

Abstract
β-Methylamino-L-alanine (BMAA) has been proposed as a global contributor to neurodegenerative diseases, including Parkinson-dementia complex (PDC) of Guam and Alzheimer’s disease (AD). The literature on the effects of BMAA is conflicting with some but not all in vitro data supporting a neurotoxic action, and experimental animal data failing to replicate the pattern of neurodegeneration of these human diseases, even at very high exposures. Recently, BMAA has been reported in human brain from individuals afflicted with PDC or AD. Some of the BMAA in human tissue reportedly is freely extractable (free) while some is protein-associated and liberated by techniques that hydrolyze the peptide bond. The latter is especially intriguing since BMAA is a non-proteinogenic amino acid that has no known tRNA. We attempted to replicate these findings with techniques similar to those used by others; despite more than adequate sensitivity, we were unable to detect free BMAA. Recently, using a novel stable isotope dilution assay we again were unable to detect free or protein-associated BMAA in human cerebrum. Here we review the development of our new assay for tissue detection of BMAA and show that we are able to detect free BMAA in liver but not cerebrum, nor do we detect any protein-associated BMAA in mice fed this amino acid. These studies demonstrate the importance of a sensitive and specific assay for tissue BMAA and seriously challenge the proposal that BMAA is accumulating in human brain.
Introduction
Parkinson-dementia complex (PDC) of the Chamorro on Guam and surrounding Mariana Islands is a unique neurodegenerative disease that remains a significant public health burden to the older members of this indigenous ethnic minority (Hirano et al., 1961; Waring, 1994; Murakami, 1999; Oyanagi and Wada, 1999; Wiederholt, 1999; Galasko et al., 2002). Previously, PDC was often co-morbid with amyotrophic lateral sclerosis (ALS); however, the incidence of ALS among Chamorros has returned to levels comparable to the rest of the world over the last 50 years (Garruto et al., 1985; Wiederholt, 1999; Plato et al., 2002; Waring et al., 2004). Current data indicate that PDC is a complex phenotype that derives significant contributions from advancing age, inherited susceptibilities, and as yet to be clarified environmental factors that broadly ultimately derive from progressive ‘westernization’ of Chamorros or consumption of initiators or promoters that are indigenous to these islands. No proposed environmental factor has withstood scrutiny as a sole cause of ALS or PDC (Garruto et al., 1984; Yanagihara et al., 1984; Spencer et al., 1987; Hudson and Rice, 1990; Duncan, 1992; Esclaire et al., 1999; Plato et al., 2002; Plato et al., 2003), leading to speculation of a yet to be identified gene-environment interaction (Morris et al., 2004).
The cycad plant was an early target of suspicion as the source of an environmental toxicant that promoted or caused ALS/PDC, since it was noted that Chamorros had relied heavily on cycads for food and medicinal purposes (Spencer et al., 1987). Arthur Bell first identified β-Methylamino-L-alanine (BMAA), a nonproteinogenic amino acid, as one of the many unusual amino acids present in cycad plants. BMAA is synthesized by cyanobacteria, and cyanobacteria are found on the roots of cycads (Cox and Banack, 2006). Indeed, cycad flour preparations contain BMAA (Kisby et al., 1992). BMAA can be toxic to neurons in some in vitro models (Allen et al., 1995; Rao et al., 2006; Lobner et al., 2007). Other groups were unable to replicate BMAA-induced in vivo neurotoxicity (Wilson et al., 2002; Cruz-Aguado et al., 2006; Santiago et al., 2006), and efforts to replicate a neurodegenerative illness similar to ALS/PDC by feeding BMAA to non-human primates were not successful (Spencer et al., 1987). Finally, it seemed impossible for humans to ingest sufficient BMAA to achieve levels expected to produce neurodegeneration (Duncan et al., 1990). For these reasons the “cycad/BMAA hypothesis” waned.
The cycad/BMAA hypothesis was resurrected in 2003 with the proposal that Chamorros are exposed to high levels of BMAA through biomagnification from cyanobacteria to cycads to flying foxes to humans (Cox et al., 2003). As a test of this proposal, others quantified extractable or free BMAA in museum specimens of flying foxes (investigation was limited by scarcity of tissue samples) and in frontal lobe specimens from Chamorros who died of PDC, Chamorro controls, patients from Western Canada who died of Alzheimer’s disease (AD), and their respective controls. While BMAA was detected in frontal lobe from all PDC patients, there was no detectable free BMAA in all thirteen frontal lobe samples from control individuals on Guam or in Western Canada who died without neurodegenerative disease (Cox et al., 2003; Banack et al., 2006). Surprisingly, two frontal lobe specimens from patients in Western Canada who died of AD had levels of free BMAA that were comparable to those observed in PDC patients from Guam. In a subsequent study, protein-associated BMAA, liberated by techniques used for peptide bond hydrolysis, was observed at many fold greater concentration than free BMAA in cerebral cortex from all six Chamorros who died of PDC, one Chamorro who died without history of neurodegenerative disease, and both Canadians who died with AD but none of thirteen Canadians who died without evidence of neurologic disease (Murch et al., 2004b). In an additional report, BMAA was quantified in frontal cortex samples as a free amino acid in 83% of Chamorro PDC patients (3–10 μg/g) and as a protein-associated amino acid in 100% of Chamorro PDC patients (149–1190 μg/g); again, both forms of BMAA were found at comparable levels in two Canadians who died of progressive neurodegenerative disease (Murch et al., 2004a). More recently, BMAA was identified as a protein-associated amino acid in the brains of humans afflicted with AD and ALS, and in low levels in one patient who died from Huntington’s Disease (HD), furthering the hypothesis that BMAA bioaccumulation is a factor in the development of several neurodegenerative diseases (Pablo et al., 2009).
