

Abstract
Hybrid QM/MM methods combine the rigor of quantum mechanical (QM) calculations with the low computational cost of empirical molecular mechanical (MM) treatment allowing to capture dynamic properties to probe critical atomistic details of enzyme reactions. Catalysis by RNA enzymes (ribozymes) has only recently begun to be addressed with QM/MM approaches and is thus still a field under development. This review surveys methodology as well as recent advances in QM/MM applications to RNA mechanisms, including those of the HDV, hairpin, and hammerhead ribozymes, as well as the ribosome. We compare and correlate QM/MM results with those from QM and/or molecular dynamics (MD) simulations, and discuss scope and limitations with a critical eye on current shortcomings in available methodologies and computer resources. We thus hope to foster mutual appreciation and facilitate collaboration between experimentalists and theorists to jointly advance our understanding of RNA catalysis at an atomistic level.
Keywords: ab initio, DFT, transition state, barrier height, ONIOM, potential of mean force, EVB, phosphoryl transfer, ribozyme, RNA self-cleavage
1. Introduction
A full understanding of enzyme catalysis is not only attractive for fundamental reasons but it is also useful in many practical applications like enzyme engineering and drug design. Therefore, numerous studies have focused on the origin of enzyme catalysis. Many contributions to enzyme catalysis have been identified and thoroughly discussed in the literature, for example, active site preorganization, substrate strain, long-range conformational dynamics, and acid-base catalysis (1-10). Despite all efforts, many open questions still limit our understanding of the principles of the enzyme catalysis. It is quite likely that each enzyme utilizes a (different) combination of catalytic strategies. Therefore, studies of enzymatic reactions are complex and the leading structural tool, X-ray crystallography, does not provide any direct insights into the transient nature of the chemical reactions catalyzed by enzymes.
The challenge in studies of RNA catalysis is even more complex. The first catalytic RNAs (ribozymes) were discovered in the early 1980ies by Altmann and Cech (11-13). Nowadays, RNAs are known to catalyze several important reactions. For instance, ribosomes are central to cellular biology and their constituent RNAs catalyze the peptide bond transfer of protein biosynthesis. Many ribozymes catalyze internal phosphoryl transfer reactions and can be divided into larger and smaller ribozymes (with a boundary at ~200 nucleotides length). Larger ribozymes catalyze the displacement of the 3’-O from an RNA 3’,5’-phosphodiester linkage. Smaller ribozymes catalyze intramolecular reactions displacing the 5’-O from the RNA phosphodiester linkage with the 2’-OH of the adjacent nucleotide that acts as the nucleophile. In addition, from in vitro selection experiments it seems likely that RNAs could have played considerably more diverse catalytic roles in the early stages of evolution (in the RNA World (14)). Many questions in this world of RNA catalysis remain unanswered, e.g., do RNA enzymes apply the same strategies as protein enzymes to achieve chemical rate acceleration? Are RNA functional groups involved in these reactions? What is the role of metal cations in these reactions? Are the reactions concerted or stepwise and what is the sequence of events at the atomistic electronic structure level? Numerous reviews are available summarizing studies aimed to elucidate these questions, mainly using experimental approaches (8, 15-22).
Given the complexity of RNA catalysis it appears likely that theoretical (computational) approaches will prove important to complement experimental tools, promising to furnish an atomistic picture of RNA catalysis and fill some of the gaps in our present knowledge. So far only few computational studies related to RNA catalysis have appeared, and a review summarizing their results is missing. More commonplace computational studies of nucleic acids are based on two types of standard techniques that, when used appropriately, provide reliable and useful data: molecular dynamics (MD) simulations with explicit inclusion of solvent and ions, and quantum chemical calculations (QM). MD simulations are based on an atomistic description where the link between molecular structure and energy is described by classic, empirically derived, molecular mechanical (MM) potentials (the Force Field, FF). The MM force field is the main approximation of MD, which can be considered a single molecule modeling technique that analyzes thermal motions of fully solvated RNA molecules on a timescale of 10-1,000 ns with ps-scale resolution (23). The simulation outcome also critically depends on the quality of the experimentally derived starting structure. MD simulations deserve some credit for their description of, for example, the structural flexibility of RNA, the effects of base substitutions, or the distribution of monovalent ions and water molecules around an RNA. However, classic force fields do not allow for the study of chemical reactions, as bond breaking and formation is not possible. In contrast, QM methods are based on solving the Schrödinger equation, taking directly into consideration the electronic structure of a molecule and therefore allow access to chemical reactions. Unfortunately, good quality QM methods are computationally very expensive, typically limiting them to studies of small model systems in vacuo. Such calculations were for example instrumental in revealing the physical chemistry nature of nucleo base stacking and in quantifying their magnitude (24). However, QM methods cannot directly be used to study enzyme reactions as it is impossible to consider sufficiently large and complete systems.
A solution to these limitations is the application of hybrid quantum mechanical/molecular mechanical (QM/MM) methods as have been extensively used in studies of protein enzymes. In QM/MM methods, the region of interest (such as the catalytic core) is described using a reliable electronic structure method that allows investigation of all participants of the chemical reaction. The rest of the molecule is included using molecular mechanics, aiming to capture the contribution of enzyme structure and dynamics to catalysis. The first hybrid QM/MM scheme for protein enzymes was developed in the mid-1980ies by Warshel and Levitt (25). Despite much progress the use of hybrid methods is still challenging as they are based on complex approximations (such as where to cut and how to couple the QM and MM regions), making them much less routine than standard MD and QM calculations. QM/MM methods still offer a very promising tool to take a holistic view of RNA catalysis, which we will review here.
2. Promise and challenges of QM/MM
What is the main justification to apply QM/MM methods? Experiments directly measure valuable information on reaction thermodynamics and kinetics. However, information about the reaction mechanism, commonly derived from a chemical, pH, ionic strength, temperature, or pressure perturbation of the enzyme, is only indirectly accessible and often too ambiguous to unequivocally pinpoint mechanistic details (8). In this respect, theoretical calculations can provide critical direct insight, allowing for a detailed description of plausible reaction pathways.
Transition state theory (TST) (26) is a vital bridge between experimental data and theoretical calculations. The transition state (TS) represents the (ensemble of) high-energy structure(s), from which the reaction has an equal probability to proceed to either the reactant or product valleys. The kinetics of the transition of reactant to product is related to the Gibbs energy difference between the TS and the reactant, ΔG‡. The rate constant k is equal to:
| (1) |
where is the transmission coefficient (27), kB and h are Boltzmann and Planck constants, respectively, and T is thermodynamic temperature. To examine the kinetics of a reaction, it is necessary to find the TS and ΔG‡. In the classical TST, TS is identified as a stationary point at a potential energy (hyper)surface (PES), which is a saddle point of the first order, and equals one as the recrossing etc. are neglected. By PES we mean the potential energy as a function of the atomic coordinates, which obviously can be very complex for large systems, as it is determined by positions of all atoms including solvent and ions. PES relevant to the chemistry is often simplified by a reaction coordinate that is expressed by a set of several key structural parameters. However, the influence of many remaining parameters can be sufficiently significant to affect the reaction energetics.
To find the correct TS it is necessary to explore PES, which is challenging even for small molecules. Therefore, this task is often reduced to a trip along a few intuitively chosen reaction coordinates, which are chemically sound. In comparison to small molecules, the exploration of a biomolecular reaction coordinate is further complicated by the fact that the barrier height is often significantly modulated by, for example, distal parts of the biopolymer (28) or the position of ions (Fig. 1). For instance, the precise positions of magnesium ions critically influences the calculated barrier height for the modeled chemical reaction of the hepatitis delta virus (HDV) ribozyme (29). Even distant changes in biomolecular conformation can influence the catalyzed reaction through conformational restraints imposed on the active site or through changes in electrostatic surface potential. The latter is of special importance in RNA due to its highly polyanionic character. Therefore, theoretical studies of small model systems extracted from large RNA molecules are of limited relevance. A relevant molecular model of biomolecular catalysis must usually account for the entire molecule, counter ions, cofactors and ligands (if present), as well as for the water solvent.
Fig 1.
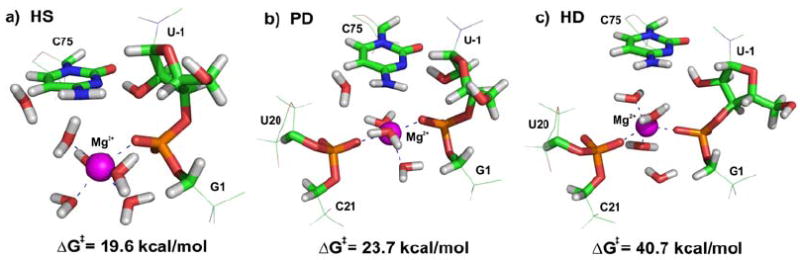
The free energy barriers of HDV ribozyme self-cleavage reaction corresponding to three different positions of Mg2+ ion: (A) hexacoordinated Mg2+ ion with one inner-shell bound phosphate, (B) pentacoordinated Mg2+ ion with two inner-shell bound phosphates, and (C) hexacoordinated Mg2+ ion with two inner-shell bound phosphates.
Furthermore, due to the complexity of a biomolecular PES and the often substantial conformational modulation of the barrier height (28, 30), a single structural snapshot might not suffice to adequately represent the conformational plasticity of a biomolecule. In principle, instead of a simple conformational search over the PES, we can apply MD simulations to a QM/MM system. However, even the shortest MD simulations drastically increase the computational expense, leading to a necessary decrease of the quality of the QM method. A possible approximate solution to this problem is to consider a wide range of possible reaction coordinates, corresponding to different microenvironments and biomolecular conformations.
Considering these challenges, a QM/MM theoretical study of biomolecular catalysis has to seek a viable compromise at three levels: (i) Construction of a suitable model that defines the sizes of the QM core and MM regions as well as how the surrounding environment is treated, including counter ions and water. (ii) Robust sampling of conformational space. (iii) Quality of the QM and MM methods used. How to achieve such a compromise is discussed in the following. We note upfront that with the presently available methods and computers all QM/MM calculations on enzymes are necessarily approximate, but can provide valuable and experimentally testable hypotheses.
3. QM/MM Methods
3.1 General considerations
Hybrid QM/MM methods divide the studied system into two (or more) parts, which are treated at different methodological levels and are allowed to communicate with each other (Fig. 2). In a typical approach, one part of the system, the QM core, describes the region where the chemical reaction takes place and is treated at the QM (ab initio or DFT), semiempirical, or Empirical Valence Bond (EVB) levels. The surrounding parts, which impose sterical and polarization constraints on the core, are treated at a less rigorous level, typically using an empirical force field. In principle, the studied system can be divided into more than two parts, which can be treated by different methods, with the largest part treated by the most approximate and computationally cheapest method (MM) and the smallest part by an accurate and expensive method (hybrid DFT or correlated ab initio level).
Fig. 2.
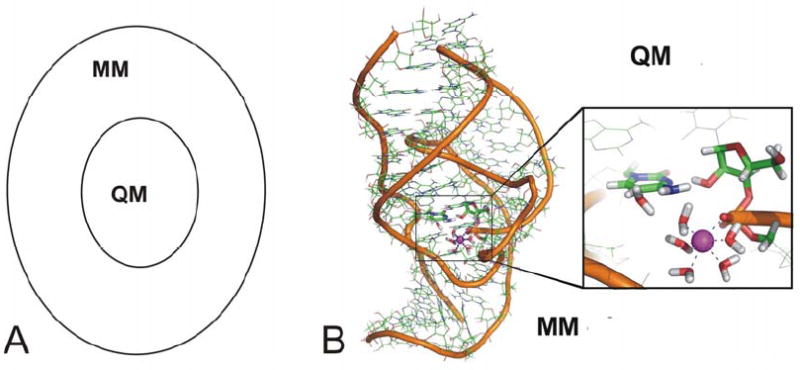
Principle of QM/MM. (A) A typical QM/MM approach divides the studied system into a QM core and an MM surrounding. (B) The MM treated part of the HDV ribozyme is shown in wire representation, the QM core is highlighted in sticks. Water molecules and counter ions of the MM part are not shown for clarity.
3.2 QM/MM Schemes
The most frequent QM/MM approach divides the system into two regions. Two alternative QM/MM schemes can be employed, known as additive and subtractive approaches. In the additive scheme, the total energy of the system is represented as a sum of the QM energy of the QM core, EQM, the MM energy of the MM region (without the QM core), EMM, and the interaction between both regions, EQM/MM:
| (2) |
The subtractive scheme (31-33) was originally called integrated molecular-orbital molecular mechanics (IMOMM) but is more frequently known as ONIOM (abbreviated from the original name: Our own N-layered Integrated molecular Orbital + molecular Mechanics) scheme. It divides the system into layers and subtracts double counted energies of the smaller layer as follows:
| (3) |
where represents energy of the whole real system (containing both the QM and MM regions) as calculated by the lower-level method, typically MM, is energy of the QM core calculated at a higher level, typically a QM method, and is the energy of the QM core calculated at the lower MM level. Subtractive and additive schemes are in principle equivalent, differing just in the technical details of their implementation.
