

Abstract
Human papillomavirus (HPV) is a non-enveloped DNA virus with an ∼8000 base pair genome. Infection with certain types of HPV is associated with cervical cancer, although the molecular mechanism by which HPV induces carcinogenesis is poorly understood. Three genes encoded by HPV16 are regarded as oncogenic- E5, E6, and E7. The role of E5 has been controversial. Expression of HPV16 E5 causes cell-cell fusion, an event that can lead to increase chromosomal instability, particularly in the presence of cell cycle checkpoint inhibitors like HPV16 E6 and E7. Using biochemical and cell biological assays to better understand HPV16 E5, we find that HPV16 E5 localizes to the plasma membrane with an intracellular amino terminus and an extracellular carboxyl terminus. Further, HPV16 E5 must be expressed on both cells for cell fusion to occur. When the extracellular epitope of HPV16 E5 is targeted with an antibody, the number of bi-nucleated cells decreases.
Introduction
Human papillomavirus (HPV) is a double stranded DNA virus comprised of a relatively small genome, ∼8000 bp. This family of virus is species specific and targets epithelial cells. Approximately 6.2 million Americans are newly diagnosed with the virus each year (CDC, 2007). Infection with certain types of HPV is associated with the development of cervical cancer and other anogenital cancers (Cogliano et al., 2005; McCance, 2005). More recently, it has been reported that significant subset of head and neck cancers are also HPV positive (Gillison et al., 2000). Understanding the role of HPV in carcinogenesis is an important first step in developing new methods for the timely diagnosis and treatment of these cancers.
One of the most prevalent and oncogenic HPVs is HPV16. HPV16 has been reported to be associated with ∼60% of cervical cancers (Clifford et al., 2003) and ∼40% of head and neck cancers (Gillison et al., 2008). The HPV16 genome is comprised of 8 genes, three of which (E5, E6, and E7) are regarded as oncogenes based on their ability to transform cells when expressed individually (McMurray et al., 2001).
Despite the strong correlative evidence linking HPV16 and cancer, the exact mechanism of transformation is not fully understood. HPV 16 E6 and E7 are well established as inhibitors of the tumor suppressor genes p53 and Rb, respectively (Jones, Alani, and Munger, 1997; Werness, Levine, and Howley, 1990). In addition, both oncogenes induce genomic instability (a hallmark of cancer), associate with a host of cellular proteins, and cause other morphological changes associated with carcinogenesis (Narisawa-Saito and Kiyono, 2007). Thus, roles for HPV16 E6 and E7 in oncogenesis are established through unregulated cell cycle progression and genomic changes.
HPV16 E5 is regarded as having weak transforming activity that has been demonstrated in a number of rodent cell lines. These assays include transformation of keratinocytes (Straight et al., 1993), anchorage independent growth of fibroblasts (Leechanachai et al., 1992; Pim, Collins, and Banks, 1992), immortalization of keratinocytes (Stoppler et al., 1996), and growth stimulation of epithelial cells (Leptak et al., 1991). When E5 is co-expressed with the E7 oncogene there is enhanced cell transformation of rodent fibroblasts (Bouvard et al., 1994; Valle and Banks, 1995).
The collaborative role of E5 with other HPV16 oncogenes is supported by data from transgenic mice. Expression of the HPV16 E5 oncogene under the direction of a basal epithelium specific promoter (K14) is sufficient to cause epidermal hyperplasia and the formation of spontaneous skin tumors (Williams et al., 2005). However, tumor size increases when the entire HPV16 genome is expressed, as compared to mice expressing only E6 and E7 (Riley et al., 2003). These in vitro and in vivo data are consistent with the notion that E5 can transform cells independently as well as enhance the transforming properties of other oncogenes.
A number of molecular mechanisms have been proposed for HPV16 E5 in carcinogenesis. These include increasing the components of lipid rafts (i.e. caveolin and ganglioside GM1) (Bravo, Crusius, and Alonso, 2005; Suprynowicz et al., 2008), attenuating apoptosis induced by ligand stimulation [i.e. Fas and TNF-α, and UV irradiation (Kabsch and Alonso, 2002; Zhang, Spandau, and Roman, 2002)], reducing cellular communication via gap junctions (Oelze et al., 1995), inhibiting major histocompatibility complex (MHC-I and II) trafficking to the plasma membrane causing an increase in antigen presentation and T-cell recognition (Zhang et al., 2003). However, the most widely accepted model is that HPV16 E5 associates with and inhibits the vacuolar ATPase of the early endosome. This model proposes that HPV16 E5 prevents acidification of the early endosome, thereby disrupting the endocytic trafficking itinerary of the epidermal growth factor receptor (EGFR), a prototypical tyrosine kinase receptor frequently overexpressed in cancer (Straight, Herman, and McCance, 1995; Thomsen et al., 2000; Tomakidi et al., 2000).
Interestingly, many of these models for HPV16 E5 function are inconsistent with the kinetics of HPV16 E5 expression. Based on studies using organotypic cultures, the synthesis of HPV16 E5 mRNA has been monitored following HPV16 infection. HPV16 E5 is expressed soon after infection and attenuates over time (Stoler et al., 1992). The in vitro studies complement other reports that the HPV16 genome is frequently integrated into the host cell’s chromosome (Schwartz et al., 1985; Stoler et al., 1992). Such cells have intact E6 and E7, but frequently E5 is not expressed.
We have recently discovered that HPV16 E5, when expressed in human keratinocytes causes a number of changes to cell morphology that include increased nuclear size, percentage of bi-nucleated cells, DNA content/cell, and tetraploidy (Hu et al., 2009). These changes are remarkable because they are characteristics of cancerous and precancerous lesions (Mittal, Chan, and Demopoulos, 1990; Prasad et al., 1993). We observe a three-fold increase in bi-nucleate cells when the entire HPV16 genome is expressed; expression of HPV16 with a frameshift mutation in the E5 gene, returns the percentage of bi-nucleate cells back to basal levels (Hu et al., 2009). Together, these data indicate that HPV16 E5 is necessary and sufficient for the formation of bi-nucleate cells.
