

Abstract
The choroid plexus, a barrier between the blood and cerebrospinal fluid (CSF), is known to accumulate lead (Pb) and also possibly function to maintain brain’s homeostasis of Aβ, an important peptide in the etiology of Alzheimer’s disease. This study was designed to investigate if Pb exposure altered Aβ levels in the blood-CSF barrier in the choroid plexus. Rats received ip injection of 27 mg Pb/kg. Twenty-four hr later, an FAM-labeled Aβ (200 pmol) was infused into the lateral ventricle and the plexus tissues removed to quantify Aβ accumulation. Results revealed a significant increase in intracellular Aβ accumulation in the Pb-exposed animals compared to controls (p<0.001). When choroidal epithelial Z310 cells were treated with 10 μM Pb for 24h and 48h, Aβ (2 μM in culture medium) accumulation was significantly increased by 1.5 fold (p<0.05) and 1.8 fold (p<0.05), respectively. To explore the mechanism, the effect of Pb on low-density lipoprotein receptor protein-1 (LRP1), an intracellular Aβ transport protein, was examined. Following acute Pb exposure with the aforementioned dose regimen, levels of LRP1 mRNA and proteins in the choroid plexus were decreased by 35 % (p<0.05) and 31.8% (p<0.05), respectively, in comparison to those of controls. In Z310 cells exposed to 10 μM Pb for 24 h and 48 h, a 33.1% and 33.4% decrease in the protein expression of LRP1 was observed (p<0.05), respectively. Knocking down LRP1 resulted in even more substantial increases of cellular accumulation of Aβ, from 31% in cells without knockdown to 72% in cells with LRP1 knockdown (p<0.05). Taken together, these results suggest that the acute exposure to Pb results in an increased accumulation of intracellular Aβ in the choroid plexus; the effect appears to be mediated, at least in part, via suppression of LRP1 production following Pb exposure.
Keywords: blood-CSF barrier, Pb, Aβ, LRP1, choroid plexus, blood-brain barrier, Alzheimer’s disease
Introduction
Lead (Pb)-induced neurotoxicity remains a major public health concern not only in developing countries but also in developed countries (Davis et al., 1993; Dingwall-Fordyce.,1963; Goyer et al., 1993;). As an indispensable metal used in the modern industry, the demand for Pb has been steadily increasing in the last decade. Despite its occupational hazard, environmental exposure to Pb in the general population continues to be a major public health issue, as the metal is widely present in air, drinking water, household products, plastic materials, paints, and other products (Staudinger et al., 1998).
While a definite relationship between Pb exposure and the pathogenesis of Alzheimer’s disease (AD) has yet to be established, some reports have suggested that Pb may be a risk factor in AD. For example, Graves et al. (1991), by re-analyzing four case-control studies, revealed a positive correlation between Pb exposure and AD. Haraguchi et al. (2001) showed the presence of high levels of Pb in diffuse neurofibrillary tangles, a form of pre-senile dementia, in 10 AD cases compared with 9 controls. More recent studies on Pb-exposed workers demonstrate an exacerbated neurodegeneration and atrophy in brain regions that are similar to those seen in AD patients (Jiang et al., 2008; Stewart et al., 2006). Pb has long been shown to affect memory in children (Counter et al., 2005) and development in animals. For example, studies conducted on mice and non-human primates suggest that Pb exposure during early development alters the expression and regulation of amyloid precursor protein (APP) with an increased aggregation of Aβ later in life (Basha et al., 2005a, b; Wu et al., 2008) and impairs certain forms of memory (Kuhlmann et al., 1997). Thus, the linkage between Pb exposure and AD etiology, particularly in disruption of Aβ metabolism in the brain, deserves further exploration (Prince 1998; White et al., 2007).
Accumulation of Aβ in brain extracellular space is often considered one of the major hallmarks of AD pathogenesis (Ogomori et al., 1989). Increased levels of Aβ in the brains of AD patients may occur by one or more processes, including overproduction of Aβ in the brain, inadequate metabolic clearance within the brain, or a disrupted transport of Aβ into and out of brain by the brain barrier system. There are two brain barriers that separate the brain parenchyma from the blood circulation. The blood-brain barrier (BBB), which is mainly composed of tightly connected cerebral capillary endothelia, separates the blood from the brain interstitial fluid. The barrier between the blood and the cerebrospinal fluid (CSF), known as the blood–CSF barrier (BCB), is located in the choroid plexus. Since there is no barrier between brain interstitial fluid and the CSF, Aβ in the brain extracellular space can readily enter into the CSF (Brody et al., 2008). In fact, the CSF concentration ratio of two major Aβ peptides (i.e., Aβ1–40 and Aβ1–42) has been suggested as a biomarker for AD diagnosis (Kanai et al., 1998). Noticeably, Aβ 1-40, a soluble form of Aβ peptides, is the major component of diffused plaques in AD patients (Mehta et al., 2000; Seubert et al., 1992; Vigo-Pelfrey et al., 1993). The regulation of Aβ at the BBB has been previously reported (Deane et al., 2004; Donahue et al., 2006). Although Aβ has been detected in the choroid plexus of AD patients (Kalaria et al., 1997; Miklossy et al., 1999), the role of the BCB in Aβ transport and metabolism remains unclear.
Available data in literature suggest that low-density lipoprotein receptor protein 1 (LRP1) may play a role in Aβ export from the brain to the blood by crossing the BBB (Goto and Tanzi, 2002; Knauer et al., 1996; Kounnas et al., 1995; Moir and Tanzi, 2005). LRP1 is a 600 kDa trans-membrane glycoprotein and is involved in receptor-mediated endocytosis and cell signaling (Hyman et al., 2000). It is subsequently cleaved by furin into an external 515 kDa α subunit and an 85 kDa β subunit which contains a transmembrane domain and a cytoplasmic tail with two NPXY motifs. These two motifs, which contain numerous tyrosine residues (Herz et al., 2001), can interact with signaling proteins and are known to participate in clearing Aβ via the BBB (Harris-White et al., 2005; Kounnas et al., 1995). At the BBB, Aβ molecules in the brain extracellular fluid can be taken up by known transport mechanisms to brain endothelial cells (Deane et al., 2004). Within the cells, the binding of Aβ to LRP1 allows the LRP1 to carry Aβ molecules to the basolateral inner membrane facing the blood with the subsequent expelling of Aβ into the blood stream (Deane et al., 2004; Donahue et al., 2006). Interestingly, Aβ has also been shown to promote proteosome induced degradation of LRP in the endothelium of the BBB, subsequently resulting in decreased LRP expression, consistent with reduced LRP levels in both patients suffering from AD and in Aβ accumulating transgenic mice (Deane et al., 2004; Deane et al., 2008). In addition, LRP1 has been found capable of binding to Aβ ligands like apolipoprotein E, APP and α2-macroglobulin (Kang et al., 2000; Knauer et al., 1996; Kounnas et al., 1995; Uden et al., 1999; 2000) thereby promoting Aβ clearance. Thus, LRP1, by either directly transporting Aβ or binding to these important proteins in regulating Aβ homeostasis, may mediate the clearance Aβ from the brain (Herz et al., 2001). Although, LRP1 has been recently identified in the choroid plexus (Johanson et al., 2006), its role in the BCB still largely remains unknown.
Human autopsy data and animal studies from this and other laboratories have established a clear relationship between Pb exposure and the ensuing accumulation of Pb in the choroid plexus (Friedheim et al., 1983; Manton et al., 1984; O’Tuama et al., 1976; Zheng et al., 1991, 1996). Accumulation of Pb in the choroid plexus has been shown to increase the leakage of the BCB (Shi and Zheng et al., 2007) and alter the production of transthyretin (TTR) and transport of thyroxin by the BCB (Zheng et al., 1996, 2001, 2003). Previous research in our laboratory has also shown that Aβ1–40 is actively transported by the choroid plexus, predominantly from the CSF towards the blood (Crossgrove et al., 2005). Since Pb exposure results in its accumulation in the choroid plexus and since Aβ is extensively transported by this tissue, it became interesting to investigate whether Pb accumulation in this barrier tissue altered the clearance of Aβ from the CSF, which may contribute to the etiology of AD.