While the choice between the additive and subtractive schemes is not critical, the description of the boundaries between the QM and MM regions as well as the interaction between them (the QM/MM coupling) is of the utmost importance. Boundary and coupling of the regions are at the heart of any QM/MM approach, but also represent its Achilles’ heel. Both of them significantly affect accuracy of the calculation. The size of the QM core and its boundaries have to be chosen carefully because the interfacial region is arbitrary. Most difficult are cases where the border between the QM and MM region cuts through covalent bonds, which is inevitable in studies of large biomolecules (enzymes). QM/MM boundaries and coupling by mechanical or electronic embedding will both be discussed further below.
We note here that many authors use the terms “additive” and “subtractive” as synonyms for electronic and mechanical embedding (see below), respectively, since the original implementation of the subtractive scheme was implemented only with mechanical embedding. However, Vreven and Morokuma have since extended their two-layer subtractive ONIOM scheme to also include electronic embedding (34-36) thus further using of these synonyms might be confusing.
3.3 QM methods
Nowadays a plethora of QM methods is available and a comprehensive description is beyond the scope of this review. Many of them have been successfully applied on RNA and DNA systems (37), and we will give a brief survey here.
3.3.1 Basic considerations and overview of standard methods
In contrast to force fields the best QM methods do not use empirical parameters, justifying the term ab initio or non-empirical methods. The best QM methods are very accurate, reaching an accuracy of ~0.5 to 1 kcal/mol for the calculated interaction energy of hydrogen bonded or stacked dimers of nucleo bases (which compares to total interaction energies of A:U and G:C Watson Crick base pairs of ~-15 and ~-30 kcal/mol, respectively (37)). Due to their computational expense they cannot be applied to QM/MM studies of enzymes but can be used, e.g., as reference methods for the uncatalyzed reaction (29). An attractive feature of ab initio QM methods is that once a certain level of quality is reached, the results start converging in a systematic manner toward the correct solution so that their accuracy can be assessed even without performing highest-level calculations. This behavior contrasts with that of empirical FFs, which can fail in an unpredictable manner when one moves away from the systems that were considered during force field parameterization (38). The threshold in ab initio QM calculations for reaching the onset of convergence (and achieving qualitatively correct results) depends on the system under study. Thus, a level of calculation that is already acceptable to describe hydrogen bonding may still not suffice for characterizing base stacking or a TS.
In general, the quality of ab initio QM calculations can be systematically improved through two factors: (i) increased inclusion of electron correlation effects (where mutual interaction of individual electrons is considered) and (ii) increased size (quality) of the basis set (set of functions) applied to the atomic orbitals to construct the molecular orbitals. To obtain high-quality results, both the inclusion of electron correlation and the size of the basis set should be increased in a balanced manner.
Since the range of available QM methods is broad, it is often difficult for the reader to assess the quality of the description of the QM core. It is certainly reasonable to assume that experienced research groups have sufficient knowledge to choose a suitable QM approach for the task at hand. However, a critical analysis of the underlying approximations and a comparison of the performance of the QM method applied to the core with that of highest-quality reference calculations for the uncatalyzed reactions ought to be included in any QM/MM study so that also the non-specialist can appreciate the quality of the QM core description. A common way to improve the quality of a QM/MM derived free energy profile is to apply a lower-level method to the computationally costly conformational search for a TS structure and then a higher-level method to the (single point) evaluation of the free energy change involved in reaching it. Both types of calculation are usually still limited by the available computer resources.
Three basic (standard) levels of ab initio quantum chemical calculations are often used to describe the QM core. First, the Hartree-Fock (HF) method with medium-quality basis sets of atomic orbitals provides a reasonable assessment of TS geometries, but is not sufficient to calculate a reaction barrier. While smaller basis sets are very deficient, extending the basis sets beyond medium size does not improve the results. Second, to improve the calculations at least for single points one can use the Second Order Moller Plesset (MP2) correlation method. MP2 should preferably employ large basis sets of atomic orbitals and can be extrapolated to the complete basis set of atomic orbitals. Third, reference calculations for the uncatalyzed reaction in small model systems can be studied by the coupled cluster method (CCSD(T)) as the highest-quality ab initio QM approach.
Standard QM methods nowadays are often replaced by various Density Functional Theory (DFT) approaches. DFT is a common abbreviation for a wide spectrum of methods of diverse quality, applicability, and computer efficiency (see also below). DFT is under intense development, especially over the past 3-4 years. For many applications carefully selected DFT methods can achieve comparable or better results than the MP2 method. When assessing DFT methods, an analysis of their performance relative to the standard HF, MP2 and CCSD(T) approaches is useful. CCSD(T) is in fact often used as reference to parameterize DFT methods.
In the following we provide more details regarding the best choice of method for the QM core in QM/MM calculations of RNA, including the HF, post-HF (including electron correlation), DFT, as well as semiempirical and EVB approaches (Table 1). The ability to predict TS barrier heights is of greatest importance for modeling reaction mechanisms and is therefore our focus. The geometry of the TS is also very important but is usually less sensitive to the level of calculation than the barrier height.
Table 1.
Overview of QM methods used for QM/MM. The description is simplified and meant as a rough guideline for an application to the nucleophilic substitution reactions in ribozymes. For more details please see the text and ref. (39).
| Basis sets | ||
|---|---|---|
| Class of basis set | Abbreviations of some common basis sets | Usual performance |
| Small | STO-3G, 3-21G | Very poor quality, unreliable results. |
| Medium | cc-pVDZ, SVP, 6-31G*, aug-cc-pVDZ | Reasonable quality for HF and DFT. For some methods, like MPW1K best results (see text). For MP2 still a small basis. |
| Large | 6-311++G(2df,2dp), TZVP, TZVPP, cc-pVTZ | Very good results for DFT and sometimes even for MP2. |
| Very large | aug-cc-pVTZ, QZVPP, cc-pVQZ, plain waves | The calculations are systematically converging with respect to the size of basis set. This does not guarantee best results for approximate (lower quality) methods like, e.g., DFT. Best available results with MP2 and CCSD(T). |
| Types of functions (atomic orbitals, AO) in basis sets | ||
| Type of AO | Abbreviation (if any) | Purpose |
| Basic AO | e.g. 6-31G, SV | Atomic orbitals (functions) of s, p and s type for second row (C, N, O) atoms and hydrogen, respectively. Just for basic description of the structure. Polarization and/or diffuse functions should be added for more accurate description (see below). |
| Polarization functions | “*” or “p”, “d”, “f” in Pople’s sets (6-31G*, 6-31G(2d,p)) “P” in SVP, TZVP, … “p” in Dunning’s cc-pVDZ, etc | Functions with higher angular momentum than s, p (i.e., d, f, g functions…) for C, N, O atoms, or higher than s for hydrogen. They describe polarization of molecules, essential for geometries and TS calculations. |
| Diffuse functions | “+” in Pople’s sets (6-31++ G*) “aug” in Dunning’s sets (aug-cc-pVDZ) | Functions reaching far from the atomic centers. They help to describe “diffuse” electrons far from the nuclei. Desirable for anions and transition state calculations. |
| Extrapolation of the results to the complete basis set | CBS | Reference quality calculations, aimed to mimic infinite basis set of AOs. |
| Plane waves | PW | Usually used with DFT in CPMD calculations. Due to errors of functional does not guarantee best results. No BSSE error. |
| Wave Function Methods | ||
| Method | Recommended quality of basis set. | Expected accuracy |
| HF | Medium basis | Poor energies, reasonable geometries |
| MP2 | Large and very large. | Very good quality, very demanding. |
| CCSD(T) | Large and very large. | With CBS provides best available results, extremely demanding. |
| DFT Methods | ||
| Class | Abbreviations of the most common functionals | Applicability |
| pure GGA | BLYP, PBE, HCTH, PW91 | Sometimes good results, but usually underestimate reaction barriers. |
| Can benefit from the density fitting (speed up). | ||
| Recommended for geometry search. | ||
| Very fast implementations allow for short molecular dynamics. | ||
| meta-GGA | TPSS | Small improvements with respect to pure GGA. |
| hybrid GGA | B3LYP, PBE0, BH&HLYP | Usually fairly good reaction barriers, often small underestimation. More demanding than pure GGA with density fitting. Very fast implementations allow for very short molecular dynamics. |
| functionals optimized (also) for kinetics | MPW1K, M06-2X | In most cases very accurate results for reaction barriers. Often, for a given purpose, comparable with CCSD(T), but much less demanding. Very fast implementations allow for very short molecular dynamics. |
| DFT-D | Empirical dispersion Correction exists for almost every functional. | Empirical correction to account for vdW forces. Usually not needed, but necessary when geometry is expected to be influenced by vdW forces (often in nonpolar systems, e.g., base stacking). |
| Other Methods | ||
| Class | Abbreviations | Applicability |
| Semiempirical methods | AM1, PM3, AM1/d-PhoT, SCC-DFTB | Relatively good results if carefully parameterized for a given type of reaction, such as AM1/d-PhoT or certain SCC-DFTB parameterizations. Otherwise very unreliable. |
| Very fast, possible molecular dynamics and sampling. | ||
| Empirical valence bond | EVB and many other abbreviations, depending on implementation. | Not a QM method. Reasonable results if very carefully parameterized. Unreliable if reaction mechanism is Somewhat different from what is assumed. Extremely fast, long molecular dynamics, sampling. |
3.3.2 Hartree Fock method
For a long time, the HF method (known also as self-consistent field, or SCF, approximation) was the method of choice in QM/MM calculations as the cheapest ab initio approximation. The HF method assumes that the exact, N-body wave-function and associated energy of a system can be approximated by a single expression of N spin-orbitals that is derived by applying the variational principle. One of the theoretical advantages of the HF method is its ab initio nature and variational character, implying that improving the quality of the basis set of atomic orbitals (see below) leads to more and more “correct” results (within the HF approximation), converging ultimately to the basis set limit of the method. However, HF is a mean-field method that is ignoring the effects arising from mutual correlation of electron motions (electron correlation), which leads at the basis set limit of the method to a significant overestimation of reaction barriers. By comparison, calculations in small and medium basis sets tend to underestimate reaction barriers compared to the basis set limit of the method. These two errors often partly cancel in HF calculations with medium basis sets, which coincidentally may lead to correct results that are in good agreement with reference values. However, relying on such a cancellation of basis set inaccuracies is not advisable as illustrated, for example, in a model reaction for RNA backbone cleavage (29), where a medium basis set gives very poor results (Fig. 3). Still, the HF method is usually fairly successful at determining molecular geometries, justifying its use in finding the TS geometry, followed by the use of a more accurate method to derive its energy. In this regard, however, the HF method has almost no advantages anymore over modern DFT and is thus increasingly displaced by the latter.
Fig. 3.
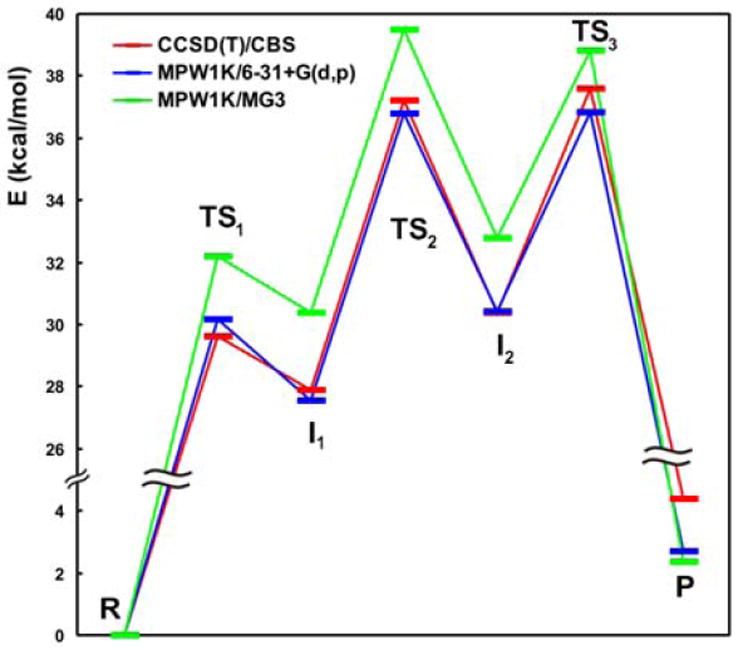
Basis set dependence of DFT results for the MPW1K DFT functional as applied to backbone self-cleavage by catalytic RNA (29). A medium basis set (6-31+G(d,p), red curve) yields results that are ~2 kcal/mol lower than those from a large basis set (MG3, green curve). For this particular functional the results for the smaller basis (MPW1K/6-31+G(d,p), red curve) are close to the reference (CCSD(T)/CBS, blue curve), because the MPW1K functional is parameterized for the 6-31+G(d,p) basis set.