The bi-nucleated cells that form with HPV16 E5 expression arise through cell-cell fusion (Hu et al., 2009). Although this is a newly reported function for HPV16 E5, the ability to induce cell fusion is a feature of many oncogenic viruses, include Hepatitis C virus, Hepatitis B virus, Epstein-Barr virus, Karposi sarcoma virus, and Human T-lymphotrophic virus (Duelli and Lazebnik, 2007). Current models for how cell fusion contributes to oncogenesis suggest that following cell fusion, most bi-nucleated cells become quiescent or apoptotic, but those that can replicate have an increased potential to mis-segregate chromosomes, as the cell is not equipped to divide twice the number of chromosomes during cell division. The probability of fused cells continuing to replicate is low, which is consistent with estimates of in vivo cell transformation. If cell cycle checkpoint inhibitors are expressed following cell fusion, the potential for chromosomal instability increases, as has been recently demonstrated using non-oncogenic viruses (Duelli et al., 2007).
Our recent data studying oncogenes of HPV16 is consistent with this model. Cells that fuse following expression of HPV16 E5 have an increased potential to undergo transformation when HPV16 E6 and E7 are expressed (Hu et al., 2009). The established roles of HPV16 E6 and E7 in the inhibition of proteins that regulate cell cycle checkpoints (Jones, Alani, and Munger, 1997; Werness, Levine, and Howley, 1990), and are postulated to help the cells with twice the chromosomal content propagate. With multiple rounds of cell division, the potential for errors in chromosomal segregation increases, and with it, the chance for cell transformation.
The incidence of cell-cell fusion occurs with low frequency in tissue culture cells and likely occurs with even lower frequency in patients. According to the Center for Disease Control, there are an estimated 20 million HPV-positive individuals in the United States, but only ∼25,000 HPV-associated cancers /year. Clearly, progression from an HPV-positive status to developing cancer is a rare event.
Understanding the molecular mechanism by which HPV16 E5 mediates cell fusion is the first step in developing potential strategies to inhibit cell-cell fusion and the initiation of carcinogenesis. In this study, we have employed a series of cell biological and biochemical assays to determine the features of HPV16 E5 that allow it to mediate cell fusion. We have found that HPV16 E5 is expressed at the plasma membrane and oriented such that the amino terminus is intracellular and the carboxyl-terminus is extracellular. For cell fusion to occur, both cells require expression of HPV16 E5. The addition of an antibody to the protein’s extracellular domain can inhibit cell fusion. These findings provide an important foundation for understanding the molecular regulation of cell fusion by HPV16 E5.
Materials and Methods
Generation of tTA-HaCaT cells
HaCaT cells were retrovirally infected with the tetracycline transactivator (tTA) (Clontech Retroviral GeneTransfer and Expression System). Neomycin resistant colonies were grown in 800 μg/ml G418. Individual colonies were isolated, amplified, and screened based on their ability to regulate expression of tetracycline regulatable adenovirus.
Parental HaCaT and tTA-HaCaT cells were maintained in Dulbecco’s Minimal Essential Media (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 units/ml streptomycin, and 2 mM glutamine. Cells stably transfected with tTA were maintained in 800 μg/ml G418. All three cell lines were maintained at 37°C in 5% CO2 (Hu et al., 2009).
Adenovirus generation/expression
Codon optimized HPV16 E5 was generated as previously described (Disbrow et al., 2003). A hemagglutinin (HA) epitope (MEYDVPDYAH) was engineered on to either the amino (HA-E5) or carboxyl (E5-HA) terminus of the protein by PCR, making the appropriate modification of stop codons. Adenoviruses were generated using Clontech Adeno-X™ Tet-off expression system. All adenoviruses were sequenced at the DNA sequencing facility at the Oklahoma Medical Research Foundation (Hu et al., 2009).
Plasmids and transfection
HA-tagged, codon optimized HPV16 E5 was generated as described above and subcloned into the pcDNA3 (Invitrogen). DNA (4 μg) were transfected into cell via nucleotransfection using the Amaxa transfector (Amaxa) according to manufacturer’s directions (Hu et al., 2009).
Adenoviral infection
Cells were plated at 75% confluency and infected at a multiplicity of infection of 20 plaque forming units/cell. Adenoviruses were cesium chloride purified and diluted in DMEM for infection. After 2 hours of incubation with adenovirus, virus containing media was removed from the cells and replaced with growth media and allowed to recover for 24-48 hours.
Indirect immunofluorescence
Indirect immunofluorescence was performed as previously described (Dinneen and Ceresa, 2004). Cells were fixed in 4% para-formaldehyde and either permeabilized with 0.01% saponin or left intact. The mouse monoclonal 12CA5 (anti-HA, Roche) (diluted 1:1000) and anti-EGFR (Ab-1, Calbiochem) (diluted 1:1000) and rabbit polyclonal antibody anti-HA (Y-11, Santa Cruz Biotechnology) (diluted 1:500), were the primary antibodies and Alexa 488-, Alexa 568-, or Alexa 647-conjugated goat anti-mouse or anti-rabbit (Molecular Probes) as the secondary antibody at a dilution of 1:250. Cells were also stained with 10 ng/ml DAPI (Sigma). Images were captured using Olympus AX70 epifluorescent microscope with Q-Capture software or Olympus Confocal FV100 microscope and analyzed using the Olympus Fluoveiw Software.