Motivated by the singular importance of an environmental contributor to neurodegenerative diseases worldwide, we previously attempted to replicate these results from human brain using a similar method as others. BMAA spiked into human brain homogenates was detectable with the range of levels reported by others and with a limit of detection for free BMAA that was an order of magnitude less than levels reported using a similar fluorescent derivatization/HPLC assay, yet no free BMAA was detected in any of the sixty samples we investigated: frontal cortex, temporal cortex, and cerebellar cortex from ten Chamorros (eight with PDC and two controls) and ten Caucasians from Seattle (five with AD and five controls) (Montine et al., 2005). There were limitations to these experiments. First, we limited our analysis to extractable (free) BMAA and did not pursue protein-associated BMAA. Second, since we did not detect BMAA in human tissue, we did not have an authentic positive control for BMAA in tissue. Recently, we have addressed these limitations. Indeed, we have reported in brief results from a new stable isotope dilution assay that we developed fro the detection of BMAA from these same human samples; no free or protein-associated BMAA was detected using this alternative method despite ample sensitivity (Snyder et al., 2009). Here, we describe in detail the development of our novel stable isotope dilution assay for BMAA, and report results from mice fed BMAA for a month at a dose that mimics proposed environmental levels (Cruz-Aguado et al., 2006).
Methods
Synthesis of D3-BMAA
Our synthetic scheme followed a previously published method (Figure 1) (Ziffer, 1990). Solid sodium hydroxide (0.88 g) was added to a frozen solution of 1.0g D3-methylamine hydrochloride (Cambridge Isotope Laboratories, 98% isotopically pure) in 4mL water. The solution was thawed slowly to room temperature and slowly distilled. The collection flask was kept in a dry ice/acetone bath. The frozen distillate was warmed to room temperature and then reacted with 0.37g of acetamidoacrylic acid (Sigma Aldrich) at 35°C for 22 hours (Ziffer, 1990). Unreacted methylamine was removed from the resulting solution using a rotovap. An excess of 3M HCl was added to the intermediate and the solution was refluxed at 100°C for 2 hours. Hydrochloric acid was removed using a Speedvac, and the resulting product was recrystallized in ethanol and water at 4°C. Crystals were then filtered, washed with cold ethanol, dried under vacuum, and stored at −20°C. This protocol yielded 180mg of product that was characterized using 1H- and 13C-NMR (Figures 2 and 3). Unlabelled (H3-) BMAA from Sigma Aldrich (97% pure) was also analyzed using NMR spectroscopy to provide a reference for the labeled BMAA synthesized in our lab. Proton NMR spectra were obtained on a Bruker 300MHz spectrometer and carbon spectra were obtained using a Bruker 500MHz spectrometer. Samples were dissolved in deuterium oxide for all NMR experiments.
Figure 1. Synthesis scheme for D3-BMAA.

Acetamidoacrylic acid was reacted with methylamine overnight, followed by acid hydrolysis of the acetyl group to achieve the final product. D3-BMAA was purified by crystallization in ethanol and water.
Figure 2. 1H-NMR spectra of H3-BMAA (top panel) and D3-BMAA (bottom panel).

The singlet peak at 2.78 ppm is absent in the deuterated isotopomer.
Figure 3. 13C-NMR spectra of H3-BMAA (top panel) and D3-BMAA (bottom panel).

The singlet peak at 37.5 ppm is absent in the deuterated isotopomer (inset shows zoom of 30 to 50 ppm domain).
Gas chromatography (GC)/mass spectrometry (MS)
Samples for GC-MS analysis were derivatized according to the procedure developed by Guo, et al. (Figure 4) (Guo et al., 2007). This one-step procedure utilizes ethyl chloroformate (ECF) that converts both amines and the carboxylic acid into esters. Derivatized solutions were extracted into dichloromethane, dried under nitrogen, and reconstituted in dichloromethane prior to GC-MS analysis on an HP 5971A GC-MS. The injector port was held at 250°C and operated in splitless mode with 1μL injections. A 60m × 0.25mm i.d. × 0.25μm Restek film RTX-5MS column was employed for chromatographic separation using helium (1.0 mL/s) as carrier gas. The initial oven temperature of 55°C was increased to 200°C at a rate of 15°C/min, then to 300°C at a rate of 5°C/min, and then by 15°C/min to 320°C and held constant for three minutes. Total run time was 34.50 minutes. Standards were run to verify fragmentation patterns for BMAA and its isotopically labeled analog, and confirmed results published by others (Guo et al., 2007). The mass spectrometer was operated in electron ionization (EI) mode with electron energy of 70 eV, and in selected ion monitoring (SIM) mode to monitor four ions: m/z 116, m/z 119, m/z 245 and m/z 248. The former two ions are used for quantification and the latter two ions used as qualifying ions. Samples were run within 72 hours of derivatization. Control standards run at the beginning and at the conclusion of each analysis showed that no significant degradation occurred even after three days.
