

Abstract
Skvorak and coworkers [1] demonstrated the therapeutic efficacy of HTx in a murine model of iMSUD, confirming significant metabolic improvement and survival. To determine the effect of HTx on extrahepatic organs, we examined the metabolic effects of HTx in brain from iMSUD animals. Amino acid analysis revealed that HTx corrected increased ornithine, partially corrected depleted glutamine, and revealed a trend toward alloisoleucine correction. For amino acid and monoamine neurotransmitters, decreased GABA was partially corrected with HTx, while the L-histidine dipeptide of GABA, homocarnosine, was decreased in iMSUD mice and hypercorrected following HTx. Elevated branched chain amino acids (BCAA; leucine, isoleucine, valine) in MSUD can deplete brain tyrosine and tryptophan (the precursors of monoamine neurotransmitters dopamine (DA) and serotonin (5-hydroxytryptamine; 5-HT)) through competition via the large neutral amino acid transporter. HTx corrected decreased DA levels and the DA metabolite, 3-methoxytyramine, and partially corrected the DA intermediate 3,4-dihydroxyphenylacetate (DOPAC) and 5-HT levels, despite normal tyrosine and tryptophan levels in iMSUD mouse brain. We further observed enhanced intracellular turnover of both DA and 5-HT in iMSUD mouse brain, both of which partially corrected with HTx. Our results suggest new pathomechanisms of neurotransmitter metabolism in this disorder and support the therapeutic relevance of HTx in iMSUD mice, while providing proof-of-principle that HTx has corrective potential in extrahepatic organs.
Keywords: Hepatocyte transplantation (HTx), monoamines, leucine, large neutral amino acids, GABA, intracellular turnover
Introduction
Maple syrup urine disease (MSUD; OMIM 248600) is a heritable amino acidemia characterized by accumulations of branched chain ketoacids (BCKAs), BCAAs and non-physiological L-alloisoleucine, the latter pathognomonic for this disorder [2]. Varying degrees of branched chain ketoacid dehydrogenase (BCKDH) deficiency lead to the subclassifications of MSUD, including classical, intermittent, intermediate and thiamine-responsive forms. Treatment strategies for MSUD are primarily dietary, focused on maintenance of normophysiological concentrations of L-leucine and other BCAAs. Additionally, BCAAs may compete with other large-neutral amino acids (LNAA; i.e.,, methionine, tyrosine, phenylalanine, tryptophan) for transport across the blood brain barrier [3]. This observation has pathophysiological relevance, since tyrosine and tryptophan are the immediate precursors of the monoamine neurotransmitters, dopamine (DA) and serotonin (5-hydroxytryptamine; 5-HT) (Fig. 1).
Fig. 1.
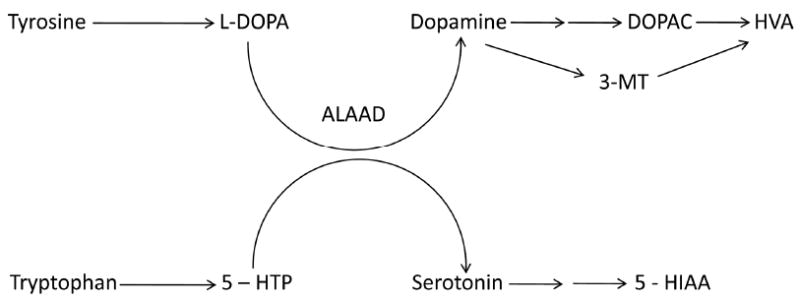
Abbreviated schematic diagram of reactions involved in the metabolism of the monoamine neurotransmitters, dopamine and serotonin (not all steps are shown). Abbreviations: L-DOPA, L-dihydroxyphenylalanine; 5-HTP, 5-hydroxytryptophan; ALAAD, aromatic L-amino acid decarboxylase; DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid; 3-MT, 3-methoxytyramine; 5-HIAA, 5-hydroxyindoleacetic acid.
Maintainenance of normophysiological leucine, isoleucine and valine levels is achieved through limiting the ingestion of the three BCAAs. Dietary control can be challenging, especially during adolescence and at times of catabolic crises. An alternative treatment approach is orthotopic liver transplantation (OLT), which has excellent therapeutic efficacy [4]. OLT effectively lowers the circulating levels of BCAAs and BCKAs. However, a limitation of OLT remains the shortage of donor livers, and the potential for prolonged immunosuppression intervention.
Successful OLT in MSUD patients suggests that hepatic cell-based treatment approaches may have clinical relevance in this disorder. Hepatocyte transplantation (HTx) has already proven to be a promising approach for long term therapy of genetic disorders with a prominent hepatic phenotype, such as ornithine transcarbamylase deficiency and glycogen storage disease [5, 6]. Accordingly, Skvorak and colleagues [1] evaluated HTx in a murine model of iMSUD [7]. These investigators demonstrated engraftment of exogenous hepatocytes into neonatal liver, a corresponding increase in liver BCKDH activity, and a dramatic improvement of blood BCAAs in weanling iMSUD-HTx mice. Despite these improvements in hepatic and peripheral metabolism, little information exists on the effect of HTx on organs beyond the liver, and few long-term studies of HTx in murine model systems have been undertaken. Thus, in the current report we have examined neurometabolic features of brains derived from iMSUD-HTx animals that were transplanted in the neonatal period.