Assessment of bi-nucleation
Images of DAPI stained nuclei were analyzed using Photoshop software. Bi-nucleated cells were calculated as the number of cells with two nuclei divided by the total cells.
Heterokaryon formation assay
tTA-HaCaT cells or parental HaCaT cells (2 × 106 cells) were transfected with pRK7-histone 2B-RFP (1.5 μg plasmid) or pRK7-histone 2B-YFP (1.5 μg plasmid) by nucleofection using the Amaxa Nucleofector I (transfection efficiency ∼70%). After 24 hours recovery, cells were mixed in a 1:1 ratio with untransfected cells and then infected with HA-E5 adenovirus (20 pfu/cell). After 72 hours, cells were fixed and observed by fluorescence microscopy for bi-nucleated cells.
YFP Cell Fusion reporter assay
tTA-HaCaT cells or parental HaCaT cells were transfected by Amaxa nucleofection with either a plasmid expressing T7 RNA polymerase (pRK-T7 RNA polymerase) or yellow fluorescent protein (YFP) driven by a T7 promoter (pT7-YFP). Following 16 hours of recovery, cells were washed, and re-plated with 200,000 cells of each population (1:1 ratio) into a well of a 12-well dish. Cells were infected with adenovirus as indicated in the figure legend using the method described above. Seventy-two hours post-infection, cells were collected, washed in PBS, and analyzed using a FACS Calibur (Becton Dickinson). The data from 10,000 cells/experiment were analyzed using Cell Quant Pro (Becton Dickinson, Version 5.2.1 software). Data were plotted as the relative increase in the total YFP intensity (percentage of positive cells multiplied by their fluorescent mean intensity) as compared to cells without virus infection (Hu et al., 2008).
Antibody inhibition
tTA-HaCaT cells were infected with either A) HPV16 HA-E5 or B) HPV16 E5-HA 3.5 ×104 cells/well of a 24-well tissue culture dish. Immediately following infection, cells were incubated with no antibody, 12CA5 (anti-HA 1:250) or 9E10 (anti-myc, 1:250). Cells were incubated additional 72 hours, fixed, and stained with Dapi. Cells were scored for the number of bi-nucleated cells.
Plasma membrane purification
The plasma membranes of HPV16 HA-E5 or E5-HA infected tTA-HaCaT cells was isolated using the non-quantitative Qproteome purification kit (Qiagen) according to the manufacturer’s directions. Briefly, cells are lysed by incubation in a hypotonic buffer containing a mild buffer, and membranes are mechanically disrupted using a 21 gauge needle and syringe. Membranes are incubated with a proprietary ligand specific for molecules on the plasma membrane. The ligand and associated plasma membranes are isolated using magnetic beads that have been engineered to capture the ligand. Fractions of the total (5%) and plasma membrane (50%) were resolved by SDS-PAGE and immunoblotted with antibodies against the EGFR (sc-03, Santa Cruz Biotechnology), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (sc-47724, Santa Cruz). The unstimulated EGFR localizes primarily to the plasma membrane and served as a positive control for purification; GADPH is a cytosolic protein and served as a negative control.
Analysis of cell surface expression of HPV16 E5
tTA-HaCaT cells were infected with nothing or adenoviruses encoding HPV16 HA-E5 or E5-HA in the absence and presence of 1 μg/ml tetracycline to regulate protein expression. After 48 hours post-infection, adherent cells were incubated in 5 mM EDTA/PBS pH 7.4 and collected. Cells were washed two times in PBS and re-suspended with the anti-HA mouse monoclonal antibody (12CA5, Roche) for one hour on ice. Unbound antibody was removed by diluting the cell suspension in PBS and gently pelleting the cells (three times). After the final wash, cells were re-suspended in PBS with a Alexa488 conjugated goat anti-mouse secondary antibody (Molecular Probes), and incubated for 1 hour on ice. After three more washes in PBS as described above, cells were re-suspended at a concentration of ∼1 ×106 cells/ml. Fluorescently tagged cells were assessed by FACS. Data are plotted as the relative level of cell associated fluorescence as compared to uninfected cells.
Results
The role of HPV16 E5 protein has been difficult to discern. Barriers to the study of this protein include low levels of protein expression, poor protein expression, and a paucity of reagents for detecting the protein. In order to overcome these obstacles, we generated tetracycline-regulatable (‘tet-off’) adenoviruses that express a hemagluttinin (HA) tagged, codon optimized form of the protein. The adenoviruses, termed HPV16 HA-E5 and HPV16 E5-HA, encode the HA epitope on either the amino- or carboxyl-terminus, respectively. These adenoviruses can only be expressed in cells that express the tetracycline transactivator (tTA), a chimeric protein that constitutively binds and activates the tetracycline promoters, except in the presence of tetracycline (Gossen and Bujard, 1992). We stably transfected the tTA in the spontaneously immortalized, human keratinocyte cell line, HaCaT cells. HaCaT cells are a HPV-negative cell line that is frequently used to study the function of HPV genes. These cells are referred to as tTA-HaCaT cells and used throughout this study. This expression system undoubtly expresses HPV16 E5 at levels that exceed those that occur following infection with the native virus. However, the progression of HPV infection into a cancer is a rare event, and overexpression of oncogenes facilitates the detection of rare, but important, cellular events.