3.3.3 Inclusion of electron correlation
Post-HF methods were developed to improve on HF method by including electron correlation as a more accurate way to account for interaction between electrons. They include perturbation Moller-Plesset (MP, usually second-order MP2), coupled clusters (CC) and quadratic configuration interaction (QCI) methods (39). These methods in principle provide better results for molecular geometries, reaction kinetics, and reaction thermodynamics. Their accuracy grows approximately in the order MP2<MP3<CCSD~QCISD<CCSD(T)~QCISD(T), in parallel to the associated time and computer costs. Thus, MP2 and CCSD(T) calculations are ~2-4 orders of magnitude slower than those based on the HF method. CCSD(T) calculations are considered a golden standard, but are currently affordable with medium basis sets when applied to molecules with less than ~30 atoms. Fortuitously, for nucleophilic substitution reactions as observed in ribozymes MP2 yields very good results comparable to those of CCSD(T) (40). We note that electron correlated ab initio calculations demand larger basis sets than the corresponding HF calculations. A survey of the accuracy of several QM methods for TS characterization can be found in Ref (40).
The computational cost of post-HF calculations can be significantly reduced (~10-fold) by using the so-called Resolution of Identity (RI) approximation, also referred to as Density Fitting (DF; leading to fast variants of MP2 abbreviated as RI-MP2 or DF-MP2, respectively). Because no integrals are neglected in the DF approximation, the accuracy of the calculation is not compromised. From a practical standpoint, an additional basis set, called fitting or auxiliary set, is needed. The latest versions of leading ab initio programs offer RI/DF as a non-default option for the MP2 level (41). Another, very different and so far less popular approximation is Local treatment of orbitals; for example, MP2 method with this local approximation is abbreviated as LMP2 (see, e.g., Ref. (42)). Methods based on local treatment of orbitals are efficient for very large molecules, but their results may be sometimes inaccurate and these methods are not yet well established for finding the TS.
3.3.4 Choice of a suitable basis set
Basis sets in standard calculations are constructed from sets of Gaussian functions. Basic atomic orbitals (basis functions) are those of s and p type for C, N, O atoms and s for hydrogen. Atomic orbitals with higher angular momentum than those of the basic atomic orbitals, i.e., d, f, g… and p, d, f,… for C,N,O atoms and hydrogen, respectively, are known as polarization functions. Any reasonable calculation requires polarization functions. Ab initio calculations of reaction barriers are sensitive to the quality (size) of the basis set (see Table 1) used, which is especially true for the electron correlated post-HF methods. This stems from the necessity to describe unusual bonding configurations (as in the TS), for which the standard basis sets are not designed. With medium basis sets, such as 6-31G**, cc-pVDZ, or SVP, inaccuracies from the basis-set superposition error (BSSE) and basis set incompleteness may be substantial. BSSE is an artifact that is related to the incompleteness of the basis set and can spoil calculations (43). The BSSE leads, e.g., to spurious stabilization of molecular complexes as the interacting monomers use each others orbitals to compensate for their own incomplete basis sets and thus artificially improve their electronic energies. To further improve the polarized basis sets, in general at least the addition of diffuse functions to cover electrons reaching far from the atomic centers is recommended (e.g., 6-31++G**, aug-cc-pVDZ, see Table 1) and in most cases the use of a large basis set is recommended.
When very high accuracy is desired, one can benefit from extrapolation schemes that allow extrapolation of calculations in two consecutive large basis sets to the Complete (infinite) Basis Set (CBS) limit (for a brief overview see, e.g., Ref (44)). Such extrapolation, however, requires the execution of two calculations with large basis sets of atomic orbitals at considerable computational cost. We have recently published series of papers reporting RI-MP2/CBS calculations complemented by evaluation of higher order electron correlation effects at the CCSD(T) level for nucleic acids base stacking and base pairing (45-47). These calculations provide the most accurate theoretical results currently available and are used as reference.
3.3.5 Density functional theory
DFT was developed in the 1960ies (48), underwent substantial expansion over the last twenty years (49), and nowadays have become one of the most widely applied classes of QM methods (50) with a confusing plethora of currently available DFT functionals. DFT determines the properties of many-electron systems by using functionals, or functions of another function, namely the spatially dependent electron density. The simplest functional Local Density Approximation (LDA, S-VWN) is no longer used in biomolecular calculations. Its successor, the generalized gradient approximation (GGA) functionals (e.g., BLYP, PBE, PW91) have proven to be surprisingly accurate in most chemical applications. However, GGA functionals have shortcomings for modeling transition states. Because GGA functionals are in the mathematical sense local, they cannot describe the intrinsically nonlocal Pauli exchange, leading to a substantial Self-Interaction Error (SIE) for TS and consequently a large, systematic underestimation of reaction barriers (49). A significant improvement was achieved by hybrid functionals that mix certain portions of the HF exchange into DFT (with the HF method overestimating barrier heights and thus partially counterbalancing the DFT). The barriers still remain somewhat underestimated (51), but hybrid functionals such as B3LYP, PBE0 or BH&H are nowadays the most popular methods for QM/MM calculations.
The next step in the development of functionals, the meta-GGA approximation (e.g., TPSS) did not persuasively improve TS description. However, we would like to direct the reader’s attention to a family of specialized empirical functionals, developed by Truhlar and coworkers, that are optimized (fitted) to, among others, the reference kinetic data. Even the older hybrid-GGA version, the “kinetic” functional MPW1K (52), is thus able to predict barrier heights with remarkable accuracy (Fig. 3, the MPW1K/6-31+G** results are in excellent agreement with those from CCSD(T)/CBS). The M06-2X functional from the most recent M06 suite of functionals by Truhlar’s group also performs well (53).
Other interesting implementations of DFT rely on the use of Plane Waves (PW) instead of the classical Gaussian basis sets. For example the CPMD program (http://www.cpmd.org/) allows for Car-Parrinello Molecular Dynamics (CPMD) (54), which propagates both coordinates and wavefunctions by using an extended Lagrange formalism. PW based calculations are free of BSSE error and naturally offer almost complete basis set results. The groups of Carloni and Rothlisberger have over time developed very advanced CPMD interfaces for biomolecular QM/MM calculations and published many excellent studies in the field of enzyme catalysis (55, 56). CPMD is in principle more efficient than classical Born-Oppenheimer Molecular Dynamics (BOMD), however, the PW basis set is exceedingly demanding for biomolecules (non-crystalline materials). This leads to a loss of the speed advantage of the Car-Parrinello method so that, in our experience, CPMD is overall slower than classical BOMD. Nevertheless, this loss can in part be compensated by the ability to very efficiently parallelize the calculations. Notably, the group of Hütter combined PW with Gaussian basis sets to overcome the slow speed of PW calculations and keep the advantages of both basis sets (57). This method is available in Quickstep as part of the program CP2K (http://cp2k.berlios.de/). A second disadvantage of PW calculations is that they are limited to pure GGA DFT functionals so that more accurate hybrid functionals cannot be routinely used. Especially the BLYP functional performs rather poorly for model ribozyme reactions as evident from the fact that the third barrier, which should separate the second intermediate from the final product, virtually disappears (0.2 kcal/mol, Fig. 4). The fact that the PW basis set is BSSE free further increases the inaccuracy of BLYP functional (data not shown). Therefore, results obtained with BLYP should be treated with care.
Fig. 4.
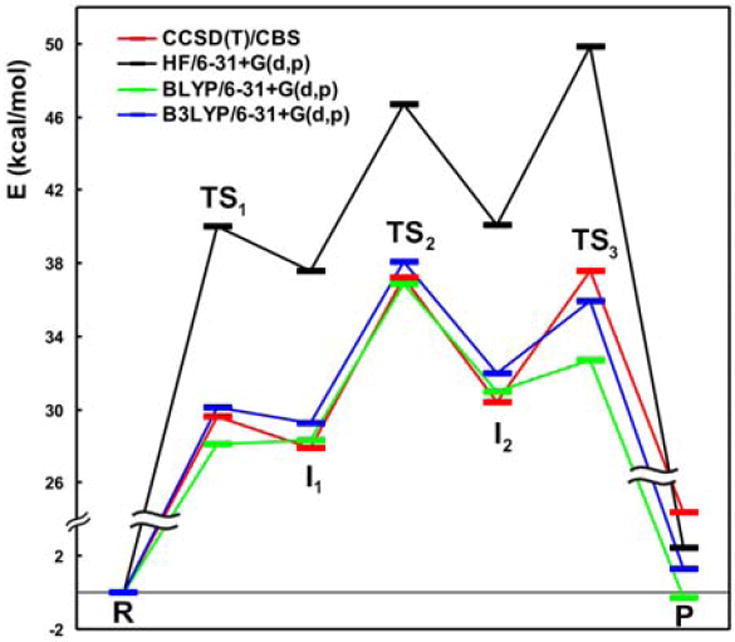
Reaction barriers of backbone self-cleavage by catalytic RNA (29) calculated using different methods in a medium 6-31+G(d,p) basis. The CCSD(T)/CBS reference data are in red. HF significantly overestimates barrier heights (blue). BLYP reproduces the reference data very poorly, especially the third TS (black).
DFT calculations have similar basis set requirements as HF calculations, i.e., they are less basis set sensitive than the electron correlated ab initio methods. Nevertheless, use of polarization functions is a must, and diffuse functions are highly recommended. Interestingly, the use of large basis sets does not improve the results for any of the functionals we tested on our model reaction (Fig. 4). For BLYP and B3LYP this is probably due to fortuitous error cancellation in the 6-31+G** basis set, while in the case of MPW1K it is a consequence of the small basis set used in the parameterization of this particular DFT functional. In this particular case, a medium quality basis set augmented with diffuse functions can thus be recommended.
When using DFT methods, special attention is needed when dispersion energy effects (London or van der Waals forces) are expected to influence geometry, because the vast majority of current GGA, meta-GGA and hybrid GGA functionals cannot describe this dispersion energy. For example, this problem may arise when stacked nucleo bases are involved in the QM region. In such a case, the simplest remedy is to include a dispersion correction through an empirically damped dispersion term (DFT-D) (58-60). Because dispersion usually influences the results only indirectly, through geometry, this correction can be omitted when the geometry is determined mainly by electrostatics.
In terms of calculation speed, the hybrid DFT is comparable with HF. Notably, pure GGA (non-hybrid) calculations can be significantly sped up by the density fitting mentioned above. However, this is true only for pure GGA functionals, and not for the more desirable hybrid functionals that do not significantly benefit from density fitting. Overall, DFT is the most economic QM method for TS calculations as it provides fairly accurate geometries (51) and, with kinetics-optimized density functionals, also reasonably accurate reaction barriers (51).
3.3.6 Semiempirical methods
Semiempirical methods historically are often considered QM methods, however, they arguably are not because they are based on more or less severe approximations and require careful parameterization. Still, they offer substantial benefit as they are considerably faster than classic QM methods, usually by 3-4 orders of magnitude. Because they are intrinsically parametrical, their reliability is seriously limited. However, with careful parameterization for a given (class of) reaction(s), they may be an attractive option. For instance, York and coworkers developed their own modification of AM1/d, well parameterized for phosphoryl transfer reactions (AM1/d-PhoT) (61). Recently, another semiempirical method derived from DFT, self-consistent-charge density functional tight-binding (SCC-DFTB) (62), was reparameterized to describe well phosphoryl transfer reactions (63). Wide utility of these methods depends, however, on the availability of easy-to-use parameterization tools to help generate semiempirical parameters for a reaction of interest.
3.3.7 Empirical valence bond
The EVB method pioneered by Warshel (64) became a very popular method due to its extremely low computer cost, allowing not only geometry optimization but also production of MD trajectories. In principle, EVB is an empirical method because its calculations are performed with an empirical potential. The form of the potential is modified in a way that allows for formation and breaking of predefined bonds. The MM Hamiltonians of the reactants and products are coupled with geometry-dependent non-diagonal elements that describe the height of the reaction barrier. Equations for these elements contain empirical parameters obtained through a fit to accurate reference calculations. The EVB method is not a genuine QM/MM method because the reaction model is treated by molecular mechanics. This approach may not be a major problem in many cases, however, when the environment of the active site is expected to significantly contribute to reaction chemistry through orbital effects (electron donation or withdrawal) or polarization, EVB may be inadequate. Certain improvements in this regard were recently suggested by Truhlar and coworkers, introducing an external field dependence in the non-diagonal coupling elements (65). Several versions of the EVB method exist and some are implemented in widely available programs like AMBER 10. However, EVB requires careful parameterization; after which MD runs can be performed and thermodynamic quantities be collected, a great advantage over the more demanding QM methods.
