli L. An evaluation of the cyclophilin inhibitor Debio 025 and its potential as a treatment for chronic hepatitis C. Expert Opin Investig Drugs. 2009;18:211–220. doi: 10.1517/13543780802651583. [DOI] [PubMed] [Google Scholar]
- 37.Watashi K, Ishii N, Hijikata M, Inoue D, Murata T, Miyanari Y, Shimotohno K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol Cell. 2005;19:111–122. doi: 10.1016/j.molcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 38.Yang F, Robotham JM, Nelson HB, Irsigler A, Kenworthy R, Tang H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J Virol. 2008;82:5269–5278. doi: 10.1128/JVI.02614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lohmann V, Körner F, Herian U, Bartenschlager R. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J Virol. 1997;71:8416–8428. doi: 10.1128/jvi.71.11.8416-8428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergsma DJ, Eder C, Gross M, Kersten H, Sylvester D, Appelbaum E, Cusimano D, Livi GP, McLaughlin MM, Kasyan K, Porter TG, Silverman C, Dunnington D, Hand A, Prichett WP, Bossard MJ, Brandt M, Levy MA. The cyclophilin multigene family of peptidyl-prolyl isomerases. Characterization of three separate human isoforms. J Biol Chem. 1991;266:23204–23214. [PubMed] [Google Scholar]
- 41.Nakagawa M, Sakamoto N, Tanabe Y, Koyama T, Itsui Y, Takeda Y, Chen CH, Kakinuma S, Oooka S, Maekawa S, Enomoto N, Watanabe M. Suppression of hepatitis C virus replication by cyclosporin A is mediated by blockade of cyclophilins. Gastroenterology. 2005;129:1031–1041. doi: 10.1053/j.gastro.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 42.Daum S, Erdmann F, Fischer G, Féaux de Lacroix B, Hessamian-Alinejad A, Houben S, Frank W, Braun M. Aryl indanyl ketones: efficient inhibitors of the human peptidyl prolyl cis/trans isomerase Pin1. Angew Chem Int Ed Engl. 2006;45:7454–7458. doi: 10.1002/anie.200601569. [DOI] [PubMed] [Google Scholar]
- 43.Braun M, Hessamian-Alinejad A, de Lacroix BF, Alvarez BH, Fischer G. Novel spiroannulated 3-benzofuranones. Synthesis and inhibition of the human peptidyl prolyl cis/trans isomerase Pin1. Molecules. 2008;13:995–1003. doi: 10.3390/molecules13040995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Erdmann F, Baumgrass R, Schutkowski M, Fischer G. Unexpected side chain effects at residue 8 of cyclosporin a derivatives allow photoswitching of immunosuppression. J Biol Chem. 2005;280:4842–4850. doi: 10.1074/jbc.M409178200. [DOI] [PubMed] [Google Scholar]
- 45.Fanghänel J, Fischer G. Thermodynamic characterization of the interaction of human cyclophilin 18 with cyclosporin A. Biophys Chem. 2003;100:351–366. doi: 10.1016/s0301-4622(02)00292-2. [DOI] [PubMed] [Google Scholar]
- 46.Janowski B, Wöllner S, Schutkowski M, Fischer G. A protease-free assay for peptidyl prolyl cis/trans isomerases using standard peptide substrates. Anal Biochem. 1997;252:299–307. doi: 10.1006/abio.1997.2330. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Jiang J, Richardson PL, Reddy RD, Johnson DD, Kati WM. A fluorescence polarization-based assay for peptidyl prolyl cis/trans isomerase cyclophilin A. Anal Biochem. 2006;356:100–107. doi: 10.1016/j.ab.2006.04.040. [DOI] [PubMed] [Google Scholar]
- 48.Wang ZX. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 1995;360:111–114. doi: 10.1016/0014-5793(95)00062-e. [DOI] [PubMed] [Google Scholar]
- 49.Zhang YL, Zhang ZY. Low-affinity binding determined by titration calorimetry using a high-affinity coupling ligand: a thermodynamic study of ligand binding to protein tyrosine phosphatase 1B. Anal Biochem. 1998;261:139–148. doi: 10.1006/abio.1998.2738. [DOI] [PubMed] [Google Scholar]
- 50.Kallen J, Spitzfaden C, Zurini MG, Wider G, Widmer H, Wüthrich K, Walkinshaw MD. Structure of human cyclophilin and its binding site for cyclosporin A determined by X-ray crystallography and NMR spectroscopy. Nature. 1991;353:276–279. doi: 10.1038/353276a0. [DOI] [PubMed] [Google Scholar]