Figure 4. BMAA derivatization for GC-MS.

Derivatization of BMAA was accomplished using a pyridine catalyzed reaction with ethylchloroformate and ethanol. This one-step reaction simultaneously converts both the amino and carboxylic acid groups.
Mice
Flash frozen liver and cerebrum samples from eight mice fed BMAA (28 mg/kg/d × 30d) and six mice fed control diet were used (Cruz-Aguado et al., 2006). No behavioral differences were observed in these BMAA fed mice (Cruz-Aguado et al., 2006). Following behavioral assessments, mice were sacrificed, perfused with ice-cold PBS, and selected tissues rapidly dissected, flash frozen, and stored at −80°C.
Tissue preparation
We prepared standard curves for quantification of extractable BMAA in each organ investigated by preparing homogenates according to the method of others (Duncan et al., 1989; Duncan et al., 1991). Briefly, tissue was thawed in ice cold HCl (1 M) containing a fixed amount of D3-BMAA and varying amounts of H3-BMAA that span the concentrations previously reported by others. Tissue was disrupted by ultrasonication, and sedimented to precipitate protein. The supernatant was transferred to a screw top test tube containing chilled chloroform (4:1 v:v). After vigorous mechanical shaking for 5 min the samples were separated into two phases by centrifugation (3000 rpm, 4°C, 10 min), the aqueous phase was transferred to a clean test tube and derivatized directly as described above using ethyl chloroformate. All samples were run in triplicate. For detection of protein-associated BMAA the protocol of others was followed (Murch et al., 2004a; Banack et al., 2006). The resulting protein pellet described above was re-homogenized in 0.1N trichloroacetic acid (TCA, 1:1), incubated for 30 min at 4°C, and sedimented for 20 min at 25,000×g. The TCA-precipitated protein pellet was washed twice with PBS (pH 7.4) and suspended in 6.0N HCI spiked with H3-BMAA of varying amounts and a fixed amount of D3-BMAA, incubated for 24 h at 110 °C to achieve total protein hydrolysis (Fountoulakis and Lahm, 1998), cooled to room temperature for 2 h prior to ultrafiltration, and evaporated to dryness using a Speedvac Concentrator. These samples were derivatized with ethyl chloroformate, extracted in dichloromethane, and analyzed as described. Again, all samples were run in triplicate. Since BMAA is a non-proteinogenic amino acid, we know of no standard to use for amino acid hydrolysis that has BMAA already incorporated into protein. Analysis showed no appreciable decay of BMAA or its isotopically labeled analog under hydrolysis conditions.
Results
1H-NMR spectrum showed that the singlet peak representing the -CH3 group at 2.78 ppm is absent for the D3-BMAA sample while other proton signals have a similar pattern of magnetic resonance as commercial H3-BMAA (inset), verifying that the target compound was achieved (Figure 2). 13C-NMR spectra for H3-BMAA and its deuterated isotopomer are shown in (Figure 3). Proton decoupled 13C-NMR showed that each peak in the reference spectrum appeared as a singlet (inset). Deuterium substitution on the labeled compound split the signal from the carbon in the methyl group.
GC retention times of ECF-derivatized H3-BMAA and D3-BMAA (Figure 4) were 19.55 min and 19.52 min, respectively. In scan mode, the MS fragmentation pattern for H3-BMAA (inset) was identical to that reported by others (Guo et al., 2007), including m/z 116 as the most abundantly detected fragment; these authors suggest a possible structure for this ion (Guo et al., 2007). As expected, D3-BMAA had similar a fragmentation pattern with a corresponding shift of 3 mass units (Figure 5) for each fragment; again, the m/z 119 was the most abundantly detected ion. We used m/z 116 and 119 for quantification, and m/z 245 and 248 as qualifying ions in selected ion monitoring (SIM) mode to generate a linear calibration curve (Figure 6) over a 100-fold concentration range (10 pg to 1000 pg) in mouse liver, mouse cerebral cortex, and human cerebral cortex; r2 = 1.00 for each seven point line in each tissue homogenate. Our limit of detection for free BMAA was 0.1 μg/g tissue, limit of quantification = 0.2 μg/g tissue. These data showed that our assay provides more than enough sensitivity to detect the 3 tom > 1,000 μg/g BMAA that has been reported in human brain (Murch et al., 2004b). Development of the assay revealed the problem of co-elution of other compounds near BMAA. We addressed this issue by using the longer 60m column, and verified compound identification by monitoring the retention times of the two ions for BMAA. Additional proof of identification was provided by the use of an internal standard, and two ions for the isotopically labeled analog were simultaneously monitored during sample analyses.