This study was designed to test the hypothesis that acute exposure to Pb interfered with Aβ1–40 regulation in the choroid plexus and this interference may result from Pb altering Aβ uptake and/or clearance via LRP1. An acute Pb exposure model with an intraperitoneal (ip) injection of 50 mg Pb-acetate/kg (i.e., 27 mg Pb/kg) was used because our previous studies using the same dose regimen have shown a substantial accumulation of Pb in the choroid plexus (Zheng et al., 1991). The purpose of this dose regimen was not to mimic real life exposure, but instead, to produce a condition in which the amount of Pb in the choroid plexus could build up significantly during a fairly short period of time.
Materials and Methods
Materials
Chemicals and assay kits were purchased from the following sources: FAM-labeled Aβ (catalog # 23514-01) from Anaspec (San Jose, CA), SOD assay kit (catalog # K335-100) from Biovision (Mountain View, CA), ELISA kit (catalog # KHB-3481) and ultra purified Aβ (1–40) (catalog # 03-138) from Biosource (Carlsbad, CA), rabbit anti LRP1 antibody (catalog # ARP32793) from Aviva (San Diego, CA), Alexa-labeled secondary antibody from Molecular Probes (Eugene, OR), enhanced chemiluminescene reagent (ECL) and ECL films from Amersham Biosciences (Piscataway, NJ), Dulbecco’s modified essential medium (DMEM), fetal bovine serum (FBS), penicillin and streptomycin, gentamycin from Gibco (Grand Island, NY), the PCR buffer, dNTP, Oligo dT and MuLV reverse transcriptase from Applied Biosystems (Foster City, CA), LDH assay kit (catalog # TOX-7), β-actin, dithiothreitol (DTT), 2-mercaptoethanol, phenylmethylsulfonyl fluoride (PMSF), polyacrylamide, tetramethyl-ethylenediamine (TEMED), and the siRNA for LRP1 and all other chemicals were from Sigma Chemicals (St. Louis, MO). TRIzol was purchased from Invitrogen (Carlsbad, CA), oligodT, MuLV from Applied Biosystems (Foster City, CA), the ABsolute QPCR SYBR green Mix kit from ABgene (Rochester, New York), the primers from Integrated DNA Technology Inc. (Coralville, IA), and transfection agent, lipofectamine from Ambion (Austin, TX). All reagents were of analytical grade, HPLC grade or the best available pharmaceutical grade.
Animals and treatment
Male Sprague-Dawley rats at the time they were used were 8–9 weeks old (250–300g). The animals were housed in a temperature-controlled, 12:12 light/dark room, and were allowed free access to tap water and food. Rats received an ip injection of 50 mg/kg Pb acetate (i.e., 27 mg Pb/kg) or an equivalent molar concentration of Na-acetate (i.e., 15 mg acetate/kg) as controls. Twenty-four hours post injection the rats were anesthetized with ketamine/xylazine (75:10 mg/mL, 1 mL/kg body weight), immobilized in a stereotaxic device and subjected to the following experimentation. Animal protocols pertinent to this study were approved by the Purdue University Animal Care and Use Committee.
Intraventricular perfusion of Aβ
A midline cutaneous incision was made from the head to the neck on the dorsal surface to expose the skull. A hole was drilled in the skull at co-ordinates determined using the Paxinos and Watson Atlas at 0.8 mm posterior to bregma and 1.4 mm lateral) followed by an insertion of a sterilized cannula at 3.5 mm ventral from the skull surface. An internal cannula connected to PE 50 tubing was inserted into the guide cannula for lateral ventricle perfusion. The other end of the PE tubing was attached to a 10 μL Hamilton syringe, which was filled with 200 pmoles of FAM-labeled Aβ diluted in artificial CSF (Yamada et al., 1998).
The Aβ solution was infused into the lateral ventricle at a rate of 12 μL/min for 0.5 min. The cannula was allowed to remain inside the ventricle for an additional 0.5 min before it was removed in order to avoid the back flow into the tubing. Twenty min post infusion, rats were euthanized with ketamine-xylazine and the brain was dissected to remove the choroid plexus. The time of Aβ incubation (15–20 minutes) was based on previous studies, which establishes a clearance of Aβ by the BBB within 30 minutes (Bell et al., 2007). The tissues were then transferred to a 35-mm dish and washed three times with an artificial CSF (aCSF). Aliquots (1–2 drops) of aCSF were then added on the tissue to prevent it from drying out; the tissue was observed immediately using an inverted laser scanning microscope (Olympus, FV1000) for live uptake.
Confocal immunofluorescence microscopy
To acquire images, the 35mm dish containing the choroid plexus specimen in artificial CSF was mounted on the stage of an Olympus, FV1000 inverted confocal laser-scanning microscope and viewed through a 40x water-immersion objective (numeric aperture=1.2), with a 488-nm laser line for excitation (Ar-ion laser). Low laser intensity was used to avoid photo bleaching. The choroid plexus was examined under reduced transmitted-light illumination and an area containing undamaged epithelium with underlying vasculature was selected. Each sample was imaged at a rate of one frame per second and care was taken to expose all the tissues for the same period of time at a reduced illumination setting to avoid photo bleaching. Confocal images (512×512×8 bits, 4 frames averaged) were acquired and saved to a disk. For each tissue sample, 4 areas of cells were selected for image collection.
The fluorescence intensity was further quantified using software ImageJ and reported in arbitrary units (a.u). Data reported, unless otherwise stated, are the results of single experiments representative of three to four replicate experiments.
Culture of choroidal epithelial Z310 cells
The characteristics of immortalized rat choroidal epithelial Z310 cells have been described in a previous publication (Zheng and Zhao, 2002). Briefly, cells were maintained in DMEM (high glucose) medium supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 40μg/mL of gentamycin in a humidified incubator with 95% air–5% CO2 at 37°C and were passaged twice a week.
Determination of Pb-induced cellular toxicity in Z310 cells
To choose the Pb concentration at which it altered Aβ transport by the choroid plexus but did not induce nonspecific cytotoxicity, three general cytotoxicity assays were used, including the methylthiazolyldiphenyl-tetrazolium bromide cell viability assay (MTT), cell membrane permeability assessment (lactate dehydrogenase or LDH assay), and cellular oxidative stress estimation (superoxide dismutase or SOD assay). MTT cell viability assay was performed by growing Z310 cells at a density of 40,000 cells/well for 2–3 days until they reached 80–85% confluence. The medium was then removed and replaced with fresh medium containing different concentrations of Pb as Pb-acetate (0, 5,10, 25, and 50 μM). The cells were incubated for an additional 24 h, followed by adding an aliquot of MTT stock solution (2 mg/mL in PBS) to each well. The absorbance of the converted dye was measured at a wavelength of 570 nm. To determine the LDH activity, Z310 cells were treated in the same way as described in MTT assays. An LDH assay was then conducted using a LDH assay kit as per protocol. The SOD activity was determined according to the instruction of the assay kit. The cells were treated with Pb at 0 or 10 μM for 24 h.