As reported previously, we have found that expression of HPV16 E5 causes cell fusion (Hu et al., 2009). This is demonstrated using a YFP-reporter assay in which tTA-HaCaT cells are transfected with either T7 RNA polymerase (pRK-T7 RNA polymerase) or YFP driven by a T7 promoter (pT7-YFP), mixed in a 1:1 ratio, infected with HPV16 E5, and examined for the presence of YFP-positive cells as measured by FACS (Schematic shown in Figure 1A) (Hu et al., 2008). This high-throughput assay allows the rapid measure of fused cells, as only when the cytosol of the two differentially transfected cells mix is the fluorescent protein produced (Figure 1B). As shown in Figure 1C, expression of the HPV16 HA-E5 or HPV16 E5-HA causes a 5-8 fold increase in the YFP fluorescence intensity. Cells infected with the HPV16 E5 adenoviruses in the presence of tetracycline do not express the protein (Figure 1D) or cause the cells to fuse (Figure 1C). Thus, cells fusion is mediated by HPV16 E5 expression and is not an indirect consequence of adenovirus infection.
Figure 1. Expression of HPV16 E5 causes cell to fuses.

A. Schematic of cell fusion assay used in this study. Separate populations of tTA-HaCaT cells were transfected with either pRK-T7 RNA polymerase or pT7-YFP, mixed in a 1:1 ratio, and infected with either adenovirus encoding HPV16 HA-E5 or HPV16 E5-HA in the present or absence of tetracycline. Cells were incubated for 72 hours. B. Following incubation, cells were fixed and stained with DAPI. Shown are representative images of YFP-positive, bi-nucleated cells generated in the cell fusion assay. Size bar = 10 μm. C. Cells were collected and sorted by FACS to assess the number of YFP-positive cells. Data are the normalized (to uninfected cells) total fluorescence intensity (percentage of positive cells times the mean fluorescence intensity). Shown are the average ± S.E.M. (n=3). D. The remaining cell lysates were collected after flow cytometry and were resolved on a 16% Tris-tricine gel and immunoblotted with the 12CA5 (anti-HA) antibody or α-tubulin as a loading control. *indicates p< 0.01 (student’s t-test).
HPV16 E5 is expressed on the plasma membrane
In order to gain insight into the molecular mechanism by which HPV16 E5 mediates cell fusion, we wanted to know where in the cell it was expressed. A common characteristic of fusogenic proteins is that they are transmembrane proteins expressed on the cell surface. To determine if this was the case for HPV16 E5, we isolated the plasma membranes of cells expressing either HPV16 HA-E5 or HPV16 E5-HA using the Qiagen Qproteome plasma membrane purification system as described in Materials and Methods.
For this experiment, we used the epidermal growth factor receptor (EGFR) as a positive control for plasma membrane purification. Glyceraldehyde-3-phosphate dehydrogenase (GADPH) is a cytosolic protein and used to monitor non-specific protein isolation (Figure 2). Both HPV16 HA-E5 and HPV16 E5-HA were present in the membrane fraction (Figure 2). Since this is a non-quantitative assay, we can not accurately estimate the percentage of HPV16 E5 that localizes to the plasma membrane. A conservative estimate indicates at least 10% of the HPV16 E5 is localized to the plasma membrane based on the relative levels of protein detected and the amounts loaded. HPV16 E5 has been reported to localize to a variety of cellular compartments, including early endosome, Golgi, and nuclear membrane (Conrad et al., 1994; Disbrow, Hanover, and Schlegel, 2005; Oelze et al., 1995; Oetke et al., 2000; Straight, Herman, and McCance, 1995). Based on the indirect immunofluorescence data (Figure 3), it is likely that the remainder of the HPV16 E5 protein has localized to one of these other locations.
Figure 2. Biochemical evidence HPV16 E5 is enriched in the plasma membrane.

tTA-HaCaT cells were infected with nothing (uninfected), HPV 16 HA-E5, or HPV16 E5-HA adenoviruses. Forty-eight hours post infection, the cells were harvested, and the plasma membrane was isolated from the infected cells (Qproteome plasma membrane protein purification, Qiagen). For the GAPDH and E5 detection, the total (Total) and plasma membrane (PM) fractions (5% and 50%, respectively) were resolved using 16% tris-tricine polyacrylamide gel and immunoblotted with antibodies against GAPDH and 12CA5. The EGFR was detected by resolving total and plasma membrane fractions (5% and 50%, respectively) on a 7.5% SDS-PAGE gel and immunoblotting with an EGFR antibody (sc-03, Santa Cruz Biotechnology). Shown is a representative experiment repeated three times.
Figure 3. Indirect immunofluorescence data localizing HPV16 E5 to the plasma membrane.

A. tTA-HaCaT cells were infected with HPV16 HA-E5, or HPV16 E5-HA. Forty-eight hours post-infection, cells were fixed and permeabilized with or without 1mg/ml saponin. Cells were processed for indirect immunofluorescence and stained with DAPI as described in Materials and Methods. Shown are representative images from an experiment repeated three times. Size bar = 10 μm. B. tTA-HaCaT cells were infected with adenovirus encoding HPV16 E5-HA, and processed for indirect immunofluorescence using antibodies against against the HA tag (Y-11, Santa Cruz Biotechnology) (green) and the EGFR (Ab-1, Calbiochem) (red). Cells were fixed and stained with DAPI (blue). Shown are the individual confocal sections of the red and green channels, as well as the merged images including the DAPI stain. For the merged image the horizontal and vertical z-stacks are shown below and to the right of the merged images. Size bar = 10 μm.
Regardless of the exact percentage of HPV16 E5 that localizes to the plasma membrane, the level is sufficient to induce cell-cell fusion, both when HPV16 E5 is expressed alone or in the context of the entire genome (Hu et al., 2008). In addition, this experiment demonstrates that the location of the HA-epitope does not affect targeting to the plasma membrane localization.
Having localized HPV16 E5 to the plasma membrane, we next wanted to determine its orientation in the membrane. To answer this question, we infected tTA-HaCaT cells with either HPV16 HA-E5 or HPV16 E5-HA. After infection, cells were subjected to indirect immunofluorescence with or without membrane permeabilization (Figure 3A). In cells that are permeabilized, both the HPV16 HA-E5 and the HPV16 E5-HA can be detected. However, in the absence of permeabilization, only the HPV16 E5-HA can be detected.