Figure 5. Fragmentation ions of commercial BMAA (top panel) and its deuterated isotopomer (bottom panel).

Electron Ionization mass spectrometric analysis of H3-BMAA shows a fragmentation pattern consistent with results reported by others. Analysis of D3-BMAA showed a shift of +3 mass units for compared to H3-BMAA.
Figure 6. Calibration curve for D3-BMAA.
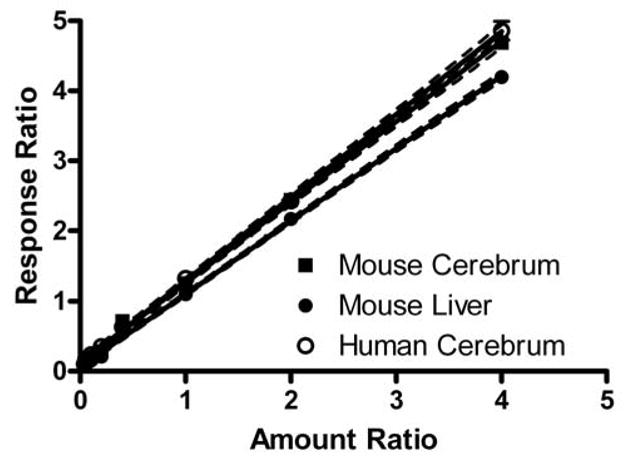
Solutions of 10 to 1000 pg/μl H3-BMAA and 250 pg/μl D3-BMAA were combined with homogenates of mouse liver or cerebrum, or human cerebrum for comparison, derivatized with ethyl chloroformate, and analyzed by GC/MS. The ratio of the amount of H3-BMAA to D3-BMAA in each sample was plotted against the ratio of relative response for m/z 116 (H3-BMAA) to m/z 119 (D3-BMAA). Linear regression analysis with 95% confidence intervals are plotted along with the seven amount ratios for each tissue homogenate (r2 = 1.00 and P < 0.0001 for each). Slope for mouse cerebrum and mouse liver were 1.15 ± 0.01 and 1.04 ± 0.01; human cerebrum yielded results virtually identical to mouse cerebrum.
Flash frozen liver and cerebral cortical samples from eight mice fed BMAA at a dose that reflects proposed environmental levels (28 mg/kg/d × 30d) and six mice fed control diet without BMAA were analyzed in triplicate. Sample analysis was blinded until completion of the study. These mice had been used in behavioral experiments that failed to detect any significant change associated with BMAA exposure (Cruz-Aguado et al., 2006). We detected free BMAA in all 24 runs of liver from mice fed BMAA (mean ± SEM = 4.8 ± 1.3 μg/g for 8 average triplicate determinations) but were unable to detect BMAA in any of the livers from mice fed control diet; the average coefficient of variation for triplicate determinations of hepatic free BMAA was 1.6 % (Figure 7). We determined that no appreciable decay of spiked BMAA occurred when samples were acid hydrolyzed for determination of protein-associated BMAA. No BMAA was detected in any of the acid hydrolyzed protein fractions from mouse liver despite our ability to detect abundant phenylalanine (migrating near BMAA) and other amino acids in our chromatographic system.
Figure 7. Mouse hepatic free BMAA after feeding.

GC-MS m/z chromatograms of stable isotope dilution assay of mouse liver. H3-BMAA (m/z 116 and 245, arrows) along with its deuterated standard (m/z 119 and 248, arrows) were detected in the extractable fraction, also called free BMAA, in mice fed BMAA (A) but none of the mice fed control diet that lacked BMAA (B). Scattergram shows average values of triplicate determinations of hepatic free BMAA as well as mean ± SEM for all eight mice fed BMAA (C). No detectable free or protein-associated BMAA was detected in any of the six mice fed control diets.
We analyzed flash frozen cerebral cortex from the same fourteen mice. Standards for a calibration curve were prepared using mouse brain homogenates from control mice that had not been exposed to BMAA. Matrix standards were run to verify a lack of BMAA in the homogenates. Standards were prepared by spiking the homogenates with a fixed amount of deuterated BMAA and varying amount of commercial BMAA to create a 7-point calibration curve. Using this data we established the following parameters for the detection of free BMAA for this assay: limit of detection = 0.1 μg/g tissue, limit of quantification = 0.2 μg/g tissue. These experiments were repeated for our analysis of protein-associated BMAA and the assay parameters were follows: limit of detection = 5.0 μg/g tissue, limit of quantification = 10.0 μg/g tissue. These limits are well within the range of levels reported in the literature, and the sensitivity and specificity of this stable isotope dilution assay makes this a suitable method for detection of BMAA in tissue. Recovery was greater than 95% for these experiments, verifying that this BMAA was not significantly degraded during sample preparation.