3.3.8 Specific advice for QM/MM calculations of RNA catalysis
The requirements for QM/MM applications to RNA catalysis are dictated by the need to describe the RNA backbone where phosphoryl transfer occurs. The RNA backbone consists of repeating sugar-phosphate motifs, where each phosphate moiety carries a negative charge. Because of the diffusiveness of the negative charge involving the phosphorus d-orbitals, a large basis set is needed for an accurate description, or at least a basis set with polarization and diffuse functions (e.g., aug-cc-pVTZ, 6-311+G(2d,p)). For description of the TS geometry the HF approach is usually sufficient, provided that dispersion interactions do not affect the geometry since the HF method neglects the dispersion energy. To obtain reliable energies, especially for the TS, a post-HF or suitable hybrid DFT method should be used (cf. also Table 1).
3.4 MM methods
MM describes RNA using classical force fields, which define the molecular potential energy as a function of the atomic coordinates. These simple analytic functions consist of a set of harmonic springs to describe covalent bonds and angles, coupled with torsional profiles for dihedral angles. Atoms are described as van der Waals (Lennard Jones) spheres with atom-centered constant point charges. The force field is pair-additive, that is, it neglects all non-additive effects such as polarization of atoms in the electric field from the surroundings. Parameterization of biomolecular force fields is a daunting task. While force fields that account for polarization are certainly possible, no such advanced force field has been successfully parameterized for nucleic acids. Polarization force fields have been under development for some time, but their future applicability to nucleic acids is difficult to predict. Despite very careful parameterization current force fields are thus far from perfect.
3.4.1 Choices of force fields for RNA
There are two force fields that have been extensively tested on RNA and DNA systems and can be viewed as established for nucleic acid simulations (38, 50, 66), namely the AMBER and CHARMM force fields. The AMBER force field family includes the original Cornell et al. parm94 (or ff94), a second-generation force field developed in the 1990ies by Kollman and coworkers (67), and its subsequent modifications, parm98 (ff98) (68), parm99 (ff99) (69), and parmbsc0 (70). The atomic point charges of ff94 were derived from the electrostatic potential calculated at the HF/6-31G* level, which overestimates the molecular dipole moments and so indirectly compensates for the missing polarization in condensed matter simulations. The ff98 and ff99 versions differ from ff94 only marginally. They were designed to improve the calculated helical twist in B-DNA, but were only partially successful in achieving that. Thus, the parm94-99 force fields are equally suitable for nucleic acids.
The parmbsc0 force field represents a more significant change. Longer (>10 ns) simulations of B-DNA with the parm94-99 variants resulted in accumulation of irreversible / backbone substates with the torsional angle (around the C4’-C5’ bond) in trans (71). This ultimately causes a severe structural deformation in simulated B-DNA (70). Parmbsc0 modifies the / dihedral profiles and thus stabilizes B-DNA simulations (70, 72, 73). In contrast, the pathological trans-substates do not accumulate in RNA with the parm94-99 versions of the force field (74, 75) so that all “parm” force field versions so far show similar and quite satisfactory RNA behavior (23, 38, 70, 76). Our parm99 simulations of A-RNA reveal ~10 to 15 % population of temporary trans- substates. This is in a meaningful agreement with the X-ray data, albeit the population of this minor A-RNA substate may be somewhat exaggerated by the force field (see ref. (75) for full discussion). Parmbsc0 (manuscript in preparation) usually does not allow to stabilize this substate, so the backbone is bouncing back immediately. Thus parmbsc0 somewhat rigidifies the trajectories compared with the parm94-99 behavior. However, until now we did not find any case of trans- substate in functional RNA which would be of functional importance and whose eventual suppression/loss would cause any structural problems.
Another force field that was rather successfully designed for nucleic acids is CHARMM27 (77, 78). This force field describes the canonical B-DNA structure relatively well, leading only to certain problems with groove widths (which are comparatively less accurate than with AMBER) and helical twist (72). Unfortunately, although it was used several times for RNA, CHARMM27 has not yet been systematically and thoroughly tested for structured RNAs. In 5-ns MD simulations of A-RNA, this CHARMM27 predicts very fast base pair breathing, which may indicate an underestimation of the strength of base pair interactions (79, 80). Similarly, instabilities of stem regions were reported for RNA kissing complexes (80, 81). Moreover, we recently performed 50-ns MD simulations of A-RNA a preliminary analysis of which revealed more obvious problems, leading to gradual A-RNA structure degradation (data not shown). In contrast, some of our data tentatively indicate that GNRA tetraloops may be more stable (rigid) when simulated using CHARMM27 compared to the AMBER family of force fields (data not shown).
3.4.2 Words of caution
We note that the currently available force fields are generally not sufficient to properly describe the various types of single stranded hairpin loop topologies found in guanine quadruplexes (G-DNA). This problem is discussed in the literature and represents a textbook example of a situation where the compensation of errors, which is necessary for the force fields to work, fails (38, 82, 83). Our extensive preliminary data indicate that the problem is widespread for G-DNA loops and that there is currently no force field available that works. We mention this here since some of our preliminary results suggest similar difficulties for single stranded RNA hairpin loops, such as GNRA tetraloops. Unfortunately, it has become common practice in the MM and MD literature to not publish such negative results, or simply to present them in a “positive” manner even if they are clearly inconsistent with experimental data. Thus, many force field problems that might already have been noticed remain unreported. Interestingly, the QM literature much more openly discusses the accuracy of calculations, which perhaps reflects the facts that QM methods can easily be cross-validated and have a large potential for rather straightforward further development. Although the MM force fields are inherently less accurate than QM methods, many MD studies do not critically dissect limitations or problems. Nevertheless, we reiterate that carefully designed and analyzed MD simulations often provide very valuable insight.
The imbalances of and differences between force fields tend to accumulate with prolongation of a simulation. They should generally be insignificant when using FFs for limited structural relaxation in QM/MM calculations or other short simulations. However, since the quality of experimental structures is also not always perfect (Ref. (37), Chapter 11), it is often necessary to use extensive MD simulations for preparing sufficiently representative structures for QM/MM. Simulations can help refine experimental structures locally (84, 85), but the limited simulation timescale together with force field imbalances does not allow for extensive corrections. If an experimental structure shows, for example, an incorrect nucleotide syn conformation a simulation will likely retain it indefinitely. As an example, in our studies of the HDV ribozyme we so far never observed a syn-anti flip of G25 in Loop 3, as suggested by comparison of the precursor and product crystal structures (74, 86). We have recently applied MD simulations for three independent and in many aspects mutually different NMR structures of HIV-1 DIS kissing complexes. The simulations were not capable on ~20 ns time scale to overcome the differences among the experimental structures, so that we could not obtain any unambiguous insights into the origin of the differences (the NMR structures in addition differ from X-ray structures that appear consistent with MD) (80). The simulations remained locked, for example, by unusual local backbone topologies associated with the bulged-in unpaired bases.
Due to the lack of polarization and charge transfer terms, FFs tend to suffer even more when describing solvent ions. Monovalent cations are described reasonably well, although obviously not perfectly. Monovalent anions like Cl- are more challenging, as such electron rich ions are highly polarizable. Divalent ions like Mg2+ are outside of the applicability of force fields and are best included only when unequivocal experimental data support specific binding sites and modes (74, 86, 87). Spurious preference of simulated Mg2+ for inner-shell binding is well documented (75), and the overall balance of forces in the first ligand shell of the divalent is not well described by force fields (88). Attempts to improve simulations through the inclusion of divalents are quite an illusion. The most frequently used ionic atmosphere in RNA or DNA simulations is therefore a net-neutralizing set of Na+ cations. There is a widespread misconception, probably partially based on a similar experience with protein simulations, that Na+ ions alone may provide a flawed description of RNA. In contrast to proteins, however, a net-neutralizing cation atmosphere for polyanionic nucleic acids results in ion concentrations of ~0.15-0.2 M (84, 89). This concentration may well resemble the experimental conditions where it is assumed that redistribution of cations and anions around an RNA molecule results in a cation cloud with roughly the excess positive charge needed for exact neutralization of the solute (90), a configuration mimicked in the periodic box of an MD simulation with net-neutralizing monovalent cations. There is not yet sufficient literature evidence to indicate that nucleic acid simulations with a modest excess of salt (e.g., both K+ and Cl-) would change the solute dynamics in any systematic way compared to the use of just a net-neutralizing Na+ atmosphere (91), although work in our laboratories is underway to test this notion for a number of RNA systems. Perhaps, the best approach that one can today recommend is to rely, in a given study, on a diversified portfolio of control simulations and test both minimal salt and modest excess salt conditions. Note that anyway single simulation is rarely sufficient to reach any definitive conclusions (74, 80).
We note that using an excess of salt carries the risk of ion clustering (salt crystallization) (92-94), although this effect can be minimized if the cation and anion parameters are carefully balanced (95, 96). It is, however, important to note that severe ion crystallization effects in earlier KCl AMBER simulations were substantially aided by the unfortunate combination of Aqvist’s parameters for cations (97), Dang’s parameters for Cl- (98) and incompatible mixing rules (95, 99). Monovalent ions in pair additive force fields are described by two adjustable parameters, the well depth and the ion radius while different combinations of the parameters may achieve similar predicted hydration energies. There is yet another problem of the pair-additive description of monovalent ions which has not been mentioned in any of the recent studies. Inevitably, once the force field parameters for monovalent cations are basically fitted to reproduce the target bulk properties (such as hydration energies) there are no more parameters available for tuning of the direct solute-cation interactions. Typically, strength of these interactions is visibly underestimated, as demonstrated on Fig. 1 in ref. (23). In any cases, ions in contemporary simulations remain to be simple Lennard-Jones spheres bearing constant point charges of +1, -1, +2… localized in their centers, so that one should not have too ambitious expectations to match exact ion conditions, investigate sodium vs. potassium differences, capture divalent ions etc. via simulations.
It should also be noted that the experimental analysis of cation locations on RNA is not unambiguous and thus not a reliable input for simulations. Careful analysis of two related RNA duplexes crystallized in the presence of each one of 13 different cations suggests that a single crystallographic study may not be sufficient to draw definitive conclusions about cation binding sites, which strikingly appear to depend on such subtle parameters as metal “softness” and size, as well as RNA hydration and crystal packing (100). Experiments can also miss important cation binding sites where ions are frequently present but fluctuate in location between several specific RNA functional groups. In such a case involving monovalents, MD simulation have been shown to help reveal the delocalized nature of the cations (101). It also cannot be ruled out that cations are sometimes incorrectly assigned in crystal structures (102). For example, a significant difference in Mg2+ binding is reported for the product and precursor structures of the HDV ribozyme. MD simulations are consistent with the precursor data, whereas some of the Mg2+ ions reported for the product structure were bound outside any significant electrostatic potential minimum (74, 86). Similarly, recent X-ray diffraction studies of the precursor in the presence of Tl- are in good agreement with the specific Na+ binding sites predicted by simulations in the catalytic pocket (103). Thus, despite their evident weaknesses MD simulations can serve as a powerful complement to better understand and predict experimental data. In summary, both experiment and theory are quite limited when revealing and/or predicting ion binding sites on RNAs, posing a formidable challenge for QM/MM studies as a chemical reaction path can be drastically affected by cation placement.
3.5 QM/MM boundaries
The weakest link of QM/MM methods lies in handling the interactions between the QM and MM regions. The interactions between the QM and MM regions can be divided into bonded and non-bonded in nature. Bonded interactions are at play when a covalent bond goes through the interfacial region (QM/MM boundary), which typically cannot be avoided in studies of enzyme and ribozyme catalysis. In general, two strategies are used for such bond cutting: (i) A link atom is used to saturate the cut bond, usually a hydrogen atom. (ii) A localized orbital is introduced at the boundary between the regions. Both strategies provide significantly more reliable results than leaving unpaired electron radicals at the boundary, and both take advantage of less demanding close-shell calculations of the QM energy. Still, both link atom and localized orbital significantly perturb the studied system, necessitating a careful choice of the boundary and its distance to the actual bond breaking or making to achieve energy convergence in the calculation. It is therefore recommended to place any QM/MM boundary at least three bonds away from the bond broken or made, otherwise a discontinuous PES may result (104). Furthermore, because charge transfer through the QM/MM boundary is neglected, it would be incorrect to cut any bonds for which charge transfer is expected, such as a conjugated or coordination bond. For instance, Wei and coworkers estimated the pKa of a water molecule coordinated to a Mg2+ ion by QM/MM using two water molecules in the QM core region and treating the rest of the system, including the Mg2+ ion, at the MM level (105). Such cutting of the Mg2+-water interaction by the QM/MM boundary will inevitably cause incorrect results. A less trivial example is found in modeling of the sugar-phosphate backbone. If we chose to cut the backbone between the O5’ oxygen and the C5’ carbon the electronic structure of the phosphate would be significantly affected, altering its pKa from ~1.0 (106) to ~1.5 (107), since the QM core contains a phosphate monoester instead of a diester. It is much more reasonable to cut the backbone between the C5’ and C4’ carbons, which results in a better description of the phosphate electronic structure and a more realistic predicted pKa of ~0.8 (107).