Materials and Methods
Methodological details concerning HTx in iMSUD mice have been recently reported [1]. Genotyping was performed using nested primers, as described [1]. All breeder pairs were mice homozygous for the two previously described transgenes [7] and heterozygous for the endogenous E2 subunit. Accordingly, wild-type (WT) mice also expressed human E2, as described [1,7]. Line A iMSUD mice were used for all experiments, on a mixed 129/C57 background [7]. Protocols for β-galactosidase staining of isolated hepatocytes, as well as real-time PCR for the murine E2 mRNA, were previously described [1]. Tissues were harvested at weaning (DOL (day of life) 19-20), for both iMSUD mice and WT control animals. Transplanted iMSUD mice (i.e., iMSUD-HTx) were harvested at DOL 35 days of age. Our rationale for later harvest of iMSUD-HTx tissues was based on our desire to estimate the level of lifespan prolongation with HTx; other tissues (WT, iMSUD) had already been harvested. By DOL 35, iMSUD-HTx animals appeared unhealthy and lethargic, and tissues were accordingly harvested at this time. At sacrifice, the skull was rapidly opened, the brain resected (sagitally) in halves and tissue immediately stored at -80 °C until analysis.
For amino acid analyses, brain tissue was extracted in ice-cold SeraPrep (Pickering Laboratories, Mountain View, CA) and clarified by centrifugation. Amino acids were quantified by ion-exchange HPLC analysis with ninhydrin post column detection [8]. GABA and homocarnosine were determined in these same extracts employing liquid chromatography-tandem mass spectrometry [9, 10]. Monoamines in brain tissue were extracted using ice-cold perchloric acid. Following centrifugation, monoamines were quantified by reversed-phase HPLC analysis with electrochemical detection [11]. Metabolic data are presented as mean ± SEM and compiled using the Prizm Graph 4.0 program. Statistical analysis was performed using one way ANOVA with Tukey post-hoc analysis. To be considered as having been “corrected” (or “normalized”) with HTx, three criteria had to be achieved for each metabolite: a) the value for iMSUD mice had to be significantly different from wild-type animals; b) the value for iMSUD-HTx animals had to be significantly different from that of iMSUD animals; and c) there was no significant difference between the values observed for iMSUD-HTx and wild-type animals. When only criteria a and c were met, the effect of HTx was considered to have been “partial correction”.
Results
Viability of isolated hepatocytes was evaluated by trypan blue exclusion, and only those preparations for which viability exceeded 80% were employed for HTx [1]. As shown in Fig. 2 (left), sectioned liver (unfixed) from a newborn ROSA26+/- (age 1 day) mouse (30 μmsection, 10×) demonstrated widespread and intense β-galactosidase staining. A punctuate blue staining pattern (Fig. 2, right) from an iMSUD mouse (age 8 days, tissue obtained 24 hours following second injection of hepatocytes) reveals dispersed regions of engrafted hepatocytes into the recipient hepatic tissue (30 μm, 10×). Engraftment of β-galactosidase positive hepatocytes into iMSUD liver is consistent with the two-fold increase in BCKDH activity in iMSUD-HTx liver extracts that was previously documented, in conjunction with the significant improvement of branched chain amino acid levels in physiological fluids from those animals [1].
Fig. 2.
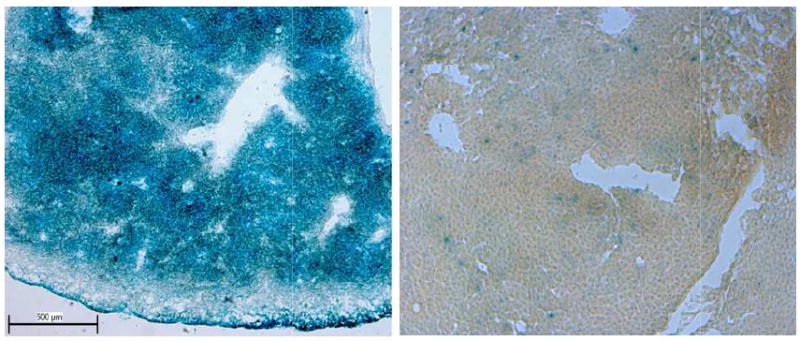
β-galactosidase staining of donor (left) and recipient (right) liver sections. Left Unfixed liver section (Rosa26+/- donor, age 1 day) stained for β-galactosidase (30 μm, 10× magnification) is depicted. Note the extensive blue staining indicative of β-galactosidase positive cells. Right. Unfixed iMSUD liver section (8 days of age, 24 hour following second administration of hepatocytes [1]) revealed a diffuse punctuate staining pattern indicative of engrafted β-galactosidase positive cells into the recipient iMSUD liver (30 μm, 10× magnification).