Detection of Aβ1–40 in Z310 Cells following Pb exposure by immunofluorescence
The effects of Pb exposure on Aβ uptake in choroidal epithelial cells were qualitatively assessed using confocal microscopy. Z310 cells were plated on 35-mm glass plates (MatTek, Ashland, MA) and grown until they reached about 85% confluence (2–3 days). The cells were treated with 10 μM Pb as Pb acetate for 24 h in DMEM medium. Following exposure, the cells were washed 3 times with phosphate buffered saline (PBS) to remove all remnants of Pb in the dish. Cells were then incubated with 200 μL of 2 μM fluorescent labeled Aβ for one hour followed by three PBS washes. Fresh cell culture medium was then added and the preparations were observed under a confocal microscope.
Quantification of Aβ1–40 accumulation in Z310 cells following Pb exposure by ELISA
Intracellular accumulation of Aβ in Z310 cells following Pb exposure was further quantified using a well established enzyme linked immunosorbent assay (ELISA) After Pb exposure at 10 μM for 4, 12, 24 or 48 h, Z310 cells were washed with PBS and incubated with 200 μL of a 2 μM solution of unlabeled ultrapure Aβ1–40 in serum free medium for 1 h. Cells were washed 3 times with PBS to remove excess Aβ, collected and sonicated to lyse the cells. The cell lysates were diluted 3:1 with diluent buffer (as per manufacturer’s instructions) before adding them to the ELISA plates. Aβ1–40 colorimetric kit (Invitrogen KHB3481) was used to determine the concentrations of Aβ1–40 in the cell lysates. This assay kit detects monomeric Aβ1–40 since this form is more likely to be transported across the barrier rather than the aggregated form. An aliquot of cell lysates was used to quantify the protein concentrations by the Bradford assay to normalize the Aβ1–40 to total protein.
Quantification of LRP1 mRNA expression by real-time RT-PCR
The transcription of the gene encoding LRP1 was quantified using real-time RT-PCR as described by Walker et al., (2001). Briefly, total RNA was isolated from Z310 cells or choroid plexus tissue using TRIzol reagent following the manufacturer’s directions. An aliquot of RNA (1 μg) was reverse-transcribed with MuLV reverse-transcriptase and oligo dT primers. The forward and reverse primers for target genes were designed using Primer Express 3.0 software. The ABsolute QPCR SYBR green Mix kit (ABgene, Rochester, New York) was used for real-time RT-PCR analyses. The amplification was carried out in the MX 3000P real-time PCR System (Stratagene, La Jolla, CA). Amplification conditions were 15 min at 95 °C, followed by 40 cycles of 30 s at 95 °C, 1 min at 55 °C and 30 s at 72 °C. A dissociation curve was used to verify that the majority of fluorescence detected could be attributed to the labeling of specific PCR products, and to verify the absence of primer-dimers and sample contamination.
All real time RT-PCR reactions were done in triplicate. Primers sequences for rat LRP1 used for real-time RT-PCR were: forward primer 5′-TTGTGCTGAGCCAAGACATC -3′ and a reverse primer 5′GGCGTGGAAGACATGTAGGT -3′ (Genbank Accession No XM_243524) and rat glyceraldehydes-3-phosphate dehydrogenase (GAPDH), used as an internal control, had a forward primer 5′-CCT GGA GAA ACC TGC CAA GTA T-3′ and a reverse primer 5′-AGC CCA GGA TGC CCT TTA GT-3′ (Genbank Accession No. NM_017008).
Quantification of LRP1 protein expression by Western blot
The choroid plexus tissues or Z310 cells were homogenized (1:10, wt/vol) on ice in a buffer containing 20 mM Tris (pH 7.5), 5 mM EGTA, 1% TritonX-100, 0.1% SDS, 50μM phenylmethylsulphonylfluoride (PMSF), 15 mM 2-mercaptoethanol and a Protease Inhibitor Cocktail containing 500 μM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), 150 nM aprotinin, 1 μM E-64, 0.5 mM EDTA, 1 μM leupeptin (Calbiochem, San Diego, CA). Samples were sonicated using a Model 500 Sonic Dismembrator (Fisher Scientific) at duty cycle 20% and output 4–6 for 30 pulses. Following centrifugation at 10,000 g at 4°C for 10 min, aliquots of supernatants were assayed for protein concentrations by the Bradford method. A volume of protein extract (40 μg of protein) was mixed with an equal volume of 2X sample buffer (0.35 M Tris-Cl, 10% SDS, 30% glycerol, 0.6 M DTT, and 0.012% bromophenol blue), loaded onto a 10% SDS-polyacrylamide gel, electrophoresed, and then transferred to a PVDF membrane. The membrane was blocked with 5% dry milk in TBST (Tris-buffered saline) at room temperature for 1 h and immunoblotted with an antibody directly against LRP1(1:250). This antibody, purchased from Aviva Systems Biology (accession number Q6PJ72), identifies the swissprot ID of LRP1 at 48 kD. The membrane was stained with a horse-radish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (1:5000) at room temperature for 1 h and developed using ECL reagent and films. The exposure time varied from 30 sec to several min depending on signal strength. β-actin (42 kD) (1:2000) was used as an loading control; the corresponding secondary antibody (1:2000) for β-actin was HRP-conjugated goat anti-mouse IgG. Band intensities were quantified using Scion Image software (Frederick, Maryland) and results were reported as a ratio of LRP1 to β-actin in the tissue or cells.
Aβ accumulation following LRP1 knockdown by RNAi
siRNA Transfection was performed as follows: Candidate sequences for LRP1 knockdown were obtained commercially from Sigma-Aldrich. The sequences used were- Forward primer: 5′ CCUAUCUUUGAGAUCCGAA 3′; reverse Primer: 5′ UUCGGAUCUCAAAGAUAGG 3. Transfection agent lipofectamine was found to work best in the Z310 cells after a series of screening. Transfection conditions were optimized according to the following variables” initial seeding density, volume of transfection agent, duration of transfection, concentration of siRNA.
Cells were seeded at a density of 1× 105 cells/well in a 6- well plate in cell culture medium. After 24 hours, the RNA/transfection system was prepared as follows: 1 ul lipofectamine was diluted in 100 uL OPTI-MEM 1 medium and incubated for 10 minutes at room temperature. siRNA was added to a separate 100 uL OPTI- MEM medium to obtain a final concentration of 50 nM per dish. The transfection agent and the siRNA were then mixed and incubated at room temperature for 45 minutes with occasional mixing. Cell culture medium was replaced with 200ul of the above mixture along with 800 uL of OPTI-MEM medium to obtain a total of 1 ml medium/well for 5 hours. An additional 1 ml of regular cell culture medium was added and the cells were grown for an additional 48 hours. The cells were transfected with either scrambled siRNA as a negative control or the siRNA sequence designed homologous to LRP1. A negative control was used to demonstrate that there was no non-specific toxicity caused by the transfection agent Knockdown was then analyzed by laser scanning cytometry, real time RT PCR and western blot analysis. Cells were exposed to Pb at 10 μM for 24 hours. Aβ uptake studies were performed as described earlier using ELISA and intracellular Aβ was normalized to total protein. The intracellular Aβ levels were normalized by the cellular total protein concentrations.
Statistical analysis
Statistical analyses of the differences between groups were carried out by a one-way ANOVA with post hoc comparisons by the Dunnett’s test or using paired t–tests (Kaleidagraph 3.6) and by using SPSS (version 30.0) to determine correlation coefficients All data are expressed as mean ± SD. Differences between two means were considered significant when p was equal or less than 0.05.