Finally, we used our new stable isotope dilution assay to determine free and protein-associated BMAA in frontal and temporal cortex samples from individuals who died of AD or PDC, as well as control samples from individuals who died without neurodegenerative disease (Snyder et al., 2009) (Table 1). Again, no free or protein-associated BMAA was detected in any of these samples with the same recovery and limits of detection and quantification as noted above for mouse cerebral cortical homogenates (Figure 8).
Table 1.
Description of individuals’ tissue that was used in this study
| Number and Gender | Age range | Tissue | ||
|---|---|---|---|---|
| Chamorros on Guam | Control | 2F: 0M | 61 and 93 | Frontal and Temporal Cortex for each |
| PDC | 5F: 3M | 64 to 89 | ||
| Residents of Seattle Area | Control | 3F: 2 M | 82 to 91 | |
| AD | 3F: 2 M | 74 to 83 |
Figure 8. Human Cerebral Cortex.

GC-MS m/z chromatograms of stable isotope dilution assay of human cerebral cortex. H3-BMAA (m/z 116 and 245, arrows) along with its deuterated standard (m/z 119 and 248, arrows) were detected in the extractable fraction, also called free BMAA, in samples spiked ex vivo with 250 pg/μl H3-BMAA (A). No H3-BMAA was detected in any human sample that was not spiked (B).
Discussion
Exposure to BMAA has been proposed as a possible global contributor to neurodegenerative diseases, including PDC and AD (Cox et al., 2003; Cox and Banack, 2006). Since these initial intriguing data and speculations about a possible role for cyanobacteria contaminating global water supplies with BMAA (Cox et al., 2003), these findings have been commented on in news and editorial sections of premier scientific and medical journals (Science, (Cox and Banack, 2006; Duncan and Marini, 2006; Miller, 2006) Nature (Whitfield, 2003), JAMA (Hampton, 2003; Kuehn, 2005)) and the national media including the Boston Globe,(2005) the Washington Post, (Stein, 2005) and the New Yorker Magazine (Weiner, 2005). There are thousands of websites dealing with this topic; one example is WaterTiger™ in British Columbia that displays a local newspaper article “Algae linked to Alzheimer’s” (Munro, 2005). Clearly, there is widespread interest and concern about this potential water-based toxicant and its possible role in neurodegeneration across the globe; thus it has become important to develop new tools to analyze and detect BMAA in tissue samples. Human tissue samples are complex in nature; therefore a straightforward assay that provides unambiguous data is necessary. This developed assay is well suited for tissue analyses and monitoring four specific ions unique to BMAA and its isotopically labeled analog allow for clear identification of the analyte of interest. Furthermore, we have addressed problems of co-elution by modifying the assay so that no other compounds that are similar to BMAA cloud the analysis. Identification is verified by running standards prior to sample analysis to confirm retention times.
We have been unable to replicate other’s finding of detectable free BMAA in human cerebral cortex (Montine et al., 2005; Snyder et al., 2009) using two assays. Since we have been unable to detect BMAA in any human sample except by ex vivo spiking, there remains the possibility that BMAA is somehow degraded in our preparations. We undertook studies to address this potential limitation here and have developed an assay with 95% recovery for the detection of BMAA in tissue. The data show clearly that this stable isotope dilution assay is capable of detecting BMAA with high sensitivity, and that free BMAA was present in liver of mice fed this compound at a dose that approximates proposed environmental exposures (Cruz-Aguado et al., 2006). Indeed, each of eight mice fed BMAA for one month had detectable levels of extractable, or free, BMAA in liver homogenates. None of six control mice that received identical diets without BMAA had detectable hepatic free BMAA. No protein-associated hepatic BMAA was detected in any of these mice using this assay. Moreover, no free or protein-associated BMAA was detected in brain samples from any of these mice. We stress the importance of these results since BMAA exposure to humans is in question. The mice that had detectable levels of BMAA were exposed to BMAA, while none of the control diet mice had detectable levels of BMAA. These data support the studies done by others, and while our results for human cerebrum contradict literature results, we acknowledge the limitations of the assay. Indeed, mice exposed to higher levels of BMAA had readily detectable levels of BMAA in brain tissue (Duncan et al., 1991) and it is likely that different feeding doses would yield different analysis results using this assay. While this assay was designed to readily detect BMAA within the range reported by others (Pablo et al., 2009), there is a need for more sensitive assays and methods to detect trace amounts of this neurotoxin. Furthermore, as mentioned before, analysis of complex samples must be verified due to the potential for misinterpretation. A thorough mass and retention time analysis was undertaken during assay development to confirm correct identification of the analyte of interest. Others have employed 15N-BMAA to assess its oral bioavailability in cynomolgous monkeys using GC/MS; following oral dosing, 80% of the administered BMAA was absorbed into the systemic circulation (Duncan et al., 1992). This same group examined the kinetics and mechanism of BMAA influx across the blood-brain barrier in a rat brain perfusion model. BMAA influx is sodium-independent, saturable, and mediated by neutral amino acid carriers (Smith et al., 1992). Our results are consistent with these earlier studies but suggest that, although possible, in vivo transport of BMAA across the blood-brain barrier in mice apparently is very limited. If BMAA was present in the cerebral samples examined using our method, then it was at levels that were below the detection limit of this assay.