Extensive amino acid profiling in extracts of whole brain revealed a number of abnormalities (Fig. 3). For those physiological amino acids not shown in Fig. 3, there were no differences with respect to genotype. Levels of alloisoleucine showed a trend toward improvement with HTx, although statistical significance was not achieved (Fig. 3). Of interest, elevated ornithine in iMSUD brain was normalized with HTx, and decreased glutamine showed partial correction (Fig. 3). Conversely, there were no improvements for the significant decreases of amino acid transmitters in iMSUD brain, including glutamate and aspartate, in conjunction with serine and alanine. We further examined both GABA and the GABA conjugate, homocarnosine (GABA:L-histidine dipeptide) (Fig. 4). Consistent with glutamate and glutamine results, GABA levels were significantly decreased and partially corrected with HTx (Fig. 4). As well, homocarnosine was significantly decreased in iMSUD brain, and corrected (actually “hypercorrected”) in iMSUD-HTx brain (Fig. 4).
Fig. 3.
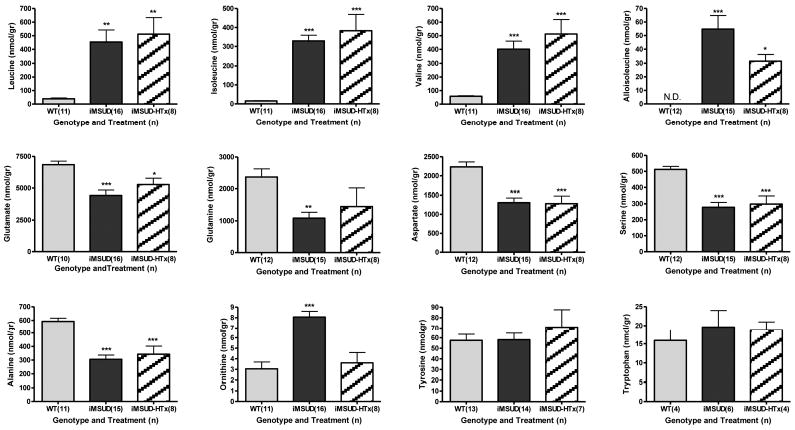
Selected whole brain amino acid concentrations in wild type (WT), iMSUD (intermediate maple syrup urine disease) and iMSUD-HTx (hepatocyte transplanted) mice. Many amino acids that were not significantly different by genotype are not depicted. Parenthetical values indicate the number of subjects studied. ND, none detected. *p<0.05; **p<0.01; ***p<0.001, one-way ANOVA with Tukey post hoc analysis as compared to WT subjects. Applying our definition of metabolic correction or partial correction (see Materials and Methods) revealed complete correction of ornithine with HTx (iMSUD vs iMSUD-HTx, p<0.001) and partial correction of glutamine concentrations.
Fig. 4.
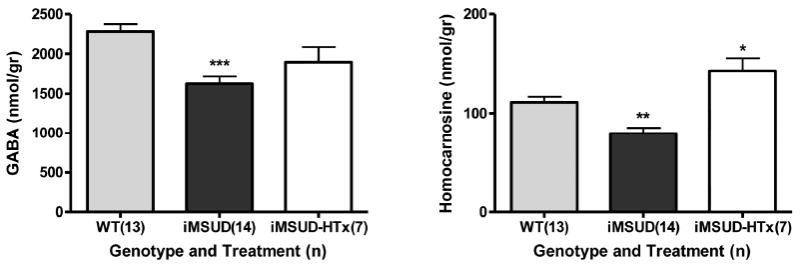
GABA and homocarnosine concentrations in whole brain. Details and statistical analyses as described in Fig. 2. Applying our definition of metabolic correction or partial correction (see Materials and Methods), GABA levels were partially corrected while homocarnosine was actually “hypercorrected” (iMSUD-HTx vs. WT, p<0.05; iMSUD-HTx vs. iMSUD, p<0.001).
Comprehensive profiling of monoamine neurotransmitters and related metabolites (Fig. 5) revealed that both dopamine and the dopamine intermediate 3-methoxytyramine (3-MT; formed exclusively in the synaptic cleft following dopamine release) were significantly depleted in iMSUD brain tissue and were corrected with HTx (Fig. 5). Similarly, HTx partially corrected decreases in the dopamine metabolic intermediate DOPAC (dihydroxyphenylacetate) and serotonin (5-HT; 5-hydroxytryptamine) levels. Conversely, significantly increased 5-hydroxyindoleacetate (5-HIAA; the end-product of 5-HT metabolism) was not responsive to HTx, and the DA end-product homovanillic acid (HVA) was not altered in iMSUD brain (Fig. 5).
Fig. 5.