Results
Increase in accumulation of intracellular Aβ1–40 in rat choroid plexus tissue following acute Pb exposure
In the current study, rats received a single ip injection of 50 mg/kg of Pb acetate (27 mg/kg Pb). The purpose of this dose regimen was to produce a significant buildup of Pb in the choroid plexus during a short period of time, as our previous study has shown that the same dose regimen produces a marked accumulation of Pb in the choroid plexus (5.8 μg/g of tissue) that is 12-fold greater than in the brain cortex (Zheng et al., 1991). Following an intraventricular infusion of FAM-labeled Aβ1–40, the fluorescent signals were evident in choroid plexus tissues (Fig. 1A); this observation confirmed our previous report that the choroid plexus possesses the capacity to acquire Aβ from the CSF (Crossgrove et al., 2005). Remarkably, Pb-treated animals showed much more abundant fluorescent labels in plexus tissues than did the controls (Fig. 1B). Moreover, the Aβ1-40 derived signals appeared to be mainly distributed in the choroidal epithelial cytoplasm but not within the nuclei in both control and Pb exposed groups. Quantification of the fluorescence by laser scanning cytometry using software Image J revealed a highly significant difference between the Pb-exposed group (342.3±49.4) and the controls (119.1 ±31.9) (p<0.001) (Fig. 1C). Fluorescence was expressed in arbitrary units (a.u).
Fig. 1.

Increased accumulation of intracellular Aβ in rat choroid plexus tissues following in vivo acute Pb exposure by confocal study. (A). Choroid plexus tissue from a control rat. (B). Rats received a single ip injection of 27 mg Pb/kg. Twenty-four hr post exposure, FAM-labeled Aβ1–40 was infused into brain ventricles for 0.5 min. The plexus tissues were then removed 20 min post infusion for the confocal study. Substantial stains were evident in the cytosol but not in nuclei of choroidal epithelia. The lower panel shows the corresponding transmission image, indicating normal morphology of plexus tissues. (C). Quantification of the fluorescent signals using Laser Scanning Cytometry. Data represent mean ± SD, n=16 (a total of 16 cells per group taken from 4 tissue samples with fluorescence averaged from 4 cells per sample). **: p<0.001 as compared to controls.
Cytotoxicity assays to determine the concentration of Pb in Z310 cells
To ensure that the Pb effect on Aβ in our subsequent in vitro studies was not due to a direct Pb-induced cytotoxicity, we first set out to screen an appropriate Pb concentration. As shown in Table 1, the MTT cell viability assay with Pb concentrations ranging between 0–50 μM revealed that exposure with 10 μM Pb yielded 96.3 % viable cells, which was not significantly different from controls. The LDH assays revealed that Pb concentrations at or below 10 μM had no significant effect on LDH release. Results from the SOD assay further showed that there was no significant oxidative stress generated by Pb at 10 μM. Based on these findings, along with previously published data from this group (Shi et al., 2007), a concentration of 10 μM Pb was chosen for the following Aβ studies.
Table 1.
Cytotoxicity test of Z310 cells following Pb exposure
| Pb (μM) | MTT Cell Viability | LDH | SOD |
|---|---|---|---|
| 0 | 1.00 ± 0.037 | 1.00 ± 0.234 | 1.00 ±0.059 |
| 5 | 0.963 ± 0.046 | 0.951 ± 0.251 | - |
| 10 | 0.944 ± 0.102 | 1.172 ±0.35 | 0.967 ± 0.058 |
| 25 | 0.888 ± 0.111 | 1.174 ±0.262 | - |
| 50 | 0.768 ± 0.04* | 1.63 ±0.312* | - |
The cells were treated with 0–50 μM Pb for 24 hr and tested for cell viability (MTT assay), membrane permeability (LDH assay) and oxidative stress (SOD assay). Data represent means ± SD, n = 5–8 as a ratio of control.
p<0.05 as compared to controls.
Increase in accumulation of intracellular Aβ1–40 in Z310 cells following acute Pb exposure
Prior to testing Aβ accumulation following Pb exposure in Z310 cells, a MTT cell viability assay was also performed to test the toxicity of Aβ to Z310 cells. Results revealed 90% viability in cells exposed to 2 μM Aβ for 1 h compared with untreated cells indicating that Aβ at this concentration was not toxic to the cells. Z310 cells were incubated with 2 μM Aβ in culture medium for 1 h and observed for Aβ derived fluorescent signals within the cells. Data in Fig. 2A demonstrated that normal Z310 cells had the capacity to take up Aβ. After the cells were exposed to 10 μM Pb for 24 h or 48 h, followed by incubation with 2 μM Aβ in the culture medium for another one hour, a stronger Aβ signal in the Pb-treated cells was observed when compared to the controls at 24 h (Fig. 2B). The elevation in Aβ signals persisted at 48 h following Pb treatment (Fig. 2C). Similar to results obtained from in vivo studies, the Aβ signals were primarily localized in the cytosol but not in nuclei.
Fig. 2.
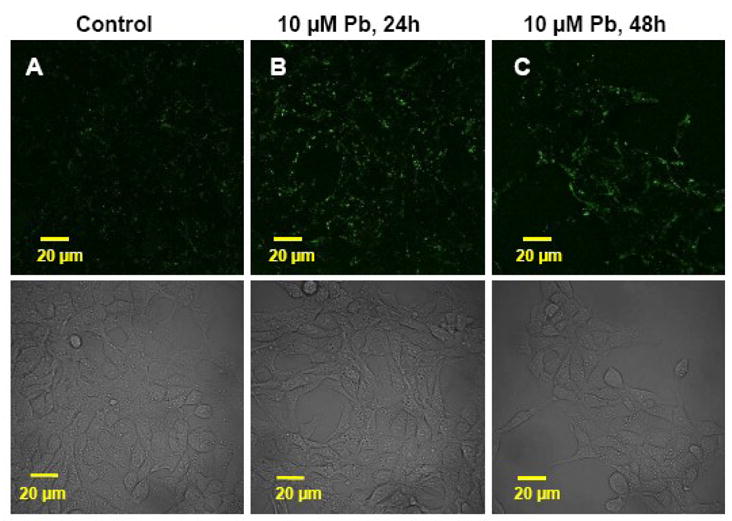
Increased accumulation of Aβ in immortalized Z310 cells following in vitro Pb exposure by confocal study. (A). Control cells. (B). Cells were incubated with 10 uM Pb for 24 hr. (C). Cells were incubated with 10 μM Pb for 48 hr. An increase in intracellular Aβ signals was evident in Pb-exposed cells as compared to controls. The lower panel shows the corresponding transmission image, indicating a normal morphology of Z310 cells.
The dose-time dependence of Pb on Aβ accumulation in Z310 cells was further investigated using an established ELISA assay. Following exposure to 0, 1, 2.5, 5, or 10 μM of Pb for 24 h, the amount of Aβ in Z310 cells appeared to be increased with increasing Pb concentrations in the range of 1–10 μM, except for Pb concentration at 2.5 μM (Fig. 3A). Exposure to 10 μM Pb produced a statistically significant increase of Aβ in the Z310 cells (r = 0.49, p<0.05). A time-course study showed a continuous increase in intracellular Aβ after the cells were exposed to 10 μM Pb for 4, 12, or 24 h (Fig. 3B). Even at 48 h, Aβ accumulation in Z310 cells was significant, about 1.8 fold higher as compared to control (r = 0.9, p<0.05).
Fig. 3.

Increases in accumulation of intracellular Aβ in Z310 cells following Pb exposure as quantified by ELISA. (A). Dose-response study. Cells were treated with Pb at the concentrations indicated for 24 hr. The cells were harvested and homogenates used for ELSIA. The value in parenthesis was excluded from the linear regression analysis. (B). Time-course study. Cells were treated with 10 μM for 4–24 h, followed by ELISA. Data represent mean ± SD, n = 4–6 wells per group. *: p<0.05.