Others have observed apparently protein-associated BMAA that exceeds free BMAA by 10- to over 100-fold, and proposed it as a “slow release” form of the putative neurotoxin (Murch et al., 2004a). We are unaware of any group that has replicated this finding. We have not been able to detect protein-associated BMAA in any human sample or in the mouse liver or cerebral samples investigated here, including the liver samples from mice fed BMAA that had detectable free hepatic BMAA. We hasten to add that BMAA is a non-proteinogenic amino acid for which there is no known tRNA, and that the conditions used to liberate protein-associated in our assay BMAA were sufficiently chaotropic to impede non-covalent interactions.
We conclude that dietary exposure to BMAA in mice results in detectable hepatic free BMAA, but not cerebral free BMAA, or protein-associated BMAA in either organ that exceeds the detection limit of 0.1μg/g tissue. Moreover, BMAA does not detectably accumulate in older adults from Guam or Seattle, regardless of the presence or absence of PDC or AD as determined by the same assay (Snyder et al., 2009). While these data cannot speak to the potential health significance of other toxins that may be present in cycads (Steele and McGeer, 2008), they seriously challenge the proposal that BMAA accumulates in brain or represents a global health concern for toxicant-induced neurodegeneration.
Acknowledgments
This work was supported by grants from the NIH (AG05136, AG029808) and the Nancy and Buster Alvord Endowment. We thank L. Kruse and M. Gelb for helpful advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Algae that produces threat to humans is more common than thought. Boston Globe 2005 [Google Scholar]
- Allen CN, Omelchenko I, Ross SM, Spencer P. The neurotoxin, beta-N-methylamino-L-alanine (BMAA) interacts with the strychnine-insensitive glycine modulatory site of the N-methyl-D-aspartate receptor. Neuropharmacology. 1995;34:651–658. doi: 10.1016/0028-3908(95)00043-6. [DOI] [PubMed] [Google Scholar]
- Banack SA, Murch SJ, Cox PA. Neurotoxic flying foxes as dietary items for the Chamorro people, Marianas Islands. J Ethnopharmacol. 2006;106:97–104. doi: 10.1016/j.jep.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Cox PA, Banack SA. A nonprotein amino acid and neurodegeneration. Science. 2006;314:1242–1242. doi: 10.1126/science.314.5803.1242a. [DOI] [PubMed] [Google Scholar]
- Cox PA, Banack SA, Murch SJ. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc Natl Acad Sci U S A. 2003;100:13380–13383. doi: 10.1073/pnas.2235808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Aguado R, Winkler D, Shaw CA. Lack of behavioral and neuropathological effects of dietary beta-methylamino-L-alanine (BMAA) in mice. Pharmacol Biochem Behav. 2006;84:294–299. doi: 10.1016/j.pbb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Duncan MW. beta-Methylamino-L-alanine (BMAA) and amyotrophic lateral sclerosis-parkinsonism dementia of the western Pacific. Ann N Y Acad Sci. 1992;648:161–168. doi: 10.1111/j.1749-6632.1992.tb24534.x. [DOI] [PubMed] [Google Scholar]
- Duncan MW, Kopin IJ, Crowley JS, Jones SM, Markey SP. Quantification of the putative neurotoxin 2-amino-3-(methylamino)propanoic acid (BMAA) in cycadales: analysis of the seeds of some members of the family Cycadaceae. J Anal Toxicol. 1989;13 doi: 10.1093/jat/13.3.169. suppl A-G. [DOI] [PubMed] [Google Scholar]
- Duncan MW, Marini AM. Debating the cause of a neurological disorder. Science. 2006;313:1737–1737. doi: 10.1126/science.313.5794.1737b. [DOI] [PubMed] [Google Scholar]
- Duncan MW, Markey SP, Weick BG, Pearson PG, Ziffer H, Hu Y, Kopin IJ. 2-Amino-3-(methylamino)propanoic acid (BMAA) bioavailability in the primate. Neurobiol Aging. 1992;13:333–337. doi: 10.1016/0197-4580(92)90047-2. [DOI] [PubMed] [Google Scholar]
- Duncan MW, Steele JC, Kopin IJ, Markey SP. 2-Amino-3-(methylamino)-propanoic acid (BMAA) in cycad flour: an unlikely cause of amyotrophic lateral sclerosis and parkinsonism-dementia of Guam. Neurology. 1990;40:767–772. doi: 10.1212/wnl.40.5.767. [DOI] [PubMed] [Google Scholar]