Since the morphology of HaCaT cells is relatively flat, it is difficult to discern whether the protein is localized on the plasma membrane using wide-field microscopy. To combat this problem, we examined the distribution of HPV16 E5-HA on HaCaT cells relative to a plasma membrane localized protein, the epidermal growth factor receptor (EGFR). HaCaT cells that had been transduced with HPV16 E5-HA adenovirus were subjected to indirect immofluorescence using antibodies against the HA-tagged E5 (red) and EGFR (green) in the absence or presence of membrane permeabilization (Figure 3B). In unpermeabilized cells, the distribution of HPV16 E5-HA is similar to that of the EGFR. However, when the cells were permeabilized, there is some staining for HPV16 E5-HA (as well as the EGFR) in the cytoplasm, indicating only a portion of the E5 is localized to the plasma membrane.
These data are corroborated by analysis of the 12CA5 (anti-HA) antibody binding to intact cells and sorting by Fluorescent Assisted Cell Sorting (FACS). There is an increase in cell-associated fluorescence in cells infected with HPV16 E5-HA, but not those expressing HPV16 HA-E5 (Figure 4A). For both proteins, fluorescence returns to background levels (uninfected cells) when tetracycline is added to ablate protein expression.
Figure 4. HPV16 E5 is orientented with an intracellular amino terminus and an extracellular caroboxyl terminus.

A. tTA-HaCaT cells were infected with nothing or either adenovirus encoding HPV16 HA-E5, or HPV16 E5-HA in the absence or presence of 1 μg/ml tetracycline, as indicated. Forty-eight hours post-infection, cells were collected, and a suspension of intact cells was incubated with the 12CA5 antibody for 1 hour at 4°C, washed extensively and incubated with an Alexa488-conjugated goat anti-mouse secondary antibody for 1 hour at 4°C, followed by more washing. Cells were sorted by FACS to determine the mean fluorescent intensity associated with the cells. Data are plotted as the fold change in fluorescent mean intensity as compared to uninfected cells. Data are plotted as the average ± S.E.M. (n=4). * indicates p< 0.01 (student’s t-test).
From these two different assays, we demonstrate that HPV16 E5 is expressed on the cell surface with the amino-terminus intracellular and the carboxyl-terminus extracellular. Based on the plasma membrane purification data, we know both proteins are expressed on the plasma membrane and we can be confident that differences in protein detection reflect the protein’s orientation.
Both cells must express HPV16 E5 for cell fusion to occur
Knowing that the HPV16 E5 is expressed on the plasma membrane, we wanted to know whether cell fusion required expression of the protein on both cells. If only one cell required protein expression that would suggest HPV16 E5 was binding a receptor on its neighboring cell. Conversely, if both cells require expression of HPV16 E5 that would be evidence that either the two proteins directly interact with one another or indirectly interact via a protein or protein complex. This distinction is important for the design of antagonists to cell fusion.
To assess whether HPV16 E5 is needed on both cells for fusion, we first used a single cell assay to monitor the formation of heterokaryons (See Figure 5A for Schematic of Experimental Design). Nuclei of different cell populations were labeled with either red fluorescent protein - histone 2B (RFP-H2B) or yellow fluorescent protein-histone 2B (YFP-H2B) and mixed in a 1:1 ratio. Cells were then infected with adenovirus encoding HPV16 HA-E5. Cells were then monitored for the formation of heterokaryons - bi-nucleated cells expressing both a red and green labeled nuclei (Figure 5B-G). tTA-HaCaT cells were used to express HPV16 E5. When it was desired that a cell population not express HPV16 E5, parental HaCaT cells were used. The parental HaCaT cells do not express HPV16 E5 following infection (Figure 5H). Shown in Figure 5B-D are representative micrographs of cells with nuclei labeled with RFP-histone2B and YFP-histone 2B with HPV16 E5 expression. Under these conditions, we observed heterokaryons (Figure 5B), as well as cells with enlarged nuclei with both fluorescent labels (Figure 5C-D). Enlarged nuclei are another characteristic of HPV16 E5-expressing cells1. Quantification of heterokaryons reveals that approximately 10% of bi-nucleated cells are heterokaryons when both cells express HPV16 E5 (Figure 5H), which is consistent with our previous findings (Hu et al., 2009). In contrast, when only one population of fluorescently-labeled cells expresses HPV16 E5, we observed cells with enlarged nuclei in the HPV16 E5 expressing cells, but no heterokaryons (Figure 5E and F). Cell populations with no HPV16 E5 have no heterokaryons or changes in nuclear size (Figure 5G).
Figure 5. Heterokaryons only form when both cells express HPV16 E5.

A. Schematic of experimental design. Briefly, tTA-HaCaT and parental HaCaT cells were transfected with either histone 2B-red fluorescent protein (H2B-RFP) or histone 2B-yellow fluorescent protein (H2B-YFP) to mark the nuclei. Combinations of tTA-HaCaT or HaCaT cells expressing H2B-RFP were mixed with cells expressing H2B-YFP. Cells were infected with HPV16 HA-E5 adenoviruses. Only cells stably transfected with the tTA expressed the adenoviral protein. After 72 hours post-infection, cells were subjected to indirect immunofluorescence using the anti-HA antibody and a Alexa 647- conjugated goat anti-mouse secondary antibody (blue). Shown are representative images from three separate experiments. B-D) Both populations of cells express HPV16 HA-E5, E) Only cells with red nuclei express HPV16 HA-E5. F) Only cells with green nuclei express HPV16 HA-E5. G) Neither cell population expresses HPV16 HA-E5. Size bar = 10 μm. H) Shown is the average percentage of bi-nuclei cells (± S.E.M.) in different cell population from three experiments (n= 690-1530 cells/condition).