LRP1 mRNA expression was decreased in vivo and in vitro following acute Pb exposure
To examine the mechanism by which Pb exposure retained intracellular Aβ, we quantified the mRNA expression of LRP1 as the consequence of Pb exposure by real time RT –PCR. The amount of mRNA was normalized using the housekeeping gene, GAPDH. Following in vivo Pb exposure (27mg/kg i.p), a significant decrease of LRP1 mRNA expression (−31.8%) in the choroid plexus was observed in comparison to controls (p<0.05, Fig 4A). In vitro exposure of Z310 cells to Pb at 10 μM for 24h also resulted in a significant reduction (−41.1%) in LRP1 mRNA expression as compared to controls (p< 0.05, Fig 4B). This reduction in LRP1 mRNA persisted even at 48 h after Pb exposure (p<0.05).
Fig. 4.

Decreased LRP1 mRNA expression following in vivo or in vitro Pb exposure. (A). Rats received ip injection of either Na-acetate (control) or Pb acetate (27 mg Pb/kg) and tissues were analyzed 24h after Pb exposure. (B). Z310 cells were treated with 10 μM Pb for 24h and 48 h. The relative mRNA levels of LRP1 and GAPDH were quantified by real-time RT-PCR and expressed as the ratio of LRP1/GAPDH. Data represent mean ± SD, n=4; *: p<0.05 as compared to control. The data are representative of triplicate experiments.
LRP1 protein expression was decreased following in vivo or in vitro Pb exposure
Western blot analysis by antibodies against LRP1 was used to quantify the effect of acute Pb exposure on LRP1 protein expression. Our results demonstrated that the protein concentrations of LRP1 in the choroid plexus were significantly lower in rats receiving acute Pb exposure (27mg Pb/kg ip for 24 hr) than those in controls (−35%) (p<0.05, Fig 5A, B). In vitro exposure of Z310 cells to 10 μM Pb for 24h and 48h, revealed a significant decrease of 33.1 % and 33.4% respectively in LRP1 protein expression compared to controls (p<0.05, Fig 5C, D).
Fig. 5.

Decreased LRP1 protein expression following in vivo or in vitro Pb exposure by Western blot analysis. (A) and (B): In vivo study. Rats received ip injection of either Na-acetate (control) or Pb acetate (27 mg Pb/kg) and tissues were analyzed 24h after Pb exposure. Data presented in (B) were estimated from the corresponding band densities in (A) and normalized to those of β-actin. (C) and (D): In vitro study. Z310 cells were treated with 10 μM Pb for 24h or 48 h. Data presented in (D) were estimated from the corresponding band densities in (C) and normalized to those of β-actin. Data represent mean ± SD, n=4; p<0.05 compared to controls.
Intracellular Aβ1–40 accumulation in Z310 cells was increased following LRP1 knockdown by siRNA
To test whether the accumulation of Aβ following Pb exposure was indeed mediated by LRP1 in the BCB, we assessed Aβ accumulation in Z310 cells following LRP1 knockdown by siRNA. The system was first optimized using real time RT-PCR, Western blot and laser scanning cytometry to determine an optimum concentration of the LRP1 siRNA which produced a knockdown of LRP1 but not affected cell’s viability. As shown in data presented in Fig. 6, introducing LRP1 siRNA to the cells caused a significant reduction of LRP1 at both mRNA (−53%) and protein (−52%) expression levels as compared to the scrambled siRNA controls (Fig. 6A, B). Confocal microscopic study coupled with laser scanning cytometry further revealed a significant reduction in fluorescent signals corresponding to LRP1 expression in LRP1 knockdown cells as compared to control cells (Fig. 6C, D). Noticeably also, LRP1 knockdown did not visibly alter the confluence or morphology of the cells, suggesting that the siRNA treatment did not produce nonspecific cell death or changes in morphology.
Fig. 6.

Optimization of LRP1 siRNA. Z310 cells were cultured with 50 nM siRNA for 48h and LRP1 knockdown was verified by three independent methods. (A). Representative bands from a Western Blot indicating a 52% knockdown in LRP1 protein expression compared to controls. (B). Reduced LRP1 mRNA expression by real-time RT-PCR analysis. There was a 53% knockdown in LRP1 mRNA. (C). Representative image from Laser scanning cytometry analysis. (D). Quantitative fluorescence intensity. LRP1(−) indicates LRP1 knockdown and control indicates scrambled siRNA There was a 50% reduction in LRP1-related fluorescent signal in the LRP1(−) group compared to controls. No evident difference in cell confluence was observed between the control and LRP1-knockdown groups. Data represent mean ± SD, n=4–6; *: p<0.05 as compared to controls. The data are representative of triplicate experiments.
Following optimization of LRP1 siRNA, the Z310 cells were then divided into four groups (Fig. 7): scrambled siRNA control without Pb treatment (Column 1), scrambled siRNA control with Pb (10 μM) exposure for 24 h (Column 2), LRP1 knockdown control without Pb treatment (Column 3), and LRP1 knockdown cells with Pb (10 μM) exposure for 24h (Column 4). Aβ accumulation was subsequently quantified as before using ELISA. Results revealed a 31% increase (p <0.01) in the Pb exposed group (no LRP1 knockdown) compared to controls as we had seen previously (Column 1 vs. 2 in Fig. 7), thus confirming the finding that Pb exposure increased cellular accumulation of Aβ. After LRP1 was knocked down and followed by Pb exposure, intracellular Aβ was further significantly increased in the Pb treated, LRP1 knocked-down cells compared to LRP1 knocked down, no Pb-treated control cells (Column 3 vs. 4 in Fig. 7, p<0.001). Pb exposure in LRP1-knocked down cells led to an accumulation of even more Aβ than did those control cells with normal LRP1 expression yet with similar Pb exposure (Column 2 vs. 4 in Fig. 7, p<0.05). These results support the hypothesis that increased accumulation in intracellular Aβ following Pb exposure may be mediated at least in part by Pb effect on LRP1.
Fig. 7.

Quantification of Aβ concentrations by ELISA. A plus sign (+) for LRP1(−) (knockdown) indicates the cells with LRP1 knockdown; a minus sign (−) for LRP1(−) indicates the control cells with scrambled siRNA. Following recovery from the transfection of LRP1 or scrambled siRNA, cells were incubated with the media in presence of 10 μM Pb (with a plus sign) or in absence of Pb (with a minus sign) for 24 h, followed by incubation with 2 μM Aβ for 1 h. Data represent mean ± SD, n = 4–6. Bars with different superscripts are significantly different from one another, p<0.05.
Discussion
Our data clearly demonstrate that exposure to Pb results in a significantly increased accumulaton of Aβ1-40 in rat choroid plexus tissue in vivo and in immortalized choroidal epithelial Z310 cells in vitro. This effect appears to be mediated at least in part, by Pb inhibiting the production of LRP1, a protein implicated in the clearance of Aβ at the blood brain barrier (Deane et al., 2003; Donahue et al., 2006).
Pb has previously been shown to accumulate in the choroid plexus of humans (Friedheim et al., 1983; Manton et al., 1984) as well as animals (O’Tuama et al., 1976; Zheng et al., 1991; 1996), suggesting that the choroid plexus is a primary target for Pb toxicity following environmental Pb exposure. Our current findings further suggest that upon entering the tissue, Pb ions are not simply sequestered in the choroid plexus, but rather act on critical cellular regulatory mechanisms that mediate Aβ clearance by this barrier structure.