Another challenge of choosing the QM/MM boundaries lies in the necessary electronic embedding calculation (see below), where the point charges of the MM region polarize the QM wavefunction. This polarization might be significantly overestimated if the simplified atom-centered MM point charges are positioned close to the QM wavefunction. This bias can cause serious inaccuracies so that extension of the QM core region should be considered in such case. In addition, it is necessary to prevent penetration of the QM electron density into the MM region. Methods that use Gaussian basis sets can take the advantage of their fundamentals since Gaussians tend to restrain the electrons to the QM core. In case of other methods, particularly those using PW functions, a special potential should be applied that avoids penetration of the plain-waves to the MM region (108). A second solution to avoid inadequate polarization of the QM wavefunction by the atom-centered point charges is found in the three-layer ONIOM scheme developed by Morokuma (104). The middle layer is described at a medium level of theory, most frequently by semi-empirical methods, and keeps the problematic QM/MM boundary far from the broken or made bonds.
3.6 QM/MM coupling
The description of the non-bonded interactions between the QM and MM regions, frequently called QM/MM coupling, is another critical point of a QM/MM calculation. The most critical part of QM/MM coupling is the handling of the electrostatic part of the non-bonded interactions between the regions. Two groups of QM/MM couplings are distinguished based on this handling: (i) Mechanical embedding calculates the electrostatic interactions between the two regions at the MM level. Mechanical embedding represents the simplest, but also most approximate QM/MM coupling. The QM calculation of the QM core is performed in the entire absence of its MM surrounding and thus the polarization of the QM wavefunction is completely neglected. (ii) In electronic embedding the polarization of the QM wavefunction by the MM part is explicitly included in one-electron QM Hamiltonian. The charge distribution of the MM region that polarizes the QM wavefunction is frequently represented by a set of the atom-centered point charges. The polarization of the QM wavefunction represents a crucial improvement of a QM/MM scheme and should always be considered. In addition to the polarization of the QM core, it is also possible to include polarizability of atoms in the MM region, however, this inclusion is less critical and significantly increases computational costs. It is therefore unclear whether an introduction of MM polarization is worthwhile, especially since robust tests of QM/MM with a polarized MM method are still lacking. Even if mutual polarization effects between the QM and MM regions is included, charge transfer between them is still neglected and thus the boundaries between the regions need to be chosen carefully.
3.7 QM/MM sampling
The choice of the extent of sampling is always a compromise between robust sampling and the demands (accuracy) of the QM/MM method. Usually, accurate QM calculations with a large QM core and explicit solvent and ions can be done with very limited sampling or only a simple energy minimization. Straight minimization has disadvantages arising from the dependence of the obtained result on the starting structure (due to trapping in local minima near the starting structure). A study using energy minimization only should therefore always be combined with another method sampling conformational space, for example, classical MD. In addition, a set of different starting geometries rather than a single snapshot should be used.
When estimating the Gibbs energy of the TS, either Monte Carlo (MC) or MD methods can be used in combination with free energy perturbation methods, but MD is more common for enzyme and ribozyme catalysis. The quality of sampling (and inevitably also the accuracy of the Gibbs energy estimate) depends on the QM level. While the most accurate QM/MM methods allow only very limited sampling from several picoseconds to tens of ps of an MD run, SE/MM (SE, semiempirical) methods currently reach up to hundreds of picoseconds. Finally, the cheapest but also most approximative EVB methods achieve the most robust sampling on a nanosecond timescale. With the advance of better computers true QM/MM MD methods are slowly replacing the significantly less accurate SE/MM MD or EVB MD methods.
Generally, in QM/MM MD there are two ways to propagate the wavefunction of the QM core (and thus perform a QM/MM ab initio MD – AIMD): (i) In Born-Oppenheimer MD (BOMD) (109, 110) the wavefunction is converged to a Born-Oppenheimer surface by SCF calculations at each step of the MD run (with a timestep usually shorter than 0.5 fs to capture motions of hydrogen atom), and the nuclei are propagated by integration of Newton’s equation of motion using, for example, the Verlet algorithm. Note that Born-Oppenheimer approximation in general separates motions of nuclei and electrons. (ii) In on-the-fly dynamics the demanding SCF calculations are avoided by propagating the wavefunction in each step directly (“on-the-fly”) using an extended Lagrangian equation (54). This fast propagation was originally developed by Car and Parinello and implemented in the program CPMD, originally developed for AIMD in the solid state and implemented using the PW basis set (whose (dis)advantages are discussed above). Subsequently the QM/MM hybrid model was added into CPMD (111), however, it is still constrained to non-hybrid DFT functionals (mainly due to the use of the PW basis set). The enormous computer demands arising from usage of the PW basis set are partly alleviated in the Quickstep method recently developed by Hütter and coworkers, which is based on using a combination of PW-Gaussians implemented in the program CP2K (57), or in the Atom-centered Density Matrix Propagation (ADMP) developed by Schlegel and Iyengar (112-114) and implemented in Gaussian03 (115). An algorithm similar to ADMP (using Gaussian basis sets) was also implemented by Head-Gordon and coworkers (116). The use of extended Lagrangian MD significantly reduces computational demands compared to BOMD and does not compromise accuracy, as underscored by the equivalence of BOMD and extended Lagrangian AIMD (117).
QM/MM AIMD is computationally still too demanding, however, to directly sample a rare event such as a chemical reaction, and thus it is not likely to achieve bond breaking or formation spontaneously. Additional techniques are needed to increase conformational space sampling. Three classes of methods are currently used to study chemical reactions, including enzyme and ribozyme catalysis: (i) A trivial approach, which is not actually MD, localizes the TS and connects it with reactant and product structures by geometry optimizations (e.g., Refs. (118, 119)); (ii) accelerated MD, such as so-called “chemical flooding” (e.g. Refs. (120, 121)); (iii) free energy sampling techniques resulting in potential of mean force (PMF) curves, e.g., perturbation methods (FEP), thermodynamic integration (TI) (122, 123),, umbrella sampling (US) (124, 125), and the adaptive biasing force (ABF) method (126).
Despite progress in computational efficiency the QM/MM AIMD timescale currently available is not still long enough to sample biomolecular conformations perpendicular to the reaction path. Experience with long classical MD simulations shows that tens of nanoseconds are often needed for the equilibration phase of the MD run, and at least hundreds of nanoseconds (and multiple simulations) are necessary for sufficient sampling of the conformational space of a biomolecule. Thus AIMD in combination with accelerated MD or FEP methods is able to sample only a limited region of conformational space around the reaction coordinate that therefore can still be biased by the starting structure. A combination of robust sampling by classical MD with simulation of the chemical reaction pathway by AIMD with, for example, a free energy perturbation method like umbrella sampling is therefore a must.
3.8 QM/MM terminology
Unfortunately, there currently exists no widely accepted consensus on the terminology to be used for QM/MM methods. Several competing terminologies are used in the literature, which may confuse or even turn readers off. At present, the abbreviation “QM/MM” is used to refer to any hybrid method combining any two methods for the QM and MM levels. Because the quality of the employed methods is an essential factor determining the accuracy of a QM/MM scheme, we suggest that it may be beneficial to distinguish several computational levels:
QM for ab initio methods;
SE for semi-empirical methods;
EVB for empirical valence bond methods; and
MM for force field calculations.
In this way it is immediately possible to distinguish, e.g., a QM/MM calculation combining an ab initio method with an empirical force field from an SE/MM method combining a semi-empirical method with a force field, which tends to be of lower accuracy. Within this terminology, the broader meaning of the abbreviation QM/MM refers to all hybrid methods, but the narrower meaning specifies a combination of ab initio and MM methods. In addition, since the QM/MM coupling also critically affects accuracy, the terms electronic or mechanical embedding should also be included in the specification of a QM/MM method. This suggested terminology would help guide the reader of QM/MM articles and provide for a quick assessment of the accuracy of a hybrid method used.
4 Applications to RNA catalysis
QM and QM/MM studies of catalytic RNAs are only beginning to emerge. In the following we summarize most available pioneering examples and discuss their scope and limitations in light of our discussion above.
4.1 The HDV ribozyme
The hepatitis delta virus (HDV) ribozyme is a catalytic RNA motif embedded in the human pathogenic HDV RNA, as well as a specific human gene (127). Similar ribozyme sequences are found in the genomic and complementary antigenomic RNA forms generated during double-rolling circle replication of the virus. Both ribozyme forms consist of ~85 nucleotides and catalyze self-cleavage, required during pathogen replication. The genomic HDV ribozyme was the first catalytic RNA motif for which structural and biochemical data suggested participation of a specific side chain, cytosine 75 (C75), in reaction chemistry (128-130). Two models were proposed for the functional role of C75. In the first model, C75 acts as the general base that deprotonates the 2’-OH group at the cleaved site, thus activating it as the nucleophile that attacks the adjacent 3’,5’-phosphodiester bond (131). The second model proposes instead that C75 protonates the 5’-oxygen of the leaving group, thereby generating the 2’,3’-cyclic phosphate and 5’-OH termini of the reaction products (132). In both models a hydrated Mg2+ ion provides the complementary general acid and base functionality, respectively, under physiological conditions. A number of biochemical studies support the hypothesis that C75 may be protonated and act as general acid (132, 133). The crystal structures of the genomic ribozyme prior to (134) and after cleavage (135) are most consistent with the general base and general acid roles, respectively, of C75. MD simulations of the genomic HDV ribozyme in the precursor state suggest that the active site readily preorganizes to favor the C75 general base mechanism (86). Despite extensive effort, a structure consistent with the C75 general acid model was not yet found by simulations (29, 74, 86), however, this does not rule out that such a conformation can be adopted after significant rearrangement from the X-ray structures. It is for example possible that some substantial rearrangement of the flexible Loop 3 region of HDV ribozyme could help in re-positioning of the C75 (136). MD simulations indeed indicated structural plasticity of the Loop 3 region and its influence on the ion binding in the catalytic pocket. However, the simulations were probably far away from fully capturing the structural dynamics of Loop 3 (74, 86).
To date, three QM or QM/MM studies have described the reaction mechanism of HDV ribozyme self-cleavage (29, 105, 137). First, Gauld et al. studied the C75 general acid mechanism by applying high-level DFT to a truncated model of the active site, lacking the remainder of the ribozyme (137). They concluded that reaction pathway with C75 as the general acid is readily accessible. Unfortunately, the truncated active site model was likely distorted as it did not correspond to any conformation observed by either X-ray crystallography or MD simulation (74, 86, 103, 134, 135). This fact seriously limits the utility of these otherwise very rigorous calculations.
Second, the work of Liu, Guo and coworkers estimated the effective concentration of the reactive C75 species adopting either the general base or acid role using the Near-inline-Attack-Conformation (NAC) concept combined with classical MD simulations. Unfortunately, the estimate for the concentration of a properly positioned C75H+ general acid is likely unreliable due to a lack of convergence of the NAC population statistics. The authors also used DFT calculations of a truncated active site model to compare the C75 general base and acid mechanisms. They concluded that the general acid mechanism is energetically favored. The authors’ QM/MM calculations did not consider, however, the cleavage reaction but were used to estimate the pKas of C75 and water molecules bound to the active site Mg2+ ion. Thus, the title of the paper suggesting that QM/MM calculations were performed is rather misleading. Furthermore, the very small selected QM regions, encompassing C75 and one water molecule for calculating the pKa of C75 and two waters in close proximity to Mg2+ for estimating the pKa of the water molecule, can hardly be viewed as reliable (105). These limitations of the study were thoroughly discussed in Ref. (29). Although it cannot be ruled out that the basic conclusion of this paper is correct, this would merely be coincidental.
Recently, a QM/MM study evaluating the plausibility of the C75 general base mechanism of the HDV genomic ribozyme was published (29). The reaction mechanism was described using QM/MM methodology in which the active site (QM region, ~80 atoms) was treated by a hybrid density functional (MPW1K (52)) in the context of the complete HDV ribozyme fold. The MM part of the system was treated by the AMBER force field (ff99). This scheme overcomes the limitations associated with the use of truncated active site models. Gibbs energy barrier heights of several experimentally supported geometries consistent with the C75 general base mechanism were estimated by scanning the reaction paths over the PES. The results exhibited good agreement with the experimentally observed reaction free energy barrier and showed that the C75 general base mechanism, in concert with a hydrated magnesium ion as general and Lewis acid, is a plausible catalytic strategy for the HDV ribozyme (29). The main limitation of the study lies in the limited sampling and indirect estimation of the Gibbs energy barrier height. Unfortunately, the C75 general acid mechanism could not be examined (or no plausible path was found) due to a lack of suitable starting geometries from either X-ray crystallography of extensive MD simulations. Considering all mechanistic data and calculations one can imagine that the HDV ribozyme may utilize either general acid or general base schemes to perform catalysis, depending on the experimental conditions.