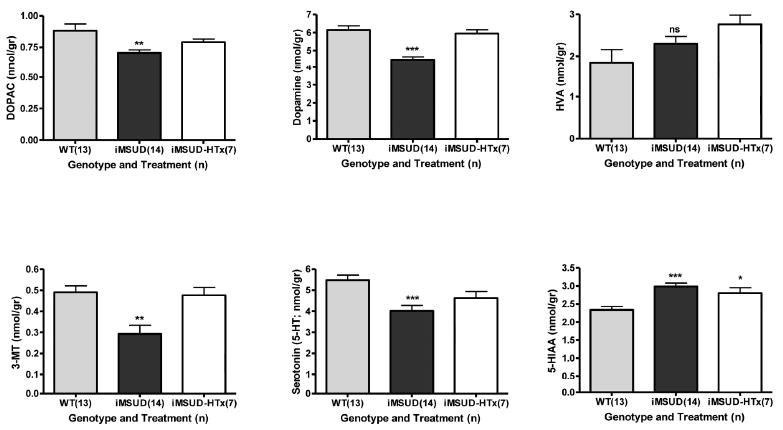
Concentrations of monoamine neurotransmitters and related metabolites in whole brain. Details of the subjects studied, and statistical analyses, as described in Fig. 2. Abbreviations as described in Fig. 1. Additional abbreviation: 5-HT, 5-hydroxytryptamine (serotonin). Applying our definition of metabolic correction or partial correction (see Materials and Methods), dopamine (iMSUD vs iMSUD-HTx, p<0.001) and 3-MT (iMSUD vs. iMSUD-HTx, p<0.001) were corrected, while DOPAC and 5-HT levels were partially corrected.
Based upon the above monoamine abnormalities, we examined indicators of neurotransmitter release and turnover in iMSUD brain. Evaluation of the 3-MT/DA ratio (an indicator of cellular dopamine release) indicated normal values in iMSUD brain (Fig. 6, left) that remained normal following HTx. Conversely, indicators of dopamine (DOPAC + HVA/DA) and serotonin (5-HIAA/5-HT) intracellular turnover revealed significantly enhanced turnover in iMSUD brain (Fig. 6, center and right), both of which were partially corrected with HTx. Metabolic trends for all intermediates in iMSUD and iMSUD-HTx mice are diplayed in Table 1.
Fig. 6.
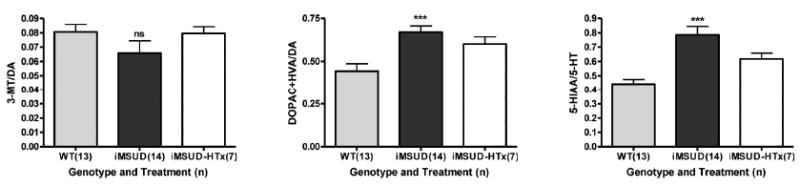
Estimates of cellular dopamine release (3-MT/DA; left), intracellular dopamine turnover (DOPAC+HVA/DA; center) and intracellular serotonin turnover (5-HIAA/5-HT) in whole brain. Details of subjects studied and statistical analyses as described in Fig. 2. Applying our definition of metabolic correction or partial correction (see Materials and Methods), ratios for DOPAC+HVA/DA and 5-HIAA/5-HT were partially corrected with HTx.
Table I.
Metabolic effects in brains of iMSUD mice following hepatocyte transplantation
| iMSUD mice | iMSUD-HTx mice | |
|---|---|---|
| Glutamate | ↓ | ↓ |
| Glutamine | ↓ | = |
| Aspartate | ↓ | ↓ |
| Serine | ↓ | ↓ |
| Alanine | ↓ | ↓ |
| Alloisoleucine | ↑ | ↑ |
| Leucine | ↑ | ↑ |
| Isoleucine | ↑ | ↑ |
| Valine | ↑ | ↑ |
| Ornithine | ↓ | = |
| GABA | ↓ | = |
| Homocarnosine | ↓ | ↑ |
| Tyrosine | = | = |
| Tryptophan | = | = |
| Dopamine | ↓ | = |
| DOPAC | ↓ | = |
| 3-MT | ↓ | = |
| HVA | = | = |
| 5-HT (serotonin) | ↓ | = |
| 5-HIAA | ↑ | ↑ |
| 3-MT/DA | = | = |
| DOPAC+HVA/DA | ↑ | = |
| 5-HIAA/5-HT | ↑ | = |
Legend to Table: See text for metabolite abbreviations. All symbols represent comparisons to WT mice. For iMSUD mice, an equals sign (=) indicates no difference from WT mice, whereas for iMSUD-HTx mice this symbol indicates either correction or partial correction (not significantly different from WT levels).