The effect of Pb on Aβ accumulation in the choroid plexus shows the following characteristics: (i) occurred in a relatively short time frame, (ii) a dependence on Pb concentration, (iii) somewhat selective to Aβ regulation in the plexus rather than a consequence of nonselective Pb cytotoxicity, and (iv) a close association with a reduced LRP1 production. A significant increase in Aβ accumulation in the choroid plexus occurred 24 hr following acute in vivo Pb exposure. Our previous studies demonstrate that under such a dose regimen, the blood Pb concentration reaches above 40 μg/dL, nearly 4 times above the current reference level of 10 μg/dL, and Pb concentration in the choroid plexus is about 22 μg/g of tissue wet weight, nearly 57 fold greater than Pb in brain cortex (Zheng et al., 1991). Remarkably, incubation of Z310 cells with 1–10 μM of Pb (about 20.7–207 μg/dL in the culture medium) produced a dose-time dependent increase in cellular accumulation of Aβ while not causing significant cell damage (i.e., normal cell viability, normal LDH and normal SOD). Hence, it seemed likely and even probable that Pb action on Aβ accumulation in the choroid plexus may be due to the selective effect of Pb on regulatory processes that mediate Aβ homeostasis in the choroid plexus.
Several mechanisms may lead to an increased Aβ level at the blood-CSF barrier: (1) a diminished expulsion of Aβ molecules from the plexus cells to the extracellular milieu, (2) an increased uptake of Aβ from the CSF, blood or both, (3) an increased synthesis of Aβ, and/or (4) a reduced metabolism or degradation of Aβ. The gene encoding LRP1 contains an Sp1-rich domain in its promoter region (Dawson et al., 1988). Pb is known to alter binding of Sp1 to its targeted DNA sequences (Zawia et al., 2003, 2000). Thus, there was reason to speculate that Pb, by interfering with Sp1 binding capacity, may interfere with gene expression of LRP1. Since extensive studies have revealed a critical role of LRP1 in expelling Aβ molecules out of the cells at the BBB (Kang et al., 2000; Hyman et al., 2000; Shibata et al., 2000), we sought to explore the impact of Pb exposure on the LRP1-mediated Aβ-clearance pathway at the BCB. The data presented in this report indicated that exposure to Pb, either in vivo or in vitro, could lead to a substantial decrease in mRNA and protein levels of LRP1 in the choroid plexus. Conceivably, a decreased LRP1 may result in a functional deficit for the choroidal epithelial cells to expel Aβ molecules from the cells and therewith an increased cellular accumulation of Aβ.
To further confirm this point, we conducted LRP1 knockdown experiments using siRNA technique. The results revealed that knocking down LRP1 in conjunction with Pb exposure exacerbated the intracellular accumulation of Aβ. Hence, it became evident that LRP1 played an important role in transporting Aβ out of the choroidal epithelial cells, and this process could be altered by Pb exposure. It is noteworthy that LRP levels are known to be significantly reduced in the mid-frontal cortex of AD patients in comparison to those of healthy age-matched control subjects (Kang et al., 2000). Contrary to what we anticipated, the LRP1 knockdown group without Pb exposure did not show an increase in Aβ accumulation. We speculate that this could be due to the partial knockdown, but not complete knockout, of LRP1 proteins. Also possible is that knocking down LRP1 expression may produce an early compensatory response, leading to a decreased Aβ uptake in the early stage. Exposure to Pb perhaps increases intracellular Aβ by a combination of its effect on LRP1 expression, shown in this report, and its action on other mechanisms including alterations in Aβ uptake. Further studies are thus needed to demonstrate the mechanism.
Our results present a number of interesting questions. First, what is the mechanism by which Pb inhibits the production of LRP1 in the choroid plexus? As mentioned above, the LRP1 gene contains a Sp1 binding domain in one of the three repeat sequences necessary for its transcription (Dawson et al., 1988; Sudhof et al., 1987). Evidence in literature supports a binding of Pb to transcription factor Sp1 (Atkins et al., 2003; Zawia et al., 1998, 2000). Since Sp1 DNA-binding is suggestively regulated by PKC (Atkins et al., 2003) and since Pb can activate PKC (Markovac and Goldstein, 1988; Murakami et al., 1987; Zhao et al., 1998), we postulate that a reduced production of LRP1 following Pb exposure may be the direct and/or indirect result of Pb interacting with PKC-Sp1-DNA regulatory pathway. Research to explore this pathway is currently in progress in this laboratory.
Second, is LRP1 the only pathway affected by Pb exposure? We should point out that this study does not imply nor is apt for the theory that LRP1 is the single most important factor in Aβ regulation at the BCB. Other mechanisms, particularly receptor-mediated Aβ uptake and clearance in the choroid plexus, must be considered. Uniquely, the choroidal epithelial cells come into contact with two entirely different body fluid compartments, i.e., the CSF and blood. The presence of Aβ in both fluid compartments has been established (Chong et al., 2007; Mehta et al., 2000). Thus, for future studies, it is important to investigate whether and how Pb exposure may affect Aβ uptake mechanisms from either interface in the BCB.
Finally, what are the consequences of Aβ accumulation in the choroid plexus following Pb exposure? Exposure to Pb has been associated with AD-like symptoms including memory deficits and neurodegeneration, both in adult animals (Wang et al., 2007) and in humans (Shih et al., 2007; Stewart et al., 2006). It still remains unknown whether these memory deficits are due to altered Aβ homeostasis in brain. As the role of the choroid plexus in cleansing Aβ via the BCB is gradually becoming evident (Crossgrove et al., 2005), it would be of interest to determine the levels of Aβ in the CSF and brain tissues as well as the ensuing pathogenic plaques in brain, once the choroid plexus’s function in cleansing brain Aβ is compromised following chronic Pb exposure.
In summary, our results suggest an increased Aβ accumulation in the blood-CSF barrier located in the choroid plexus after acute Pb exposure in intact animals as well as in cultured BCB cells. Pb-induced Aβ accumulation in the choroid plexus appears to be due to its inhibition of LRP1, a key intracellular Aβ transport protein in the choroid plexus. Since the choroid plexus is anatomically adjacent to the hippocampus, a region known to be involved in memory deficits in AD patients, the implication of the altered homeostasis of Aβ in the CSF due to Pb accumulation in the choroid plexus is of interest for future investigation.