- Duncan MW, Villacreses NE, Pearson PG, Wyatt L, Rapoport SI, Kopin IJ, Markey SP, Smith QR. 2-Amino-3-(Methylamino)-Propanoic Acid (Bmaa) Pharmacokinetics and Blood-Brain-Barrier Permeability in the Rat. Journal of Pharmacology and Experimental Therapeutics. 1991;258:27–35. [PubMed] [Google Scholar]
- Esclaire F, Kisby G, Spencer P, Milne J, Lesort M, Hugon J. The Guam cycad toxin methylazoxymethanol damages neuronal DNA and modulates tau mRNA expression and excitotoxicity. Exp Neurol. 1999;155:11–21. doi: 10.1006/exnr.1998.6962. [DOI] [PubMed] [Google Scholar]
- Fountoulakis M, Lahm HW. Hydrolysis and amino acid composition analysis of proteins. Journal of Chromatography A. 1998;826:109–134. doi: 10.1016/s0021-9673(98)00721-3. [DOI] [PubMed] [Google Scholar]
- Galasko D, Salmon DP, Craig UK, Thal LJ, Schellenberg G, Wiederholt W. Clinical features and changing patterns of neurodegenerative disorders on Guam, 1997–2000. Neurology. 2002;58:90–97. doi: 10.1212/wnl.58.1.90. [DOI] [PubMed] [Google Scholar]
- Garruto RM, Fukatsu R, Yanagihara R, Gajdusek DC, Hook G, Fiori CE. Imaging of calcium and aluminum in neurofibrillary tangle-bearing neurons in parkinsonism-dementia of Guam. Proc Natl Acad Sci U S A. 1984;81:1875–1879. doi: 10.1073/pnas.81.6.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garruto RM, Yanagihara R, Gajdusek DC. Disappearance of high-incidence amyotrophic lateral sclerosis and parkinsonism-dementia on Guam. Neurology. 1985;35:193–198. doi: 10.1212/wnl.35.2.193. [DOI] [PubMed] [Google Scholar]
- Guo T, Geis S, Hedman C, Arndt M, Krick W, Sonzogni W. Characterization of Ethyl Chloroformate Derivative of beta-Methylamino-l-alanine . J Am Soc Mass Spectrom. 2007;18:817–825. doi: 10.1016/j.jasms.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Hampton T. Food chain of evidence points to brain toxin. Jama-Journal of the American Medical Association. 2003;290:2788–2789. doi: 10.1001/jama.290.21.2788. [DOI] [PubMed] [Google Scholar]
- Hirano A, Kurland LT, Krooth RS, Lessell S. Parkinsonism-dementia complex, an endemic disease on the island of Guam. I. Clinical features. Brain. 1961;84:642–661. doi: 10.1093/brain/84.4.642. [DOI] [PubMed] [Google Scholar]
- Hudson AJ, Rice GP. Similarities of guamanian ALS/PD to post-encephalitic parkinsonism/ALS: possible viral cause. Can J Neurol Sci. 1990;17:427–433. doi: 10.1017/s0317167100031024. [DOI] [PubMed] [Google Scholar]
- Kisby GE, Ellison M, Spencer PS. Content of the neurotoxins cycasin (methylazoxymethanol beta-D-glucoside) and BMAA (beta-N-methylamino-L-alanine) in cycad flour prepared by Guam Chamorros. Neurology. 1992;42:1336–1340. doi: 10.1212/wnl.42.7.1336. [DOI] [PubMed] [Google Scholar]
- Kuehn BM. Environmental neurotoxin may pose health threat. Jama-Journal of the American Medical Association. 2005;293:2460–2462. doi: 10.1001/jama.293.20.2460. [DOI] [PubMed] [Google Scholar]
- Lobner D, Piana PM, Salous AK, Peoples RW. Beta-N-methylamino-L-alanine enhances neurotoxicity through multiple mechanisms. Neurobiol Dis. 2007;25:360–366. doi: 10.1016/j.nbd.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. Neurodegenerative disease. Guam’s deadly stalker: on the loose worldwide? Science. 2006;313:428–431. doi: 10.1126/science.313.5786.428. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Li K, Perl DP, Galasko D. Lack of beta-methylamino-l-alanine in brain from controls, AD, or Chamorros with PDC. Neurology. 2005;65:768–769. doi: 10.1212/01.wnl.0000174523.62022.52. [DOI] [PubMed] [Google Scholar]
- Morris HR, Steele JC, Crook R, Wavrant-De Vrieze F, Onstead-Cardinale L, Gwinn-Hardy K, Wood NW, Farrer M, Lees AJ, McGeer PL, Siddique T, Hardy J, Perez-Tur J. Genome-wide analysis of the parkinsonism-dementia complex of Guam. Arch Neurol. 2004;61:1889–1897. doi: 10.1001/archneur.61.12.1889. [DOI] [PubMed] [Google Scholar]
- Munro M. Algae toxin linked to Alzheimer’s. Victoria Times Colonist 2005 [Google Scholar]
- Murakami N. Parkinsonism-dementia complex on Guam - overview of clinical aspects. J Neurol. 1999;246(Suppl 2):II16–18. doi: 10.1007/BF03161077. [DOI] [PubMed] [Google Scholar]
- Murch SJ, Cox PA, Banack SA. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc Natl Acad Sci U S A. 2004a;101:12228–12231. doi: 10.1073/pnas.0404926101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murch SJ, Cox PA, Banack SA, Steele JC, Sacks OW. Occurrence of beta-methylamino-l-alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol Scand. 2004b;110:267–269. doi: 10.1111/j.1600-0404.2004.00320.x. [DOI] [PubMed] [Google Scholar]