Discussion
HTx has been an effective therapeutic intervention for several inherited metabolic liver diseases, include ornithine transcarbamylase (OTC) deficiency, alpha-1-antitrypsin deficiency, glycogen storage disease, infantile Refsum disease and Crigler–Najjar syndrome type 1 [6, 15, 16]. HTx studies in murine models have mirrored the positive outcomes observed in clinical trials. HTx has been effective in mouse models of phenylketonuria, tyrosinemia type I and Wilson disease [17-19] and other small animal model systems [20,21]. Jiang and coworkers [22] recently employed HTx for correction of murine hyperoxaluria type I. However, these investigators employed tissue preparative regimens (including irradiation) that might be unlikely to obtain IRB-approval in the clinic. Conversely, Skvorak and coworkers [1] demonstrated that HTx could be achieved in neonatal iMSUD animals without preconditioning, leading to significant metabolic and hepatic improvement with only ∼3% engraftment. These improvements manifested as decreased total BCAA/alanine ratios in bloodspots of iMSUD-HTx mice at weaning, associated with a 2-fold increase in liver branched-chain ketoacid dehydrogenase activity measured at the same age [1]. However, it is unknown if hepatic correction can lead to metabolic improvements in other organs, including brain, or if this metabolic correction is time-constrained.
Our data on amino acids and neurotransmitters in iMSUD brain suggested a pattern of global energy disruption (Figs. 3 & 4; Table I), as indicated by the significant decrease of amino acid neurotransmitters including glutamate, aspartate and GABA, in addition to alanine and serine. Results for glutamate, glutamine, aspartate and GABA were consistent with those presented by Dodd and coworkers in the bovine MSUD model [12], and with the recently published report by Zinnanti and colleagues on iMSUD animals [13]. Dodd and coworkers analyzed brain cortex from neonatal MSUD calves and found that glutamate, aspartate and GABA were uniformly reduced. We speculate that global energy dysfunction in iMSUD brain is correlated with disrupted nitrogen balance induced by leucine accumulation (Fig. 7), consistent with the hypotheses recently proposed by Zinnanti and colleagues [13]. This hypothesis is also consistent with the leucine-glutamate cycle as proposed by Yudkoff [23], which posits that elevated leucine disrupts the glutamate-glutamine-GABA cycle via increased nitrogen flux associated with transamination. Increased nitrogen loads result in 2-oxoglutarate consumption following transamination to glutamate (see Fig. 7). Enhanced production of 2-oxoisocaproate (the leucine transamination product) is further predicted to alter pyruvate and 2-oxoglutarate dehydrogenase complex activity, with resulting disruption of the oxidation/reduction potential [24-26]. Our finding of enhanced brain ornithine levels (Fig. 3) may represent an attempt to correct depleted levels of 2-oxoglutarate (Fig. 7) linked to dysfunction of the 2-oxoglutarate dehydrogenase complex. This hypothesis would be consistent with the elegantly described hypothesis of brain injury in MSUD due to altered bioenergetics, recently put forth by Zinnanti and coworkers [13]. Overall, it remains possible that disturbed glutamate/2-oxoglutarate homeostasis underscores the acute encephalopathy seen in MSUD patients who decompensate [27].
Fig. 7.
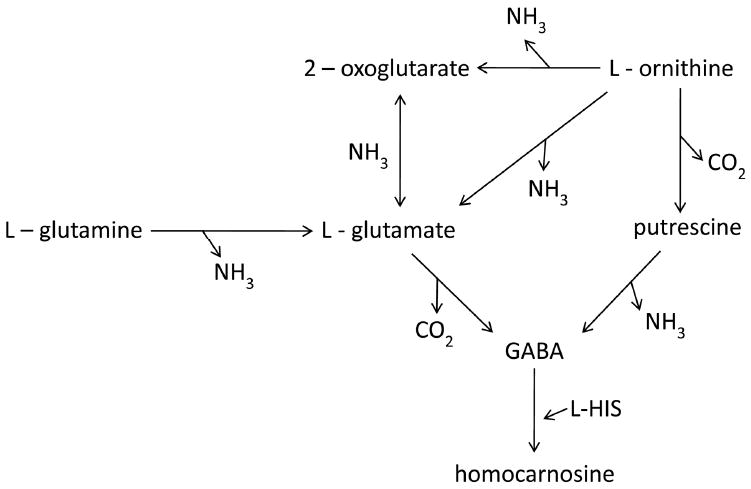
Schematic diagram of interrelationships for selected amino acid abnormalities identified in iMSUD mice in the current study. Note that putrescine (1,4-diaminobutane) and 2-oxoglutarate were not determined in the current study. We speculate that enhanced ammonia production from leucine transamination in brain would disrupt the kinetics of many of the reactions depicted that utilize ammonia in catalysis, in accord with the leucine-glutamate cycle proposed by Yudkoff (1997).
Concentrations of BCAAs were not improved with HTx (Fig. 3; Table 1), consistent with blood BCAA levels previously reported [1]. As previously suggested [1], we believe that iMSUD-HTx animals had become catabolic, and thus protein turnover in muscle likely resulted in significant transport of BCAAs across the blood brain barrier in DOL 35 iMSUD-HTx animals. Two observations support this hypothesis: 1) hyperglycinemia in iMSUD-HTx animals [1]; and 2) improved levels of alloisoleucine both in blood [1] and brain (current study) in iMSUD-HTx mice. Alloisoleucine is a non-protein amino acid that would not be released during muscle protein catabolism. The cumulative findings in Table 1 would tend to support our hypothesis of hypercatabolism in iMSUD-HTx mice. Table 1 reveals that the major biochemical improvements in iMSUD-HTx animals were in monoamine catabolic pathways, which in neural tissue might be more compartmentalized and perhaps less affected by catabolic crises as might be expected for amino acids such as glutamate, aspartate and alanine.