4.2 The hairpin ribozyme
The hairpin ribozyme is a self-cleaving or -ligating RNA enzyme, found in the minus strand of the satellite RNA associated with the tobacco ringspot virus, where it promotes double-rolling circle replication (138-140). The hairpin ribozyme can be engineered to catalyze reversible, site-specific phosphodiester bond cleavage on an external, complementary RNA substrate (141). The hairpin ribozyme achieves rate acceleration similar to other metal-dependent ribozymes (142), but it does not require a specific metal ion for catalysis (141). This clear lack of a potentially catalytic metal ion makes the hairpin ribozyme especially useful in probing the direct role of nucleobases in RNA catalysis.
A range of structural and biochemical data have implicated guanine 8 (G8) and adenine 38 (A38) as catalytic participants in cleavage and ligation. Abasic substitution of A38 impairs catalysis more then 10,000-fold, while exogenous nucleobase rescue experiments indicate that the protonation state of A38(N1) plays a direct role in catalysis (143, 144). Transition-state analog crystal structures place A38(N1) close to the 5’-oxygen leaving group, implicating A38 as the general acid (145-148). It has also been suggested that the catalytic roles of A38 are to align reactive groups and to stabilize negative charge in the TS (143, 144). Recent MD simulations are consistent with A38 functioning as the general acid. The MD simulations started from crystal structures wherein a catalytically inhibitory 2’-O-methyl capping group on the attacking 2’-OH nucleophile was removed showed that the 2’-methoxy group distorts the active site. In all MD simulations the reintroduced 2’-OH systematically moved toward the A38 Watson-Crick edge (84, 85). This finding raises the possibility that A38 shuttles a proton from the 2’-OH nucleophile to the 5’-oxygen leaving group (85). The conserved G8 is positioned near the active site and mutation or deletion at this site impairs activity ~1,000-fold without significantly disrupting the global structure of the ribozyme (149, 150). The position of G8 near the reactive 2’-OH observed in crystal structures suggested the possibility that an unprotonated N1 acts as general base (145, 149, 151, 152). In contrast, exogenous nucleobase rescue experiments suggested that the catalytic role of G8 lies in charge stabilization of the transition state and/or alignment of the reactive groups (15, 17, 150, 153). The recent MD simulations are consistent with the latter model as they find that G8 could facilitate catalysis through stabilizing both the developing charge on the scissile phosphate and the strained backbone conformations adopted along the reaction pathway (85).
The simulations also highlighted an interesting difference between the HDV and hairpin ribozymes. Both ribozymes develop a pocket of very deeply negative electrostatic surface potential, however, they may utilize different strategies for using it in chemistry. The catalytic pocket of the HDV ribozyme appears to be optimally suited to be filled by cations (in the absence of divalent cations there are typically two monovalents continuously occupying the pocket when C75 is not protonated (74)). In contrast, the pocket of the hairpin ribozyme does not appear to be as suitable for ion binding due to steric reasons. Thus, its RNA functional groups and long-residency water molecules are fully exposed to the negative electrostatic surface potential (84, 85). However, we cannot rule out trapping of a cation in the sheltered catalytic core pocket as a rare event not readily sampled by MD. Nevertheless, it is clear that hairpin ribozyme should at least experience long time periods where no ions are present at its catalytic pocket, which is very unlikely to happen with the HDV ribozyme and its open catalytic pocket.
The potential of mean force (PMF) of the reaction catalyzed by the hairpin ribozyme has been extensively studied in recent years by means of MD-QM/MM methods (MD-SE/MM in our terminology). The SE/MM scheme employed a reparameterized AM1 semiempirical method (AM1/d-PhoT) for the QM core and a CHARMM27 potential for the MM part. The SE/MM boundaries were treated by the generalized hybrid orbital method (154). The QM core contained the scissile phosphodiester bond and part of the A-1 and G+1 ribose rings. This SE/MM scheme is a reasonable compromise between quality and demands allowing for sufficient sampling of configuration space to generate Gibbs energy estimates. However, the approach is limited in reliability by the AM1/d-PhoT method, while the CHARMM27 force field is of limited applicability to RNA simulations, although deficiencies would not become apparent at the nanosecond timescale used in SE/MM dynamics. The scheme allowed for production of one- and two-dimensional PMF profiles under various conditions, which suggested that the catalytic rate enhancement of the hairpin ribozyme is due to its electrostatic field rather than any direct involvement of nucleobases as either general acid or base catalysts. This finding implies that G8 mainly plays a structural role. Accordingly, the role of general base was proposed to be played by the nonbridging phosphoryl oxygens. The role of A38 has not been fully resolved because two mechanism, where either A38 is involved in hydrogen bonding or the protonated A38H+ acts as the general acid, were found to be plausible (155, 156).
Considering the quality of the method and robustness of sampling, the results of the latter studies (155, 156) may also be interpreted such that catalysis proceeds via multiple alternative pathways whose barriers are energetically similar. The RNA enzyme follows a particular pathway depending on environmental conditions (ionic strength, pH etc.). Such a multi-channel mechanism has been also suggested for the HDV ribozyme (29). Such multi-dimensional mechanisms underscore the importance of robust sampling not only at the QM/MM level, nowadays still seriously limited to timescales of several nanoseconds during which large conformational changes hardly occur, but also at the MD level to provide representative structures as starting points for the QM/MM calculations.
4.3 The hammerhead ribozyme
The hammerhead ribozyme was originally derived from a group of small, self-cleaving RNA satellites of various plant viruses including, for example, the tobacco Ringspot virus where a hammerhead ribozyme in the plus strand complements a hairpin ribozyme in the minus strand to effect double-rolling circle replication (157, 158). It was recently also found to be embedded in certain mammalian mRNAs (159). The hammerhead ribozyme was the first ribozyme for which crystal structures were resolved (160, 161). It catalyzes reversible self-cleavage of its own backbone, where 2’-OH of cytosine 17 (C17) attacks the adjacent scissile phosphate (162). The detailed catalytic mechanism has been extensively studied, highlighting possible roles of two specific guanosines (G8 and G12), as well as of divalent metal ions, water molecules, and the global conformation in catalysis (163-165).
The necessity for relevant QM/MM studies of the catalytic mechanism of the hammerhead ribozyme has been repeatedly advocated (166, 167). The catalytic role of Mg2+ in particular has been studied by QM methods on model systems cut from the structure of a minimal hammerhead ribozyme that lacks distal interacting loops now known to be important for a proper catalytic core conformation (165). The study of Leclerc and Karplus concluded that a double-metal mechanism, where the divalent ions work not only as Lewis acids but also as general acids/bases, is plausible (166). Earlier work by Torres and Lovell stated that only one Mg2+ ion is needed as Lewis acid and general base to provide sufficient lowering of the barrier height (168). The plausibility of this mechanism was recently reassessed by classical MD and QM/MM-MD simulations of the proper catalytic core conformation found in an extended hammerhead ribozyme (167, 169). These data showed that the mono-metal mechanism can be considered plausible for hammerhead self-cleavage. Moreover, a catalytic mechanism without any direct participation of metal ions has also recently been suggested based on X-ray crystallography and MD simulation data (164).
The role of Mg2+ ions has also been studied by the QM/MM-CPMD method. Unfortunately, the resultant free energies are likely significantly overestimated due to sampling of an incorrect reaction pathway (170). The only currently available relevant QM/MM study was designed to test the idea that the catalytic step is coupled with long-range conformational motions of the minimal hammerhead ribozyme (30). The applied QM/MM method combines the B3LYP/6-311G** DFT method for the QM core with the CHARMM27 force field for the remaining atoms. The calculated Gibbs energy surface shows that the reaction starting from a minimal crystal-like structure follows a pathway that is not consistent with the experimental data. In contrast, when started from a global conformation found by MD simulation the reaction proceeds via a proper in-line attack mechanism and leads to reliable TS and product structures, which differ from the crystal structure of the minimal hammerhead ribozyme. These results imply that the minimal hammerhead undergoes a conformational change prior to cleavage, as predicted by a comparison of the available crystal structures (165). In summary, however, the precise mechanism of the hammerhead ribozyme remains elusive, despite significant effort to elucidate it.
4.4 The ribosome
The ribosome is a complex molecular machine consisting of several ribosomal RNAs and numerous proteins (171, 172). It is widely accepted that the ribosome is a ribozyme where the RNA catalyzes peptide bond formation during translation. Considering the size of the system and complexity of the catalyzed reaction, the ribosome goes beyond the scope of the current review, however, there are several QM/MM studies that deserve to be mentioned. In fact, QM/MM methods have been widely used to elucidate the mechanism of polypeptide elongation and termination catalyzed by the ribosome (173-177). QM/MM with the QM core described by EVB (EVB/MM in our terminology) is the cheapest hybrid method allowing reasonably robust conformational space sampling by umbrella sampling and free energy perturbation (178). This method has been used by the groups of Åqvist (173) and Warshel (177) on the ribosome. Both groups calculated a similar lowering of the activation barrier of the reaction catalyzed by ribosome (by ~7 kcal/mol), which agrees well with the experimental value (~6 kcal/mol) (179). The applied method also allowed a dissection of the catalytic power of the ribosome, identifying active site preorganization as the main source of rate enhancement, whereas the previous proposal that the catalytic power of the ribosome lies in substrate alignment (i.e., in reducing substrate conformational space) was unequivocally rejected. The same strategy to achieve catalysis is also employed by many other enzymes. It is too early to tell for sure whether RNA enzymes in general use the same strategies as protein enzymes, have a more limited repertoire, or utilize additional tricks.
5. Concluding remarks
QM/MM calculations, when properly applied, have a high potential for elucidating important atomistic details of the mechanisms involved in RNA catalysis. QM/MM studies thus complement structural and mechanistic probing. However, QM/MM results must be considered with care because of the limitations of sampling and accuracy, sometimes masked by a confusing terminology. In the near term, it is unlikely that QM/MM techniques will become user friendly black-box applications that can be readily employed by non-experts, because of the technical subtleties and requirements surveyed in this review. The field of ribozyme catalysis is still very vibrant with no unequivocal consensus reached on any single mechanism so that significant effort will have to be expended before substantial progress can be expected. Cooperation of experimentalists and theorists is therefore highly desirable and will require an open mind, a positive attitude, and at least a basic level of understanding of each other’s methods.
Acknowledgments
This work was supported by grants from the Grant Agency of the Czech Republic (203/09/H046 and 203/09/1476), the Academy of Sciences of the Czech Republic (IAA400040802 and 1QS500040581), the Czech Ministry of Education, Youth, and Sports (LC512 and MSM6198959216), the Academy of Sciences of the Czech Republic (AV0Z50040507 and AV0Z50040702) and the National Institutes of Health (GM62357 to N.G.W.)