- Oyanagi K, Wada M. Neuropathology of parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam: an update. J Neurol. 1999;246(Suppl 2):II19–27. doi: 10.1007/BF03161078. [DOI] [PubMed] [Google Scholar]
- Pablo J, Banack SA, Cox PA, Johnson TE, Papapetropoulos S, Bradley WG, Buck A, Mash DC. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol Scand. 2009 doi: 10.1111/j.1600-0404.2008.01150.x. [DOI] [PubMed] [Google Scholar]
- Plato CC, Galasko D, Garruto RM, Plato M, Gamst A, Craig UK, Torres JM, Wiederholt W. ALS and PDC of Guam: forty-year follow-up. Neurology. 2002;58:765–773. doi: 10.1212/wnl.58.5.765. [DOI] [PubMed] [Google Scholar]
- Plato CC, Garruto RM, Galasko D, Craig UK, Plato M, Gamst A, Torres JM, Wiederholt W. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam: changing incidence rates during the past 60 years. Am J Epidemiol. 2003;157:149–157. doi: 10.1093/aje/kwf175. [DOI] [PubMed] [Google Scholar]
- Rao SD, Banack SA, Cox PA, Weiss JH. BMAA selectively injures motor neurons via AMPA/kainate receptor activation. Exp Neurol. 2006;201:244–252. doi: 10.1016/j.expneurol.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Santiago M, Matarredona ER, Machado A, Cano J. Acute perfusion of BMAA in the rat’s striatum by in vivo microdialysis. Toxicol Lett. 2006;167:34–39. doi: 10.1016/j.toxlet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Smith QR, Nagura H, Takada Y, Duncan MW. Facilitated transport of the neurotoxin, beta-N-methylamino-L-alanine, across the blood-brain barrier. J Neurochem. 1992;58:1330–1337. doi: 10.1111/j.1471-4159.1992.tb11346.x. [DOI] [PubMed] [Google Scholar]
- Snyder LR, Cruz-Aguado R, Sadilek M, Galasko D, Shaw CA, Montine TJ. Lack of cerebral bmaa in human cerebral cortex. Neurology. 2009;72:1360–1361. doi: 10.1212/WNL.0b013e3181a0fed1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer PS, Nunn PB, Hugon J, Ludolph AC, Ross SM, Roy DN, Robertson RC. Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin. Science. 1987;237:517–522. doi: 10.1126/science.3603037. [DOI] [PubMed] [Google Scholar]
- Steele JC, McGeer PL. The ALS/PDC syndrome of Guam and the cycad hypothesis. Neurology. 2008;70:1984–1990. doi: 10.1212/01.wnl.0000312571.81091.26. [DOI] [PubMed] [Google Scholar]
- Stein R. Algae Toxin Is Widespread. Washington Post 2005 [Google Scholar]
- Waring SC. School of Public Health. University of Texas Health Science Center; Houston: 1994. Amyotrophic Lateral Sclerosis and Parkinsonism-Dementia Complex of Guam: Descriptive Epidemiology, Secular Trends, and Birth Cohort Effects on Incidence, 1950–1989. [Google Scholar]
- Waring SC, Esteban-Santillan C, Reed DM, Craig UK, Labarthe DR, Petersen RC, Kurland LT. Incidence of amyotrophic lateral sclerosis and of the parkinsonism-dementia complex of Guam, 1950–1989. Neuroepidemiology. 2004;23:192–200. doi: 10.1159/000078505. [DOI] [PubMed] [Google Scholar]
- Weiner J. The Tangle. The New Yorker. 2005:42. [Google Scholar]
- Whitfield J. Bug makes bats bad for brains. Nature.com 2003 [Google Scholar]
- Wiederholt WC. Neuroepidemiologic research initiatives on Guam: past and present. Neuroepidemiology. 1999;18:279–291. doi: 10.1159/000026223. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Khabazian I, Wong MC, Seyedalikhani A, Bains JS, Pasqualotto BA, Williams DE, Andersen RJ, Simpson RJ, Smith R, Craig UK, Kurland LT, Shaw CA. Behavioral and neurological correlates of ALS-parkinsonism dementia complex in adult mice fed washed cycad flour. Neuromolecular Med. 2002;1:207–221. doi: 10.1385/NMM:1:3:207. [DOI] [PubMed] [Google Scholar]
- Yanagihara R, Garruto RM, Gajdusek DC, Tomita A, Uchikawa T, Konagaya Y, Chen KM, Sobue I, Plato CC, Gibbs CJ., Jr Calcium and vitamin D metabolism in Guamanian Chamorros with amyotrophic lateral sclerosis and parkinsonism-dementia. Ann Neurol. 1984;15:42–48. doi: 10.1002/ana.410150108. [DOI] [PubMed] [Google Scholar]
- Ziffer YZaH. Synthesis and Optical Resolution of the Neurotoxin 2-amino-3-([15N]-methylamino)propanoic (BMAA) Journal of Labelled Compounds and Radiopharmaceuticals. 1990;28:581–586. [Google Scholar]