These data are consistent with our findings using the YFP-reporter assay (Figure 6A). This assay allows us to look at large populations of cells and to quantify changes in cell fusion. Much like what was seen in the microscopy based assays, we detect increased cell fusion only when both cells express HPV16 E5. When only one cells expresses HPV16 E5, the change in cell fusion is not statistically different from control (HPV16 E5 negative) cells.
Figure 6. Cell fusion requires expression of the HPV16 E5 on both cells.

As a secondary approach to quantify cell-cell fusion, a derivation of the approach shown in Figure 5A was used. tTA-HaCaT cells and parent HaCaT cells were transfected with either pRK T7-RNA polymerase or pT7-YFP. Either tTA-HaCaT or HaCaT cells expressing pRK T7-RNA polymerase were mixed with either tTA-HaCaT or HaCaT cells expressing pT7- YFP, such that all four possible combinations of cells were mixed. Cells were infected with HPV16 HA-E5 adenoviruses and only tTA-HaCaT cells expressed the HPV16 HA-E5, thus allowing HPV16 HA-E5 to be differentially co-expressed with pRK T7-RNA polymerase or pT7-YFP. Cells engineered to express HPV16 HA-E5 are indicated by “+E5”, whereas those that do not are marked as “-E5”. After 72 hours, cells were collected and sorted by FACS to assess the number of YFP-positive cells (indicating mixing of cytosols). A) Data are plotted as the fold increase in Total Fluorescence Intensity (percentage of positive cells times the mean fluorescence intensity). Shown are the average ± S.E.M. (n=3). * indicates p< 0.01 (student’s t-test) as compared to cells expressing HPV16 E5 in only one cell. B) Cells collected from (A) were lysed, resolved by 16% Tris-tricine gel, and immunoblotted for the presence of HPV16 HA-E5 using an anti-HA antibody (12CA5) or α-tubulin (Sigma) as a loading control.
Together, these data indicate that either HPV16 E5 is forming a dimer with itself or the two proteins are interacting via a bridge protein or protein complex. Importantly, these data eliminate the possibility that HPV16 E5 binds to a receptor on its neighboring cell. It has been shown by others (Gieswein, Sharom, and Wildeman, 2003), and repeated by us (data not shown) that HPV16 E5 forms oligomeric complexes with itself. We have not been able to demonstrate that HPV16 E5 on neighboring cells can form a complex. That will be the focus of future studies.
We observe a disproportionate decrease in protein expression when only one population of cells expresses HPV16 E5 (Figure 6B). Based on our indirect immunofluorescence analysis of HA-positive cells, this is not a function of the initial level of HPV16 HA-E5 expression. One explanation for the differences is the growth advantage that is conferred to unfused, HPV16 E5-negative cells. We have observed previously that bi-nucleate cells that form following HPV16 HA-E5 expression fail to proliferate (Hu et al., 2009). Thus, despite the two different populations of cells being plated and infected at a 1:1 ratio, 72 hours after introduction of HPV16 E5, the HPV16 E5-negative cells will have grown more. When HPV16 HA-E5 expression is assayed based on loading equivalent amounts of protein, <50% of the cells are HPV16 E5 positive. When both populations express HPV16 E5, neither population has a growth advantage over the other and the level of HPV16 HA-E5 expression is higher relative to total protein.
However, these differences in growth among the two cell populations do not effect the interpretation of the data, as ratio of the two cell populations (based on cell counting and infection efficiency) is 1:1 initially. Only after cell fusion begins to occur do the ratios change.
Cell fusion can be inhibited by antibody binding to the extracellular domain of HPV16 E5
To determine if HPV16 E5-mediated cell fusion could be inhibited, we expressed either HPV16 HA-E5 or HPV16 E5-HA in HaCaT cells. Following infection, cells were incubated with the 12CA5 (anti-HA) antibody for 72 hours, fixed, DAPI stained, and examined under the microscope for the presence of bi-nucleated cells (Figure 7). As observed in the past, there was a 4-fold increase in the percentage of bi-nucleated cells with HPV16 E5-HA expression (Hu et al., 2009). The addition of the 12CA5 antibody attenuated the formation of bi-nucleated cells to levels observed when E5 expression was abrogated by the addition of tetracycline. The addition of the 9E10 (anti-myc epitope) had no effect on the percentage of bi-nucleated cells. Importantly, in cells expressing HPV16 HA-E5, the addition of the 12CA5 antibody did not alter the levels of bi-nucleated cells. Thus, antibody binding to the extracellular portion of HPV16 E5 prevents fusion, presumably through sterically hindering an interaction of the protein with the requisite molecules for cell fusion. It is not know if the interacting protein is another HPV16 E5 molecule or a bridging intermediate. Regardless of the exact mechanism, this finding provides proof of principle that HPV16 E5-mediated cell fusion can be inhibited by the addition of extracellular molecules.
Figure 7. Antibody inhibition of HPV16 E5-HA cell fusion.
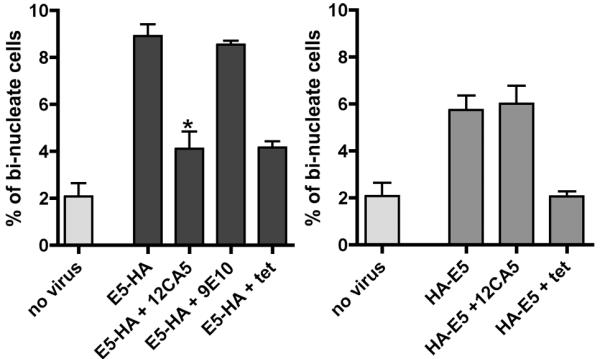
tTA-HaCaT cells were infected with either A) HPV16 E5-HA or B) HPV16 HA-E5. Immediately following infection, cells were incubated with no antibody, 12CA5 (anti-HA 1:250) or 9E10 (anti-myc, 1:250). Cells were incubated additional 72 hours, fixed, and stained with Dapi. Cells were scored for the number of bi-nucleated cells. Data are plotted as the percentage of bi-nucleated cells found in each population (the average ± S.E.M.; n= 3). * indicates p< 0.01 (student’s t-test) as compared to cells without the 12CA5 antibody.