- 51.Klebe G, Böhm HJ. Energetic and entropic factors determining binding affinity in protein-ligand complexes. J Recept Signal Transduct Res. 1997;17:459–473. doi: 10.3109/10799899709036621. [DOI] [PubMed] [Google Scholar]
- 52.Damsker JM, Bukrinsky MI, Constant SL. Preferential chemotaxis of activated human CD4+ T cells by extracellular cyclophilin A. J Leukoc Biol. 2007;82:613–618. doi: 10.1189/jlb.0506317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang XJ, Etzkorn FA. Peptidyl-prolyl isomerase inhibitors. Biopolymers. 2006;84:125–146. doi: 10.1002/bip.20240. [DOI] [PubMed] [Google Scholar]
- 54.Hart SA, Etzkorn FA. Cyclophilin Inhibition by a (Z)-Alkene cis-Proline Mimic. J Org Chem. 1999;64:2998–2999. doi: 10.1021/jo990409a. [DOI] [PubMed] [Google Scholar]
- 55.Wang HC, Kim K, Bakhtiar R, Germanas JP. Structure-activity studies of ground- and transition-state analogue inhibitors of cyclophilin. J Med Chem. 2001;44:2593–2600. doi: 10.1021/jm010009r. [DOI] [PubMed] [Google Scholar]
- 56.Demange L, Moutiez M, Dugave C. Synthesis and evaluation of Glypsi(PO(2)R-N)Pro-containing pseudopeptides as novel inhibitors of the human cyclophilin hCyp-18. J Med Chem. 2002;45:3928–3933. doi: 10.1021/jm020865i. [DOI] [PubMed] [Google Scholar]
- 57.Fanghänel J, Fischer G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front Biosci. 2004;9:3453–3478. doi: 10.2741/1494. [DOI] [PubMed] [Google Scholar]
- 58.Mikol V, Kallen J, Walkinshaw MD. X-ray structure of a cyclophilin B/cyclosporin complex: comparison with cyclophilin A and delineation of its calcineurin-binding domain. Proc Natl Acad Sci U S A. 1994;91:5183–5186. doi: 10.1073/pnas.91.11.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Price ER, Zydowsky LD, Jin MJ, Baker CH, McKeon FD, Walsh CT. Human cyclophilin B: a second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc Natl Acad Sci U S A. 1991;88:1903–1907. doi: 10.1073/pnas.88.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neri P, Gemmecker G, Zydowsky LD, Walsh CT, Fesik SW. NMR studies of [U-13C]cyclosporin A bound to human cyclophilin B. FEBS Lett. 1991;290:195–199. doi: 10.1016/0014-5793(91)81258-a. [DOI] [PubMed] [Google Scholar]
- 61.Olsson TS, Williams MA, Pitt WR, Ladbury JE. The thermodynamics of protein-ligand interaction and solvation: insights for ligand design. J Mol Biol. 2008;384:1002–1017. doi: 10.1016/j.jmb.2008.09.073. [DOI] [PubMed] [Google Scholar]
- 62.Weber PC, Wendolowski JJ, Pantoliano MW, Salemme FR. Crystallographic and thermodynamic comparison of natural and synthetic ligands bound to streptavidin. J Am Chem Soc. 1992;114:3197–3200. [Google Scholar]
- 63.Velazquez-Campoy A, Kiso Y, Freire E. The binding energetics of first and second generation HIV-1 protease inhibitors: implications for drug design. Arch Biochim Biophys. 2001;390:169–175. doi: 10.1006/abbi.2001.2333. [DOI] [PubMed] [Google Scholar]
- 64.Wildemann D, Erdmann F, Alvarez BH, Stoller G, Zhou XZ, Fanghanel J, Schutkowski M, Lu KP, Fischer G. Nanomolar inhibitors of the peptidyl prolyl cis/trans isomerase Pin1 from combinatorial peptide libraries. J Med Chem. 2006;49:2147–2150. doi: 10.1021/jm060036n. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Daum S, Wildemann D, Zhou XZ, Verdecia MA, Bowman ME, Lucke C, Hunter T, Lu KP, Fischer G, Noel JP. Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem Biol. 2007;2:320–328. doi: 10.1021/cb7000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ke H, Mayrose D, Belshaw PJ, Alberg DG, Schreiber SL, Chang ZY, Etzkorn FA, Ho S, Walsh CT. Crystal structures of cyclophilin A complexed with cyclosporin A and N-methyl-4-[(E)-2-butenyl]-4,4-dimethylthreonine cyclosporin A. Structure. 1994;15:33–44. doi: 10.1016/s0969-2126(00)00006-x. [DOI] [PubMed] [Google Scholar]
- 67.Fesik SW, Neri P, Meadows R, Olejniczak ET, Gemmecker G. A model of the cyclophilin/cyclosporin A (CSA) complex from NMR and X-ray data suggests that CSA binds as a transition-state analog. J Am Chem Soc. 1992;114:3165–3166. [Google Scholar]