List of abbreviations
- ADMP
atomic center density matrix propagator
- AIMD
ab initio MD
- BOMD
Born-Oppenheimer MD
- BSSE
basis set superposition error
- CC
coupled cluster
- CPMD
Car-Parrinello MD
- GGA
generalized gradient approximation
- DF
density fitting
- DFT
density functional theory
- DFTB
density functional tight-binding
- EVB
empirical valence bond
- FF
force field (empirical potential)
- FEP
free energy perturbation
- HDV
hepatitis delta virus
- HF
Hartree-Fock
- IMOMM
integrated molecular-orbital molecular mechanics
- LDA
local density approximation
- MD
molecular dynamics
- MM
molecular mechanic
- MPn
Moller-Plesset perturbation theory of n-th order
- NAC
near attack conformation
- ONIOM
our own n-layered integrated molecular orbital + molecular mechanics
- PES
potential energy (hyper)surface
- PMF
potential of mean force
- PW
plane wave(s)
- RI
resolution of identity
- SCC-DFTB
self-consistent charge DFTB
- SE
semiempirical
- SIE
self-interaction error
- QCI
quadratic configuration interaction
- QM
quantum mechanics/quantum chemistry
- TS
transition state
- TST
transition state theory
- vdW
van der Waals
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Science. 2002;295:1520–23. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 2.Olsson MHM, Parson WW, Warshel A. Chem Rev. 2006;106:1737–56. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 3.Warshel A, Sharma PK, Kato M, Xiang Y, Liu HB, Olsson MHM. Chem Rev. 2006;106:3210–35. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Viloca M, Gao J, Karplus M, Truhlar DG. Science. 2004;303:186–95. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- 5.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Nature. 2005;438:117–21. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 6.Daniel RM, Dunn RV, Finney JL, Smith JC. Annu Rev Biohys Biomol Struct. 2003;32:69–92. doi: 10.1146/annurev.biophys.32.110601.142445. [DOI] [PubMed] [Google Scholar]
- 7.Bruice TC. Acc Chem Res. 2002;35:139–48. doi: 10.1021/ar0001665. [DOI] [PubMed] [Google Scholar]
- 8.Kraut DA, Carroll KS, Herschlag D. Annu Rev Biochem. 2003;72:517–71. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 9.Gao JL, Ma SH, Major DT, Nam K, Pu JZ, Truhlar DG. Chem Rev. 2006;106:3188–209. doi: 10.1021/cr050293k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao JL, Fan Y, Ma SH, Truhlar DG. FASEB J. 2007;21:A151–A51. [Google Scholar]
- 11.Cech TR, Zaug AJ, Grabowski PJ. Cell. 1981;27:487–96. doi: 10.1016/0092-8674(81)90390-1. [DOI] [PubMed] [Google Scholar]
- 12.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Cell. 1982;31:147–57. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 13.Guerriertakada C, Gardiner K, Marsh T, Pace N, Altman S. Cell. 1983;35:849–57. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 14.Gilbert W. Nature. 1986;319:618–18. [Google Scholar]
- 15.Cochrane JC, Strobel SA. Acc Chem Res. 2008;41:1027–35. doi: 10.1021/ar800050c. [DOI] [PubMed] [Google Scholar]
- 16.Strobel SA, Cochrane JC. Curr Opin Chem Biol. 2007;11:636–43. doi: 10.1016/j.cbpa.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walter NG. Mol Cell. 2007;28:923–29. doi: 10.1016/j.molcel.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott WG. Curr Opin Struct Biol. 2007;17:280–86. doi: 10.1016/j.sbi.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Bevilacqua PC. Biochemistry. 2003;42:2259–65. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- 20.Bevilacqua PC, Yajima R. Curr Opin Chem Biol. 2006;10:455–64. doi: 10.1016/j.cbpa.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 21.Bevilacqua PC, Blose JM. Annu Rev Phys Chem. 2008;59:79–103. doi: 10.1146/annurev.physchem.59.032607.093743. [DOI] [PubMed] [Google Scholar]
- 22.Harris ME, Cassano AG. Curr Opin Chem Biol. 2008;12:626–39. doi: 10.1016/j.cbpa.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDowell SE, Spackova N, Sponer J, Walter NG. Biopolymers. 2007;85:169–84. doi: 10.1002/bip.20620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sponer J, Leszczynski J, Hobza P. Biopolymers. 2001;61:3–31. doi: 10.1002/1097-0282(2001)61:1<3::AID-BIP10048>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 25.Warshel A, Levitt M. J Mol Biol. 1976;103:227–49. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 26.Truhlar DG, Garrett BC, Klippenstein SJ. J Phys Chem. 1996;100:12771–800. [Google Scholar]
- 27.Pu JZ, Gao JL, Truhlar DG. Chem Rev. 2006;106:3140–69. doi: 10.1021/cr050308e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klahn M, Braun-Sand S, Rosta E, Warshel A. J Phys Chem B. 2005;109:15645–50. doi: 10.1021/jp0521757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banas P, Rulisek L, Hanosova V, Svozil D, Walter NG, Sponer J, Otyepka M. J Phys Chem B. 2008;112:11177–87. doi: 10.1021/jp802592z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radhakrishnan R. Biophys J. 2007;93:2391–99. doi: 10.1529/biophysj.107.104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maseras F, Morokuma K. J Comp Chem. 1995;16:1170–79. [Google Scholar]
- 32.Svensson M, Humbel S, Morokuma K. J Chem Phys. 1996;105:3654–61. [Google Scholar]
- 33.Froese RDJ, Musaev DG, Morokuma K. J Am Chem Soc. 1998;120:1581–87. [Google Scholar]
- 34.Vreven T, Morokuma K, Farkas O, Schlegel HB, Frisch MJ. J Comp Chem. 2003;24:760–69. doi: 10.1002/jcc.10156. [DOI] [PubMed] [Google Scholar]
- 35.Vreven T, Morokuma K. Theor Chem Acc. 2003;109:125–32. [Google Scholar]
- 36.Vreven T, Byun KS, Komaromi I, Dapprich S, Montgomery JA, Morokuma K, Frisch MJ. J Chem Theor Comp. 2006;2:815–26. doi: 10.1021/ct050289g. [DOI] [PubMed] [Google Scholar]
- 37.Sponer J, Lankas F. Computational studies of RNA and DNA. Springer; Dordrecht: 2006. [Google Scholar]
- 38.Sponer J, Spackova N. Methods. 2007;43:278–90. doi: 10.1016/j.ymeth.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Encyclopedia of Computational Chemistry. John Wiley and Sons; New York: 1998. [Google Scholar]
- 40.Zhao Y, Gonzalez-Garcia N, Truhlar DG. J Phys Chem A. 2005;109:2012–18. doi: 10.1021/jp045141s. [DOI] [PubMed] [Google Scholar]
- 41.Kendall RA, Fruchtl HA. Theor Chem Acc. 1997;97:158–63. [Google Scholar]
- 42.Werner HJ, Manby FR, Knowles PJ. J Chem Phys. 2003;118:8149–60. [Google Scholar]
- 43.Boys SF, Bernardi F. Mol Phys. 1970;19:553. [Google Scholar]
- 44.Min SK, Lee EC, Lee HM, Kim DY, Kim D, Kim KS. J Comp Chem. 2008;29:1208–21. doi: 10.1002/jcc.20880. [DOI] [PubMed] [Google Scholar]
- 45.Sponer J, Jurecka P, Hobza P. J Am Chem Soc. 2004;126:10142–51. doi: 10.1021/ja048436s. [DOI] [PubMed] [Google Scholar]
- 46.Jurecka P, Hobza P. J Am Chem Soc. 2003;125:15608–13. doi: 10.1021/ja036611j. [DOI] [PubMed] [Google Scholar]
- 47.Sponer J, Jurecka P, Marchan I, Luque FJ, Orozco M, Hobza P. Chem Eur J. 2006;12:2854–65. doi: 10.1002/chem.200501239. [DOI] [PubMed] [Google Scholar]
- 48.Kohn W, Sham LJ. Phys Rev. 1965;140:1133. [Google Scholar]
- 49.Koch W, Holthausen MC. A Chemist’s Guide to Density Functional Theory. Wiley-VCH; Weinheim: 2001. [Google Scholar]
- 50.Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157–67. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 51.Lynch BJ, Truhlar DG. J Phys Chem A. 2001;105:2936–41. [Google Scholar]
- 52.Lynch BJ, Fast PL, Harris M, Truhlar DG. J Phys Chem A. 2000;104:4811–15. [Google Scholar]
- 53.Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215–41. [Google Scholar]
- 54.Car R, Parrinello M. Phys Rev Lett. 1985;55:2471–74. doi: 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- 55.Carloni P, Rothlisberger U, Parrinello M. Acc Chem Res. 2002;35:455–64. doi: 10.1021/ar010018u. [DOI] [PubMed] [Google Scholar]
- 56.Dal Peraro M, Ruggerone P, Raugei S, Gervasi FL, Carloni P. Curr Opin Struct Biol. 2007;17:149–56. doi: 10.1016/j.sbi.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 57.VandeVondele J, Krack M, Mohamed F, Parrinello M, Chassaing T, Hutter J. Comp Phys Commun. 2005;167:103–28. [Google Scholar]
- 58.Elstner M, Hobza P, Frauenheim T, Suhai S, Kaxiras E. J Chem Phys. 2001;114:5149–55. [Google Scholar]
- 59.Jurecka P, Cerny J, Hobza P, Salahub DR. J Comp Chem. 2007;28:555–69. doi: 10.1002/jcc.20570. [DOI] [PubMed] [Google Scholar]
- 60.Grimme S. J Comp Chem. 2004;25:1463–73. doi: 10.1002/jcc.20078. [DOI] [PubMed] [Google Scholar]
- 61.Nam K, Cui Q, Gao JL, York DM. J Chem Theor Comp. 2007;3:486–504. doi: 10.1021/ct6002466. [DOI] [PubMed] [Google Scholar]
- 62.Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim T, Suhai S, Seifert G. Phys Rev B. 1998;58:7260–68. [Google Scholar]
- 63.Yang Y, Yu HB, York D, Elstner M, Cui Q. J Chem Theor Comp. 2008;4:2067–84. doi: 10.1021/ct800330d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warshel A. Computer Modeling of Chemical Reactions in Enzymes and Solutions. John Wiley and Sons; New York: 1991. [Google Scholar]
- 65.Higashi M, Truhlar DG. J Chem Theor Comp. 2008;4:790–803. doi: 10.1021/ct800004y. [DOI] [PubMed] [Google Scholar]
- 66.Mackerell AD. J Comp Chem. 2004;25:1584–604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 67.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179–97. [Google Scholar]
- 68.Cheatham TE, Cieplak P, Kollman PA. J Biomol Struct Dynam. 1999;16:845–62. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 69.Wang JM, Cieplak P, Kollman PA. J Comp Chem. 2000;21:1049–74. [Google Scholar]
- 70.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, Laughton CA, Orozco M. Biophys J. 2007;92:3817–29. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Varnai P, Zakrzewska K. Nuc Acids Res. 2004;32:4269–80. doi: 10.1093/nar/gkh765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perez A, Lankas F, Luque FJ, Orozco M. Nuc Acids Res. 2008;36:2379–94. doi: 10.1093/nar/gkn082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perez A, Luque FJ, Orozco M. J Am Chem Soc. 2007;129:14739–45. doi: 10.1021/ja0753546. [DOI] [PubMed] [Google Scholar]
- 74.Krasovska MV, Sefcikova J, Reblova K, Schneider B, Walter NG, Sponer J. Biophys J. 2006;91:626–38. doi: 10.1529/biophysj.105.079368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reblova K, Lankas F, Razga F, Krasovska MV, Koca J, Sponer J. Biopolymers. 2006;82:504–20. doi: 10.1002/bip.20503. [DOI] [PubMed] [Google Scholar]
- 76.Spackova N, Sponer J. Nuc Acids Res. 2006;34:697–708. doi: 10.1093/nar/gkj470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Foloppe N, MacKerell AD. J Comp Chem. 2000;21:86–104. [Google Scholar]
- 78.MacKerell AD, Banavali NK. J Comp Chem. 2000;21:105–20. [Google Scholar]
- 79.Pan YP, MacKerell AD. Nuc Acids Res. 2003;31:7131–40. doi: 10.1093/nar/gkg941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reblova K, Fadrna E, Sarzynska J, Kulinski T, Kulhanek P, Ennifar E, Koca J, Sponer J. Biophys J. 2007;93:3932–49. doi: 10.1529/biophysj.107.110056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sarzynska J, Reblova K, Sponer J, Kulinski T. Biopolymers. 2008;89:732–46. doi: 10.1002/bip.21001. [DOI] [PubMed] [Google Scholar]
- 82.Fadrna E, Spackova N, Svozil D, Sponer J. J Biomol Struct Dynam. 2007;24:709–09. [Google Scholar]
- 83.Fadrna E, Spackova N, Stefl R, Koca J, Cheatham TE, Sponer J. Biophys J. 2004;87:227–42. doi: 10.1529/biophysj.103.034751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rhodes MM, Reblova K, Sponer J, Walter NG. Proc Natl Acad Sci USA. 2006;103:13380–85. doi: 10.1073/pnas.0605090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ditzler MA, Sponer J, Walter NG. RNA. 2009;15:560–75. doi: 10.1261/rna.1416709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Krasovska MV, Sefcikova J, Spackova N, Sponer J, Walter NG. J Mol Biol. 2005;351:731–48. doi: 10.1016/j.jmb.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 87.Gresh N, Sponer JE, Spackova N, Leszczynski J, Sponer J. J Phys Chem B. 2003;107:8669–81. [Google Scholar]