Discussion
In this manuscript, we demonstrate that the fusogenic HPV16 E5 is expressed on the plasma membrane of cells and is oriented with its amino-terminus inside the cell and the carboxyl-terminus outside the cell. Our data show that both cells must express HPV16 E5 for cell-cell fusion to occur, indicating that E5 can not induce cell-cell fusion through an interaction with another protein (receptor) on a neighboring cell, but rather forms an E5-E5 dimer or a complex containing at least two HPV16 E5 molecules. These findings provide important insight into how the fusogenic process is mediated.
Cell-cell fusion is a common occurrence in biology. It is observed in diverse processes from fertilization, muscle and bone development, immune response, and tissue regeneration (Chen and Olson, 2005). Despite the well-established roles for cell-cell fusion in these processes, very little is understood regarding how this process is mediated.
HPV16 E5 has all the characteristics of fusogenic proteins. These include localization to the plasma membrane, high level of hydrophobicity, and the ability for form dimers (Gieswein, Sharom, and Wildeman, 2003; Martin and Ryuysschaert, 2000). However, mechanistically, little is known how fusion is mediated. Our data regarding the localization and orientation of the HPV16 E5 protein will be useful for elucidating the molecular details regarding how cell-cell fusion occurs.
Despite the knowledge that many oncogenic viruses are able to promote cell-cell fusion, a definitive link between cell-cell fusion and carcinogenesis has only been established recently (Duelli and Lazebnik, 2007; Duelli et al., 2007). Based on recent findings and the subsequent models, it is easy to see why delineating a role for cell-cell fusion in oncogenesis has been difficult. Under most conditions, fused cells become quiescent or undergo apoptosis (Hu et al., 2009) and are irrelevant to cancer progression as they are not involved in tumor formation. Those cells that escape quiescence/apoptosis and do divide appear to be mononucleated. Thus, the bi-nucleated cells are transient. These mononucleated cells are characterized by an increased incidence of chromosomal abnormalities - gain or loss of chromosomes or structural abnormalities (Duelli et al., 2007). Such chromosomal changes, or aneuploidy, are hallmarks of cancer cell. Thus, since cell-cell fusion is rare event coupled with the fact that the presence of bi-nucleated cells are transient, observing, tracking, and understanding these events has been difficult. Knowing the basic cell biology of the HPV16 E5 protein will facilitate the design of reagents that will inhibit cell-cell fusion, enhance cell fusion, and understand the fate of cell that have fused.
In order to better understand the role of cell-cell fusion in developmental and cancer biology, it is critical that cell biological and biochemical properties of these proteins be better understood. Having identified the cellular location, orientation, and a strategy for inhibiting HPV16 E5-mediated cell-cell fusion, we provide “proof of principle” evidence that E5 can serve as a target to inhibit cell-cell fusion. These data will be the basis for future studies that target the native HPV16 E5 protein to study and prevent cell transformation.
Supplementary Material
Expression of HPV16 E5 causes an increase in cell fusion as measured by the fluorescence reporter assay.
Shown are representative data from one of three experiments used to generate Figure 1C. Separate populations of tTA-HaCaT cells were transfected with either pRK-T7 RNA polymerase or pT7-YFP, mixed in a 1:1 ratio, and infected with either adenovirus encoding HPV16 HA-E5 or HPV16 E5-HA in the present or absence of tetracycline. After 72 hours, cells were collected and sorted by FACS to assess the number of YFP-positive cells. Shown are representative FACS images from three experiments. Typically, 0.1 - 0.5% of cells that were not infected with any HPV16 E5 had a low level of autofluorescence.