Results for amino acid neurotransmitters (e.g., glutamate, aspartate and GABA) and the GABA dipeptide homocarnosine, were of interest. Glutamine showed partial correction with HTx, and since glutamine is a precursor of GABA in neural tissue, this may correlate with partial GABA correction (Fig. 4; Table 1). However, homocarnosine significantly improved (Fig. 4). The reason for this remains unclear, but it may relate to the observation that homocarnosine is a GABA:L-histidine dipeptide. Although we found no differences in blood or brain concentrations of histidine, it remains possible that HTx significantly bolstered brain concentrations of histidine within different compartments, and this might explain the “hypercorrection” of homocarnosine in the face of only “partial” GABA correction. Along these lines, histidine is known to be increased in phenylketonuric body fluids [28], although the exact mechanism of this increase remains obscure, and we have recently documented increased histidine in blood and brain of Pah-/- mice [29]. It is possible that histidine may be similarly elevated in iMSUD brain (in discrete regions), and this may potentially explain the “hypercorrection” of homocarnosine levels.
The values obtained in the current study for alanine, tyrosine and tryptophan contrasted with data obtained by Zinnanti and coworkers [13] in the same model. The latter group found alanine increased in iMSUD animals, whereas we found this amino acid significantly depleted in brain (Fig. 3). This discrepancy is unlikely to have been associated with diet (18% protein [13] vs. 22% protein in the current study) but may have been related to age (42 days [13] vs. 20 for our animals). Moreover, the absolute values of alanine measured in brain were comparable between both studies (∼400-600 nmol/g wet weight). Conversely, if iMSUD animals were catabolic even at weaning, it is possible that alanine would be utilized to generate pyruvate via transamination; the latter would yield acetyl-CoA and oxalacetate via the pyruvate dehydrogenase and carboxylase reactions, respectively, which would stimulate Krebs cycle function. With respect to tyrosine and tryptophan, the discrepancies are more challenging to explain, yet our normal levels for brain tyrosine (Fig. 3) were consistent with normal amounts detected in blood of iMSUD mice, and overall the absolute concentrations we detected were about 2-fold higher than those measured by Zinnanti (∼55-70 nmol/gr wet weight vs. 25-35) for tyrosine. Concentrations of tryptophan, however, were quite comparable between our study and the findings of Zinnanti and coworkers [13].
The data for monoamine metabolism in iMSUD brain reveals new features of pathophysiology in this disorder (Figs. 5, 6). Despite normal tyrosine and tryptophan concentrations in brain, significant alterations of dopamine, DOPAC, 3-MT, 5-HT and 5-HIAA were observed (Fig. 5). HTx corrected or partially corrected four of these. Conversely, abnormalities in 5-HIAA levels were not corrected by HTx (Fig. 5). Puglisi-Allegra and coworkers [14] reported significant alterations in brain monoamines in a murine model of PKU, a model in which a large neutral amino acid (e.g., phenylalanine) accumulates as for iMSUD mice with respect to BCAAs, although Puglisi-Allegra and coworkers did not report levels of tyrosine and tryptophan in the brains of their subjects. Zinnanti and colleagues [13] also found that dopamine levels were depleted in iMSUD brain. Our results are consistent with the findings of these investigators. Lastly, although dopamine release was normal in iMSUD brain (Fig. 6), intracellular turnover of dopamine and serotonin was significantly enhanced (Fig. 6) and showed partial correction with HTx. The monoamine disturbances in iMSUD animals occurred in the face of normal tyrosine and tryptophan concentrations, however, which remains paradoxical. Metabolite quantitation occurred in whole brain, however, and it remains possible that regional levels (hippocampus, cortex, cerebellum, etc) of tyrosine and tryptophan may have been altered prior to and after HTx.
In conclusion, the current report provides new insight into metabolic pathophysiology associated with MSUD, and proof-of-principle that HTx can result in selected metabolic improvements in extrahepatic organs (Table I). As well, our findings for abnormal 5-HT levels represent the first demonstration of serotoninergic anomalies associated with MSUD. The current investigation was designed as a pilot to assess the global effect of HTx on brain neurometabolites in the short term. Clearly, longer-term studies of iMSUD-HTx animals, following the level of hepatocyte engraftment for several months, while assessing neurometabolites in brain using a regional dissection approach, are needed and planned for the near future. Nonetheless, based upon the success thus far with OLT in MSUD [4, 30] and our positive preliminary results in iMSUD mice [1], we propose to continue our evaluation of HTx in this model in order to generate preclinical outcome data that could eventually support a move for this methodology to the clinic.