Acknowledgments
This work was supported in part by NIH/National Institute of Environmental Health Sciences Grants Numbers ES008146 and ES017055.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atkins DS, Basha MR, Zawia NH. Intracellular signaling pathways involved in mediating the effects of lead on the transcription factor Sp1. International Journal of Developmental Neuroscience. 2003;21:235–244. doi: 10.1016/s0736-5748(03)00067-4. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005a;25:823–9. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha MR, Murali M, Siddiqi HK, Ghosal K, Siddiqi OK, Lashuel HA, Ge YW, Lahiri DK, Zawia NH. Lead (Pb) exposure and its effect on APP proteolysis and Aβ aggregation. The FASEB Journal express article. 2005b;10:1–16. doi: 10.1096/fj.05-4375fje. [DOI] [PubMed] [Google Scholar]
- Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. Journal of Cerebral Blood Flow & Metabolism. 2007;27:909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM. Amyloid-β dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J, Wolley D, Vijayan V, Overmann S. Morphometric effects of postnatal lead exposure on hippocampal development of the 15-day old rat. Developmental Brain Research. 1982;3:595–612. doi: 10.1016/0165-3806(82)90056-6. [DOI] [PubMed] [Google Scholar]
- Chong MS, Lim WS, Sahadevan S. Biomarkers in preclinical Alzheimer’s disease. Current Opinion in Investigational Drugs. 2006;7:600–607. [PubMed] [Google Scholar]
- Counter SA, Buchanan LH, Ortega F. Neurocognitive impairment in lead-exposed children of Andean lead glazing workers. J Occup Environ Med. 2005;47:306–12. doi: 10.1097/01.jom.0000155717.45594.65. [DOI] [PubMed] [Google Scholar]
- Crossgrove JS, Li GJ, Zheng W. The choroid plexus removes beta-amyloid from the cerebrospinal fluid. Exp Biol Med. 2005;230:771–776. doi: 10.1177/153537020523001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Elias RW, Grant LD. Current issues in human lead exposure and regulation of lead. Neurotoxicology. 1993;14:15–28. [PubMed] [Google Scholar]
- Dawson PA, Hofmann SL, van der Westhuyzen DR, Siidhof TC, Brown MS, Goldstein JL. Sterol-dependent Repression of Low Density Lipoprotein Receptor Promoter Mediated by 16-Base Pair Sequence Adjacent to Binding Site for Transcription Factor Spl. The Journal of Biological Chemistry. 1988;263:3372–3379. [PubMed] [Google Scholar]
- Deane R, Sagare A, Zlokovic BV. The role of the cell surface LRP and soluble LRP in blood-brain barrier a clearance in Alzheimer’s Disease Current Pharmaceutical Design. 2008;14:1601–1605. doi: 10.2174/138161208784705487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Yan SD, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/Amyloid β-Peptide Interaction Mediates Differential Brain Efflux of Aβ Isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Deane R, Du YS, Submamaryan R, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt A, Armstrong D, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nature Medicine. 2003;9:907–91. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Dingwall-Fordyce I, Lane RE. A following up study of lead workers. British Journal of Industrial Medicine. 1963;20:313. doi: 10.1136/oem.20.4.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue JE, Flaherty SL, Johanson CE, Duncan JA, Silverberg GD, Miller MC, Tavares R, Yang W, Wu Q, Sabo E, Hovanesian V, Stopa EG. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease Acta. Neuropathologica. 2006;112:405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- Friedheim E, Corvi C, Graziano J, Donnelli T, Breslin D. Choroid Plexus as Protective Sink for Heavy Metals? Lancet. 1983;8331:981–982. doi: 10.1016/s0140-6736(83)92099-8. [DOI] [PubMed] [Google Scholar]
- Goto JJ, Tanzi RE. The role of the low-density lipoprotein receptor-related protein (LRP1) in Alzheimer’s Abeta generation: development of a cell-based model system. J Mol Neurosci. 2002;19:37–41. doi: 10.1007/s12031-002-0008-4. [DOI] [PubMed] [Google Scholar]
- Goyer RA. Lead toxicity: current concerns. Environmental Health Perspectives. 1993;100:177–87. doi: 10.1289/ehp.93100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves AB, van Duijn CM, Chandra V, Fratiglioni L, Heyman A, Jorm AF, Kokmen E, Kondo K, Mortimer JA, Rocca WA. Occupational exposures to solvents and lead as risk factors for Alzheimer’s disease: a collaborative re-analysis of case-control studies. Int J Epidemiol. 1991;20(Suppl 2):S58–61. doi: 10.1093/ije/20.supplement_2.s58. [DOI] [PubMed] [Google Scholar]
- Haraguchi T, Ishizu H, Takehisa Y, Kawai K, Yokota O, Terada S, Tsuchiya K, Ikeda K, Morita K, Horike T, Kira S, Kuroda S. Lead content of brain tissue in diffuse neurofibrillary tangles with calcification (DNTC): the possibility of lead neurotoxicity. Neuroreport. 2002;13(1) doi: 10.1097/00001756-200112210-00006. inside back cover. (Response to Neuroreport. 2001 Dec 21;12, 3887–3890. [DOI] [PubMed] [Google Scholar]
- Harris-White ME, Frautschy SA. Low Density Lipoprotein Receptor-Related Proteins (LRPs), Alzheimer’s and Cognition. Current Drug Targets - CNS & Neurological Disorders. 2005;4:469–480. doi: 10.2174/156800705774322102. [DOI] [PubMed] [Google Scholar]
- Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. The Journal of Clinical Investigation. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Yang J, Tanaka S, Gonias SL, Mars WM, Liu Y. Tissue-type plasminogen activator acts as a cytokine that triggers intracellular signal transduction and induces Matrix Metalloproteinase-9 gene expression. The Journal of Biological Chemistry. 2009;281:2120–2127. doi: 10.1074/jbc.M504988200. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Strickland D, Rebeck GW. Role of the low-density lipoprotein receptor-related protein in β-amyloid metabolism and Alzheimer disease. Archives of Neurology. 2000;57:646–650. doi: 10.1001/archneur.57.5.646. [DOI] [PubMed] [Google Scholar]
- Jiang YM, Long LL, Zhu XY, Zheng H, Fu X, Ou SY, Wei DL, Zhou HL, Zheng W. Evidence for altered hippocampal volume and metabolites in workers occupationally exposed to lead: A study by magnetic resonance imaging and 1H magnetic resonance spectroscopy. Toxicology Letters. 2008;181:118–125. doi: 10.1016/j.toxlet.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson C, Flaherty S, Messier A, Duncan J, III, Silverberg G. Expression of the beta-amyloid transporter, LRP-1, in aging choroid plexus: implications for the CSF-brain system in NPH and Alzheimer’s disease, Oral Presentation. Cerebrospinal Fluid Research. 2006;3(Suppl 1):S29. [Google Scholar]
- Kalaria RN, Premkumar DRD, Pax AB, Cohen DL, Lieberburg I. Production and increased detection of amyloid β protein and amyloidogenic fragments in brain microvessels, meningeal vessels and choroid plexus in Alzheimer’s disease. Molecular Brain Research. 1996;35:58–68. doi: 10.1016/0169-328x(95)00180-z. [DOI] [PubMed] [Google Scholar]
- Kanai M, Matsubara E, Isoe K, Urakami K, Nakashima K, Arai H, Sasaki H, Abe K, Iwatsubo T, Kosaka T, Watanabe M, Tomidokoro Y, Shizuka M, Mizushima K, Nakamura T, Igeta Y, Ikeda Y, Amari M, Kawarabayashi T, Ishiguro K, Harigaya Y, Wakabayashi K, Okamoto K, Hirai S, Shoji M. Longitudinal study of cerebrospinal fluid levels of tau, A beta1–40, and A beta1–42(43) in Alzheimer’s disease: a study in Japan. Ann Neurol. 1998;44:17–26. doi: 10.1002/ana.410440108. [DOI] [PubMed] [Google Scholar]
- Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo1 EH. Modulation of amyloid b-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor–related protein pathway. The Journal of Clinical Investigation. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauer MF, Orlando RA, Glabe CG. Cell surface APP751 forms complexes with protease nexin 2 ligands and is internalized via the low density lipoprotein receptor-related protein (LRP) Brain Research. 1996;740:6–14. doi: 10.1016/s0006-8993(96)00711-1. [DOI] [PubMed] [Google Scholar]