- 88.Sponer J, Sabat M, Gorb L, Leszczynski J, Lippert B, Hobza P. J Phys Chem B. 2000;104:7535–44. [Google Scholar]
- 89.Cheatham TE, Kollman PA. Annu Rev Phys Chem. 2000;51:435–71. doi: 10.1146/annurev.physchem.51.1.435. [DOI] [PubMed] [Google Scholar]
- 90.Chu VB, Bai Y, Lipfert J, Herschlag D, Doniach S. Curr Opin Chem Biol. 2008;12:619–25. doi: 10.1016/j.cbpa.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Razga F, Zacharias M, Reblova K, Koca J, Sponer J. Structure. 2006;14:825–35. doi: 10.1016/j.str.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 92.Auffinger P, Westhof E. J Mol Biol. 2000;300:1113–31. doi: 10.1006/jmbi.2000.3894. [DOI] [PubMed] [Google Scholar]
- 93.Auffinger P, Cheatham TE, Vaiana AC. J Chem Theor Comp. 2007;3:1851–59. doi: 10.1021/ct700143s. [DOI] [PubMed] [Google Scholar]
- 94.Degreve L, da Silva FLB. J Chem Phys. 1999;111:5150–56. [Google Scholar]
- 95.Chen AA, Pappu RV. J Phys Chem B. 2007;111:11884–87. doi: 10.1021/jp0765392. [DOI] [PubMed] [Google Scholar]
- 96.Joung IS, Cheatham TE. J Phys Chem B. 2008;112:9020–41. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aqvist J. J Phys Chem. 1990;94:8021–24. [Google Scholar]
- 98.Smith DE, Dang LX. J Chem Phys. 1994;100:3757–66. [Google Scholar]
- 99.Aqvist J. J Phys Chem. 1994;98:8253–55. [Google Scholar]
- 100.Ennifar E, Walter P, Dumas P. Nuc Acids Res. 2003;31:2671–82. doi: 10.1093/nar/gkg350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Reblova K, Spackova N, Sponer JE, Koca J, Sponer J. Nuc Acids Res. 2003;31:6942–52. doi: 10.1093/nar/gkg880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Auffinger P, Bielecki L, Westhof E. Structure. 2004;12:379–88. doi: 10.1016/j.str.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 103.Ke A, Ding F, Batchelor JD, Doudna JA. Structure. 2007;15:281–7. doi: 10.1016/j.str.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 104.Morokuma K, Wang QF, Vreven T. J Chem Theor Comp. 2006;2:1317–24. doi: 10.1021/ct600135b. [DOI] [PubMed] [Google Scholar]
- 105.Wei K, Liu L, Cheng YH, Fu Y, Guo QX. J Phys Chem B. 2007;111:1514–16. doi: 10.1021/jp070120u. [DOI] [PubMed] [Google Scholar]
- 106.Bunton CA, Llewellyn DR, Oldham KG, Vernon CA. J Chem Soc. 1958:3574–87. [Google Scholar]
- 107.Bunton CA, Mhala MM, Oldham KG, Vernon CA. J Chem Soc. 1960:3293–301. [Google Scholar]
- 108.Laio A, VandeVondele J, Rothlisberger U. J Phys Chem B. 2002;106:7300–07. [Google Scholar]
- 109.Helgaker T, Uggerud E, Jensen HJA. Chem Phys Lett. 1990;173:145–50. [Google Scholar]
- 110.Uggerud E, Helgaker T. J Am Chem Soc. 1992;114:4265–68. [Google Scholar]
- 111.Laio A, VandeVondele J, Rothlisberger U. J Chem Phys. 2002;116:6941–47. [Google Scholar]
- 112.Schlegel HB, Millam JM, Iyengar SS, Voth GA, Daniels AD, Scuseria GE, Frisch MJ. J Chem Phys. 2001;114:9758–63. [Google Scholar]
- 113.Schlegel HB, Iyengar SS, Li XS, Millam JM, Voth GA, Scuseria GE, Frisch MJ. J Chem Phys. 2002;117:8694–704. [Google Scholar]
- 114.Iyengar SS, Schlegel HB, Millam JM, Voth GA, Scuseria GE, Frisch MJ. J Chem Phys. 2001;115:10291–302. doi: 10.1063/1.1944720. [DOI] [PubMed] [Google Scholar]
- 115.Frisch MJ, Frisch A, Foresman JB. Gaussian, Inc.; Pittsburgh: 2003. [Google Scholar]
- 116.Herbert JM, Head-Gordon M. J Chem Phys. 2004;121:11542–56. doi: 10.1063/1.1814934. [DOI] [PubMed] [Google Scholar]
- 117.Iyengar SS, Schlegel HB, Voth GA, Millam JM, Scuseria GE, Frisch MJ. Isr J Chem. 2002;42:191–202. [Google Scholar]
- 118.Henkelman G, Jonsson H. J Chem Phys. 2000;113:9978–85. [Google Scholar]
- 119.Fischer S, Karplus M. Chem Phys Lett. 1992;194:252–61. [Google Scholar]
- 120.Iannuzzi M, Laio A, Parrinello M. Phys Rev Lett. 2003;90 doi: 10.1103/PhysRevLett.90.238302. [DOI] [PubMed] [Google Scholar]
- 121.Muller EM, de Meijere A, Grubmuller H. J Chem Phys. 2002;116:897–905. [Google Scholar]
- 122.Ciccotti G, Ferrario M. J Mol Liq. 2000;89:1–18. [Google Scholar]
- 123.Ciccotti G, Ferrario M. Mol Simul. 2004;30:787–93. [Google Scholar]
- 124.Torrie GM, Valleau JP. J Comp Phys. 1977;23:187–99. [Google Scholar]
- 125.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. J Comp Chem. 1992;13:1011–21. [Google Scholar]
- 126.Sprik M, Ciccotti G. J Chem Phys. 1998;109:7737–44. [Google Scholar]
- 127.Salehi-Ashtiani K, Luptak A, Litovchick A, Szostak JW. Science. 2006;313:1788–92. doi: 10.1126/science.1129308. [DOI] [PubMed] [Google Scholar]
- 128.Fedor MJ, Williamson JR. Nat Rev Mol Cell Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 129.Doudna JA, Lorsch JR. Nat Struct Mol Biol. 2005;12:395–402. doi: 10.1038/nsmb932. [DOI] [PubMed] [Google Scholar]
- 130.Been MD. Curr Top Microbiol Immunol. 2006;307:47–65. doi: 10.1007/3-540-29802-9_3. [DOI] [PubMed] [Google Scholar]
- 131.Perrotta AT, Shih I, Been MD. Science. 1999;286:123–6. doi: 10.1126/science.286.5437.123. [DOI] [PubMed] [Google Scholar]
- 132.Nakano S, Chadalavada DM, Bevilacqua PC. Science. 2000;287:1493–97. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 133.Das SR, Piccirilli JA. Nat Chem Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 134.Ke AL, Zhou KH, Ding F, Cate JHD, Doudna JA. Nature. 2004;429:201–05. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- 135.Ferre-D’Amare AR, Zhou K, Doudna JA. Nature. 1998;395:567–74. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 136.Koo S, Novak T, Piccirilli J. In: Ribozymes and RNA Catalysis. Lilley DMJ, Eckstein F, editors. The Royal Society of Chemistry; Cambridge: 2008. pp. 92–122. [Google Scholar]
- 137.Liu HN, Robinet JJ, Ananvoranich S, Gauld JW. J Phys Chem B. 2007;111:439–45. doi: 10.1021/jp064292n. [DOI] [PubMed] [Google Scholar]
- 138.Buzayan JM, Hampel A, Bruening G. Nuc Acids Res. 1986;14:9729–43. doi: 10.1093/nar/14.24.9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Buzayan JM, Feldstein PA, Segrelles C, Bruening G. Nuc Acids Res. 1988;16:4009–23. doi: 10.1093/nar/16.9.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.van Tol H, Buzayan JM, Feldstein PA, Eckstein F, Bruening G. Nuc Acids Res. 1990;18:1971–75. doi: 10.1093/nar/18.8.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Walter NG, Burke JM. Curr Opin Chem Biol. 1998;2:24–30. doi: 10.1016/s1367-5931(98)80032-x. [DOI] [PubMed] [Google Scholar]
- 142.Fedor MJ. J Mol Biol. 2000;297:269–91. doi: 10.1006/jmbi.2000.3560. [DOI] [PubMed] [Google Scholar]
- 143.Kuzmin YI, Da Costa CP, Cottrell JW, Fedor MJ. J Mol Biol. 2005;349:989–1010. doi: 10.1016/j.jmb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 144.Cottrell JW, Kuzmin YI, Fedor MJ. J Biol Chem. 2007;282:13498–507. doi: 10.1074/jbc.M700451200. [DOI] [PubMed] [Google Scholar]
- 145.Rupert PB, Massey AP, Sigurdsson ST, Ferre-D’Amare AR. Science. 2002;298:1421–24. doi: 10.1126/science.1076093. [DOI] [PubMed] [Google Scholar]
- 146.Torelli AT, Krucinska J, Wedekind JE. RNA. 2007;13:1052–70. doi: 10.1261/rna.510807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Macelrevey C, Salter JD, Krucinska J, Wedekind JE. RNA. 2008;14:1600–16. doi: 10.1261/rna.1055308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Torelli AT, Spitale RC, Krucinska J, Wedekind JE. Biochem Biophys Res Commun. 2008;371:154–58. doi: 10.1016/j.bbrc.2008.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pinard R, Hampel KJ, Heckman JE, Lambert D, Chan PA, Major F, Burke JM. EMBO J. 2001;20:6434–42. doi: 10.1093/emboj/20.22.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kuzmin YI, Da Costa CP, Fedor MJ. J Mol Biol. 2004;340:233–51. doi: 10.1016/j.jmb.2004.04.067. [DOI] [PubMed] [Google Scholar]
- 151.Rupert PB, Ferre-D’Amare AR. Nature. 2001;410:780–86. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 152.Salter J, Krucinska J, Alam S, Grum-Tokars V, Wedekind JE. Biochemistry. 2006;45:686–700. doi: 10.1021/bi051887k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lebruska LL, Kuzmine II, Fedor MJ. Chem Biol. 2002;9:465–73. doi: 10.1016/s1074-5521(02)00130-8. [DOI] [PubMed] [Google Scholar]
- 154.Gao JL, Amara P, Alhambra C, Field MJ. J Phys Chem A. 1998;102:4714–21. [Google Scholar]
- 155.Nam K, Gao JL, York DM. RNA. 2008;14:1501–07. doi: 10.1261/rna.863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nam KH, Gao JL, York DM. J Am Chem Soc. 2008;130:4680–91. doi: 10.1021/ja0759141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Forster AC, Symons RH. Cell. 1987;50:9–16. doi: 10.1016/0092-8674(87)90657-x. [DOI] [PubMed] [Google Scholar]
- 158.Scott WG. Quart Rev Biophys. 1999;32:241–84. doi: 10.1017/s003358350000353x. [DOI] [PubMed] [Google Scholar]
- 159.Martick M, Horan LH, Noller HF, Scott WG. Nature. 2008;454:899–U57. doi: 10.1038/nature07117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pley HW, Flaherty KM, Mckay DB. Nature. 1994;372:68–74. doi: 10.1038/372068a0. [DOI] [PubMed] [Google Scholar]
- 161.Scott WG, Finch JT, Klug A. Cell. 1995;81:991–1002. doi: 10.1016/s0092-8674(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 162.Wedekind JE, McKay DB. Annu Rev Biohys Biomol Struct. 1998;27:475–502. doi: 10.1146/annurev.biophys.27.1.475. [DOI] [PubMed] [Google Scholar]
- 163.Blount KE, Uhlenbeck OC. Annu Rev Biohys Biomol Struct. 2005;34:415–40. doi: 10.1146/annurev.biophys.34.122004.184428. [DOI] [PubMed] [Google Scholar]
- 164.Martick M, Lee TS, York DM, Scott WG. Chem Biol. 2008;15:332–42. doi: 10.1016/j.chembiol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nelson JA, Uhlenbeck OC. RNA. 2008;14:605–15. doi: 10.1261/rna.912608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Leclerc F, Karplus M. J Phys Chem B. 2006;110:3395–409. doi: 10.1021/jp053835a. [DOI] [PubMed] [Google Scholar]
- 167.Lee TS, Silva-Lopez C, Martick M, Scott WG, York DM. J Chem Theor Comp. 2007;3:325–27. doi: 10.1021/ct6003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Torres RA, Himo F, Bruice TC, Noodleman L, Lovell T. J Am Chem Soc. 2003;125:9861–67. doi: 10.1021/ja021451h. [DOI] [PubMed] [Google Scholar]
- 169.Lee TS, Lopez CS, Giambasu GM, Martick M, Scott WG, York DM. J Am Chem Soc. 2008;130:3053–64. doi: 10.1021/ja076529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Boero M, Park JM, Hagiwara Y, Tateno M. J Phys -Cond Matter. 2007;19:365217–25. doi: 10.1088/0953-8984/19/36/365217. [DOI] [PubMed] [Google Scholar]
- 171.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. Science. 2000;289:905–20. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 172.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. Science. 2000;289:920–30. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 173.Trobro S, Aqvist J. Proc Natl Acad Sci USA. 2005;102:12395–400. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Trobro S, Aqvist J. Biochemistry. 2006;45:7049–56. doi: 10.1021/bi0605383. [DOI] [PubMed] [Google Scholar]
- 175.Trobro S, Aqvist J. Mol Cell. 2007;27:758–66. doi: 10.1016/j.molcel.2007.06.032. [DOI] [PubMed] [Google Scholar]
- 176.Trobro S, Aqvist J. Biochemistry. 2008;47:4898–906. doi: 10.1021/bi8001874. [DOI] [PubMed] [Google Scholar]
- 177.Sharma PK, Xiang Y, Kato M, Warshel A. Biochemistry. 2005;44:11307–14. doi: 10.1021/bi0509806. [DOI] [PubMed] [Google Scholar]
- 178.Aqvist J, Warshel A. Chem Rev. 1993;93:2523–44. [Google Scholar]
- 179.Sievers A, Beringer M, Rodnina MV, Wolfenden R. Proc Natl Acad Sci USA. 2004;101:12397–98. doi: 10.1073/pnas.0402488101. [DOI] [PMC free article] [PubMed] [Google Scholar]