Acknowledgments
These studies were supported in part by NIH NS 40270, HD 58553, HD 57864 and the support of a grant from the Pediatric Neurotransmitter Disease Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Skvorak KJ, Paul HS, Dorko K, Marongiu F, Ellis E, Chace D, Ferguson C, Gibson KM, Homanics GE, Strom SC. Hepatocyte transplantation improves phenotype and extends survival in a murine model of intermediate maple syrup urine disease. Mol Ther. doi: 10.1038/mt.2009.99. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chuang DT, Chuang JL, Wynn RM. Lessons from genetic disorders of branched-chain amino acid metabolism. J Nutr. 2006;136(1 Suppl):243S–249S. doi: 10.1093/jn/136.1.243S. [DOI] [PubMed] [Google Scholar]
- 3.Zielke HR, Zielke CL, Baab PJ, Collins RM. Large neutral amino acids auto exchange when infused by microdialysis into the rat brain: implication for maple syrup urine disease and phenylketonuria. Neurochem Int. 2002;40:347–54. doi: 10.1016/s0197-0186(01)00077-8. [DOI] [PubMed] [Google Scholar]
- 4.Strauss KA, Mazariegos GV, Sindhi R, Squires R, Finegold DN, Vockley G, Robinson DL, Hendrickson C, Virji M, Cropcho L, Puffenberger EG, McGhee W, Seward LM, Morton DH. Elective liver transplantation for the treatment of classical maple syrup urine disease. Am J Transplant. 2006;6:557–64. doi: 10.1111/j.1600-6143.2005.01209.x. [DOI] [PubMed] [Google Scholar]
- 5.Puppi J, Tan N, Mitry RR, Hughes RD, Lehec S, Mieli-Vergani G, Karani J, Champion MP, Heaton N, Mohamed R, Dhawan A. Hepatocyte transplantation followed by auxiliary liver transplantation--a novel treatment for ornithine transcarbamylase deficiency. Am J Transplant. 2008;8:452–7. doi: 10.1111/j.1600-6143.2007.02058.x. [DOI] [PubMed] [Google Scholar]
- 6.Lee KW, Lee JH, Shin SW, Kim SJ, Joh JW, Lee DH, Kim JW, Park HY, Lee SY, Lee HH, Park JW, Kim SY, Yoon HH, Jung DH, Choe YH, Lee SK. Hepatocyte transplantation for glycogen storage disease type Ib. Cell Transplant. 2007;16:629–37. doi: 10.3727/000000007783465019. [DOI] [PubMed] [Google Scholar]
- 7.Homanics GE, Skvorak K, Ferguson C, Watkins S, Paul HS. Production and characterization of murine models of classic and intermediate maple syrup urine disease. BMC Med Genet. 2006;7:33. doi: 10.1186/1471-2350-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slocum RH, Cummings JG. Amino acid analysis of physiological samples. In: Hommes FA, editor. Techniques in Diagnostic Human Biochemical Genetics. Wiley-Liss; New York: 1991. pp. 87–126. [Google Scholar]
- 9.Jansen EE, Gibson KM, Shigematsu Y, Jakobs C, Verhoeven NM. A novel, quantitative assay for homocarnosine in cerebrospinal fluid using stable-isotope dilution liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;830:196–200. doi: 10.1016/j.jchromb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 10.Kok RM, Howells DW, van den Heuvel CC, Guérand WS, Thompson GN, Jakobs C. Stable isotope dilution analysis of GABA in CSF using simple solvent extraction and electron-capture negative-ion mass fragmentography. J Inherit Metab Dis. 1993;16:508–12. doi: 10.1007/BF00711667. [DOI] [PubMed] [Google Scholar]
- 11.Ogburn KD, Bottiglieri T, Wang Z, Figueiredo-Pereira ME. Prostaglandin J2 reduces catechol-O-methyltransferase activity and enhances dopamine toxicity in neuronal cells. Neurobiol Dis. 2006;22:294–301. doi: 10.1016/j.nbd.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 12.Dodd PR, Williams SH, Gundlach AL, Harper PA, Healy PJ, Dennis JA, Johnston GA. Glutamate and gamma-aminobutyric acid neurotransmitter systems in the acute phase of maple syrup urine disease and citrullinemia encephalopathies in newborn calves. J Neurochem. 1992;59:582–90. doi: 10.1111/j.1471-4159.1992.tb09409.x. [DOI] [PubMed] [Google Scholar]
- 13.Zinnanti WJ, Lazovic J, Griffin K, Skvorak KJ, Paul HS, Homanics GE, Bewley MC, Cheng KC, Lanoue KF, Flanagan JM. Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain. 2009 doi: 10.1093/brain/awp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puglisi-Allegra S, Cabib S, Pascucci T, Ventura R, Cali F, Romano V. Dramatic brain aminergic deficit in a genetic mouse model of phenylketonuria. Neuroreport. 2000;11:1361–4. doi: 10.1097/00001756-200004270-00042. [DOI] [PubMed] [Google Scholar]