- Kounnas MZ, Moir RD, Rebeck GW, Nush AI, Argraves WS, Tanzi RE, Hyman BT, Strickland DK. LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted β-amyloid precursor protein and mediates its degradation. Cell. 1995;82:331–340. doi: 10.1016/0092-8674(95)90320-8. [DOI] [PubMed] [Google Scholar]
- Kuhlmann AC, McGlothan JL, Guilarte TR. Developmental lead exposure causes spatial learning deficits in adult rats. Neuroscience Letters. 1997;233:101–104. doi: 10.1016/s0304-3940(97)00633-2. [DOI] [PubMed] [Google Scholar]
- Manton WI, Kirkpatrick JB, Cook JD. Does the choroid plexus really protect the brain from lead? Lancet ii. 1984;(8398):351. doi: 10.1016/s0140-6736(84)92719-3. [DOI] [PubMed] [Google Scholar]
- Markovac J, Goldstein GW. Picomolar concentrations of lead stimulate brain protein kinase C. Nature. 1988;334:71–73. doi: 10.1038/334071a0. [DOI] [PubMed] [Google Scholar]
- Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and Cerebrospinal Fluid Levels of Amyloid β Proteins 1–40 and 1–42 in Alzheimer Disease. Archives of Neurology. 2000;57:100–105. doi: 10.1001/archneur.57.1.100. [DOI] [PubMed] [Google Scholar]
- Miklossy J, Taddei K, Martins R, Escher G, Kraftsik R, Pillevuit O, Lepori D, Campiche M. Alzheimer Disease: Curly Fibers and Tangles in Organs Other Than Brain. Journal of Neuropathology & Experimental Neurology. 1999;58:803–814. doi: 10.1097/00005072-199908000-00003. [DOI] [PubMed] [Google Scholar]
- Moir RD, Tanzi RE. LRP-mediated clearance of Abeta is inhibited by KPI-containing isoforms of APP. Current Alzheimer Research. 2005;2:269–73. doi: 10.2174/1567205053585918. [DOI] [PubMed] [Google Scholar]
- Murakami K, Feng G, Chen SG. Inhibition of brain protein kinase C subtypes by lead. Journal of Pharmacology and Experimental Therapeutics. 1987;264:757–761. [PubMed] [Google Scholar]
- Ogomori K, Kitamoto T, Tateishi J, Sato Y, Suetsugu M, Abe M. β-Protein Amyloid Is Widely Distributed in the Central Nervous System of Patients with Alzheimer’s Disease. American Journal of Pathology. 1989;134:243–251. [PMC free article] [PubMed] [Google Scholar]
- O’Tuama LA, Kim CS, Gatzy JT, Krigman MR, Mushak P. The distribution of inorganic lead in Guinea pig brain and neural barrier tissues in control and lead-poisoned animals. Toxicology and Applied Pharmacology. 1976;36:1–9. doi: 10.1016/0041-008x(76)90021-1. [DOI] [PubMed] [Google Scholar]
- Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Brit Med J. 1978;2:1457–1459. doi: 10.1136/bmj.2.6150.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince M. Is chronic low-level lead exposure in early life an etiologic factor in Alzheimer’s disease? Epidemiology. 1998;9:618–21. [PubMed] [Google Scholar]
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- Shi LZ, Zheng W. Early lead exposure increases the leakage of the blood-cerebrospinal fluid barrier, in vitro. Human & Experimental Toxicology. 2007;26:159 – 167. doi: 10.1177/0960327107070560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih RA, Hu H, Weisskopf MG, Schwartz BS. Cumulative Lead Dose and Cognitive Function in Adults: A Review of Studies That Measured Both Blood Lead and Bone Lead. Environmental Health Perspectives. 2007;115:483–492. doi: 10.1289/ehp.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger KC, Roth VS. Occupational Lead Poisoning. American family Physician. 1998;57:1–15. [PubMed] [Google Scholar]
- Stewart WF, Schwartz BS, Davatzikos C, Shen D, Liu D, Wu X, Todd AC, Shi W, Bassett S, Youssem D. Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology. 2006;66:1476–1484. doi: 10.1212/01.wnl.0000216138.69777.15. [DOI] [PubMed] [Google Scholar]
- Uden EV, Sagara Y, Uden JV, Orlando R, Mallory M, Rockenstein E, Masliah E. A Protective Role of the Low Density Lipoprotein Receptor-related Protein against Amyloid b-Protein Toxicity. The Journal Of Biological Chemistry. 2000;275:30525–30530. doi: 10.1074/jbc.M001151200. [DOI] [PubMed] [Google Scholar]
- Uden EV, Carlson G, St George-Hyslop P, Westaway D, Orlando R, Mallory M, Rockenstein E, Masliah E. Aberrant Presenilin-1 Expression Downregulates LDL Receptor-Related Protein (LRP): Is LRP Central to Alzheimer’s Disease Pathogenesis? Molecular and Cellular Neuroscience. 1999;14:129–140. doi: 10.1006/mcne.1999.0772. [DOI] [PubMed] [Google Scholar]
- Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of beta-amyloid peptide from human cerebrospinal fluid. Journal of Neurochemistry. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- Wang M, Chen WH, Zhu DM, She JQ, Ruan DY. Effects of carbachol on lead-induced impairment of the long-term potentiation/depotentiation in rat dentate gyrus in vivo. Food and Chemical Toxicology. 2007;45:412–418. doi: 10.1016/j.fct.2006.08.025. [DOI] [PubMed] [Google Scholar]
- White LD, Cory-Slechta DA, Gilbert ME, Tiffany-Castiglioni E, Zawia NH, Virgolini M, Rossi-George A, Lasley SM, Qian YC, Basha MR. New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol. 2007;225:1–27. doi: 10.1016/j.taap.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Wu J, Basha R, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, Zawia NH. Alzheimer’s disease (AD)-Like Pathology in Aged Monkeys after Infantile Exposure to Environmental Metal Lead (Pb): Evidence for a Developmental Origin and Environmental Link for AD. The Journal of Neuroscience. 2008;28:3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Wozniak DF, Cirrito J, Cam JA, Osaka H, Bales KR, Zhuo M, Paul SM, Holtzman DM, Bu G. Increased soluble amyloid- β peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. PNAS. 2004;101:1075–80. doi: 10.1073/pnas.0305803101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawia NH, Sharan R, Brydie M, Oyama T, Crumpton T. Sp1 as a target site for metal-induced perturbations of transcriptional regulation of developmental brain gene expression. Developmental Brain Research. 1998;107:291–298. doi: 10.1016/s0165-3806(98)00023-6. [DOI] [PubMed] [Google Scholar]
- Zawia NH, Crumpton T, Brydie M, Reddy GR, RazmiAfshari M. Disruption of the zinc finger domain: a common target that underlies many of the effects of lead. Neurotoxicology. 2000;21:1–11. [PubMed] [Google Scholar]
- Zhao Q, Slavkovich V, Zheng W. Lead exposure promotes translocation of protein kinase C activities in rat choroid plexus in vitro but not in vivo. Toxicol Appl Pharmacol. 1998;149:99–106. doi: 10.1006/taap.1997.8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W. Toxicology of choroid plexus: Special reference to metal-induced neurotoxicities. Microscopy Research and Techniques. 2001;52:89–103. doi: 10.1002/1097-0029(20010101)52:1<89::AID-JEMT11>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Aschner M, Ghersi-Egea JF. Brain barrier systems: a new frontier in metal neurotoxicological research. Toxicol Applied Pharmacology. 2003;192:1–11. doi: 10.1016/s0041-008x(03)00251-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Zhao Q. The blood-CSF barrier in culture. Development of a primary culture and transepithelial transport model from choroidal epithelial cells. Methods Mol Biol. 2002;188:99–114. doi: 10.1385/1-59259-185-X:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Perry DF, Nelson DL, Aposhian HV. Protection of cerebrospinal fluid against toxic metals by the choroid plexus. FASEB J. 1991;5:2188–2193. doi: 10.1096/fasebj.5.8.1850706. [DOI] [PubMed] [Google Scholar]
- Zheng W, Shen H, Blaner WS, Zhao Q, Ren X, Graziano JH. Chronic Lead Exposure Alters Transthyretin Concentration in Rat Cerebrospinal Fluid: The Role of the Choroid Plexus. Toxicology and Applied Pharmacology. 1996;139:445–450. doi: 10.1006/taap.1996.0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Lu YM, Lu GY, Zhao Q, Cheung O, Blaner WS. Transthyretin, thyroxin, and retinol-binding protein in human cerebrospinal fluid: Effect of lead exposure. Toxicological Sciences. 2001;61:107–114. doi: 10.1093/toxsci/61.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Deane R, Redzic Z, Preston JE, Segal MB. Transport of L-[125I]Thyroxine by in-situ perfused ovine choroid plexus: Inhibition by lead exposure. J Toxicology and Environmental Health. 2003;66:435–451. doi: 10.1080/15287390306451. [DOI] [PMC free article] [PubMed] [Google Scholar]