- 15.Hansen K, Horslen S. Metabolic liver disease in children. Liver Transpl. 2008;14:713–33. doi: 10.1002/lt.21520. [DOI] [PubMed] [Google Scholar]
- 16.Lysy PA, Najimi M, Stephenne X, Bourgois A, Smets F, Sokal EM. Liver cell transplantation for Crigler-Najjar syndrome type I: update and perspectives. World J Gastroenterol. 2008;14:3464–70. doi: 10.3748/wjg.14.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamma K, Clark H, Montini E, Al-Dhalimy M, Grompe M, Finegold M, Harding CO. Low therapeutic threshold for hepatocyte replacement in murine phenylketonuria. Mol Ther. 2005;12:337–44. doi: 10.1016/j.ymthe.2005.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, Finegold M, Grompe M. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1996;12:266–73. doi: 10.1038/ng0396-266. [DOI] [PubMed] [Google Scholar]
- 19.Allen KJ, Cheah DM, Wright PF, Gazeas S, Pettigrew-Buck NE, Deal YH, Mercer JF, Williamson R. Liver cell transplantation leads to repopulation and functional correction in a mouse model of Wilson's disease. J Gastroenterol Hepatol. 2009;19:1283–90. doi: 10.1111/j.1440-1746.2004.03451.x. [DOI] [PubMed] [Google Scholar]
- 20.Seppen J, Filali EE, Elferink RO. Small animal models of hepatocyte transplantation. Methods Mol Biol. 2009;481:75–82. doi: 10.1007/978-1-59745-201-4_7. [DOI] [PubMed] [Google Scholar]
- 21.Weber A, Groyer-Picard MT, Franco D, Dagher I. Hepatocyte transplantation in animal models. Liver Transpl. 2009;15:7–14. doi: 10.1002/lt.21670. [DOI] [PubMed] [Google Scholar]
- 22.Jiang J, Salido EC, Guha C, Wang X, Moitra R, Liu L, Roy-Chowdhury J, Roy-Chowdhury N. Correction of hyperoxaluria by liver repopulation with hepatocytes in a mouse model of primary hyperoxaluria type-1. Transplantation. 2008;85:1253–60. doi: 10.1097/TP.0b013e31816de49e. [DOI] [PubMed] [Google Scholar]
- 23.Yudkoff M. Brain metabolism of branched-chain amino acids. Glia. 1997;21:92–8. doi: 10.1002/(sici)1098-1136(199709)21:1<92::aid-glia10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 24.Bridi R, Braun CA, Zorzi GK, Wannmacher CM, Wajner M, Lissi EG, Dutra-Filho CS. Alpha-keto acids accumulating in maple syrup urine disease stimulate lipid peroxidation and reduce antioxidant defences in cerebral cortex from young rats. Metab Brain Dis. 2005;20:155–67. doi: 10.1007/s11011-005-4152-8. [DOI] [PubMed] [Google Scholar]
- 25.Funchal C, Gottfried C, De Almeida LM, Wajner M, Pessoa-Pureur R. Evidence that the branched-chain alpha-keto acids accumulating in maple syrup urine disease induce morphological alterations and death in cultured astrocytes from rat cerebral cortex. Glia. 2004;48:230–40. doi: 10.1002/glia.20072. [DOI] [PubMed] [Google Scholar]
- 26.Barschak AG, Marchesan C, Sitta A, Deon M, Giugliani R, Wajner M, Vargas CR. Maple syrup urine disease in treated patients: biochemical and oxidative stress profiles. Clin Biochem. 2008;41:317–24. doi: 10.1016/j.clinbiochem.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 27.Zielke HR, Huang Y, Tildon JT, Zielke CL, Baab PJ. Elevation of amino acids in the interstitial space of the rat brain following infusion of large neutral amino and keto acids by microdialysis: alpha-ketoisocaproate infusion. Dev Neurosci. 1996;18:420–5. doi: 10.1159/000111436. [DOI] [PubMed] [Google Scholar]
- 28.Snyderman SE, Sansaricq C, Castro PM, Castro JV. Plasma and cerebrospinal fluid amino acid concentrations in phenylketonuria during the newborn period. J Pediatr. 1981;99:63–67. doi: 10.1016/s0022-3476(81)80958-4. [DOI] [PubMed] [Google Scholar]
- 29.Arning E, Bottiglieri T, Sun Q, Jansen EEW, Jakobs C, Lin B, Stetson L, Harding CO, Gibson KM. Metabolic profiling in phenylalanine hydroxylase-deficient (Pah-/-) mouse brain reveals decreased amino acid neurotransmitters and preferential alterations of the serotoninergic system. Molec Genet Metab. in press. [Google Scholar]
- 30.Khanna A, Hart M, Nyhan WL, Hassanein T, Panyard-Davis J, Barshop BA. Domino liver transplantation in maple syrup urine disease. Liver Transpl. 2006;12:876–82. doi: 10.1002/lt.20744. [DOI] [PubMed] [Google Scholar]