Acknowledgements
This work was funded by OCAST Grant HR03-014, The Mary Kay Ash Foundation, and NIH Grant Number P20 RR 017703 from the COBRE Program of the National Center for Research Resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bouvard V, Matlashewski G, Gu ZM, Storey A, Banks L. The Human Papillomavirus type 16 E5 gene cooperates with the E7 gene to stimulate proliferation of primary cells and increases viral gene expression. Virol. 1994;203:73–80. doi: 10.1006/viro.1994.1456. [DOI] [PubMed] [Google Scholar]
- Bravo IG, Crusius K, Alonso A. The E5 protein of the human papillomavirus type 16 modulates composition and dynamics of membrane lipids in keratinocytes. Arch Virol. 2005;150(2):231–246. doi: 10.1007/s00705-004-0420-x. [DOI] [PubMed] [Google Scholar]
- CDC . In: Genital HPV Infection - CDC Fact Sheet. Disease I, editor. 2007. [Google Scholar]
- Chen EH, Olson EN. Unveiling the Mechanisms of Cell-Cell Fusion. Science. 2005;308:369–373. doi: 10.1126/science.1104799. [DOI] [PubMed] [Google Scholar]
- Clifford GM, Smith JS, Plummer M, Munoz N, Franceschi S. Human papillomavirus types in invasive cervical cancer worldwide: a meta-analysis. Brit J of Cancer. 2003;88(1):63–73. doi: 10.1038/sj.bjc.6600688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, El Chissassi F. Carcinogenicity of human papillomaviruses. Lancet Oncology. 2005;6:204. doi: 10.1016/s1470-2045(05)70086-3. [DOI] [PubMed] [Google Scholar]
- Conrad M, Goldstein D, Andresson T, Schlegel R. The E5 Protein of HPV-6, but not HPV-16, Associates Efficiently with Cellular Growth Factor Receptors. Virol. 1994;200:796–800. doi: 10.1006/viro.1994.1244. [DOI] [PubMed] [Google Scholar]
- Dinneen JL, Ceresa BP. Constitutive activation of rab5 results in a ligand independent redistribution of the EGFR and attenuates its ability to signal. Traffic. 2004;5(8):606–615. doi: 10.1111/j.1398-9219.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- Disbrow GL, Hanover J, Schlegel R. Endoplasmic Reticulum-Localized Human Papillomavirus Type 16 E5 Protein Alters Endosomal pH but Not trans-Golgi pH. J. Virol. 2005;79(9):5839–5846. doi: 10.1128/JVI.79.9.5839-5846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disbrow GL, Sunitha I, Baker CC, Hanover J, Schlegel R. Codon Optimization of HPV-16 E5 Gene Enhances Protein Expression. Virol. 2003;311:105–114. doi: 10.1016/s0042-6822(03)00129-6. [DOI] [PubMed] [Google Scholar]
- Duelli D, Lazebnik Y. Cell-to-cell fusion as a link between viruses and cancer. Nat Rev Cancer. 2007;7:968–976. doi: 10.1038/nrc2272. [DOI] [PubMed] [Google Scholar]
- Duelli DM, Padilla-Nash HM, Berman D, Murphy KM, Ried T, Lazebnik Y. A Virus Causes Cancer by Inducing Massive Chromosomal Instability through Cell Fusion. Curr Biol. 2007;17:1–7. doi: 10.1016/j.cub.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Gieswein CE, Sharom FJ, Wildeman AG. Oligomerization of the E5 protein of human papillomavirus type 16 occurs through multiple hydrophobic regions. Virol. 2003;313:415–426. doi: 10.1016/s0042-6822(03)00296-4. [DOI] [PubMed] [Google Scholar]
- Gillison ML, D’Souza G, Westra W, Sugar E, Xiao W, Begum S, Viscidi R. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100(6):407–20. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal asociation between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92(9):709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci (U.S.A) 1992;89(12):5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Plafker K, Henthorn J, Ceresa BP. A Non-invasive Technique for Quantifying and Isolating Fused Cells. Cytotechnology. 2008;58(3):113–118. doi: 10.1007/s10616-009-9186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Plafker K, Vorozhoko V, Zuna RE, Hanigan MH, Gorbsky GJ, Plafker SM, Angeletti PC, Ceresa BP. Human Papillomavirus 16 E5 Induces Polyploidy by Cell Fusion. Virology. 2009;384(1):125–134. doi: 10.1016/j.virol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Alani RM, Munger K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21 Cip1-mediated inhibition of cdk2. Genes Dev. 1997;111:2101–2111. doi: 10.1101/gad.11.16.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch K, Alonso A. The human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J. Virol. 2002;76:12162–12172. doi: 10.1128/JVI.76.23.12162-12172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leechanachai P, Banks L, Moreau F, Matlashewski G. The E5 gene from human papillomavirus type 16 is an oncogene which enhances growth factor-mediated signal transduction to the nucleus. Oncogene. 1992;7(1):19–25. [PubMed] [Google Scholar]
- Leptak C, Ramon y Cajal S, Kulke R, Horwitz BH, Riese DJ, Dotto GP, DiMaio D. Tumorigenic Transformation of Murine Keratinocytes by the E5 Genes of Bovine Papillomavirus Type 1 and Human Papillomavirus Type 16. J. Virol. 1991;65(12):7078–7083. doi: 10.1128/jvi.65.12.7078-7083.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Ryuysschaert J-M. Common Properties of Fusion Peptides from Diverse Systems. Biosci Reports. 2000;20(6):483–500. doi: 10.1023/A:1010454803579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCance DJ. Human Papillomaviruses and Cell Signaling. Science STKE. 2005;29:29. doi: 10.1126/stke.2882005pe29. [DOI] [PubMed] [Google Scholar]
- McMurray HR, Nguyen D, Westbrook TF, McCance DJ. Biology of Papillomaviruses. J Exp Path. 2001;82:15–33. doi: 10.1046/j.1365-2613.2001.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal KR, Chan W, Demopoulos RL. Sensitivity and specificity of various morphological features of cervical condylomas. Arch Pathol Lab Med. 1990;114:1038. [PubMed] [Google Scholar]
- Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci. 2007;98(10):1505–11. doi: 10.1111/j.1349-7006.2007.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelze I, Kartenbeck J, Crusius K, Alonso A. Human papillomavirus type 16 E5 protein affects cell-cell communication in an epithelial cell line. J Virol. 1995;69:4489–4494. doi: 10.1128/jvi.69.7.4489-4494.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oetke CE, Auvinen E, Pawlita M, Alonso A. Human Papillomavirus type 16 E5 protein localizes to the Golgi apparatus but does not grossly affect cellular glycosylation. Arch Virol. 2000;145:2183–2191. doi: 10.1007/s007050070048. [DOI] [PubMed] [Google Scholar]
- Pim D, Collins M, Banks L. Human Papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene. 1992;7(1):27–32. [PubMed] [Google Scholar]
- Prasad CJ, Sheets E, Selig AM, McArthur MC, Crum CP. The bi-nucleated Squamous Cell: Histologic Spectrum and Relationship to Low-grade Squamous Intraepithelial Lesions. Mod Pathol. 1993;6(3):313–317. [PubMed] [Google Scholar]
- Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63(48624871) [PubMed] [Google Scholar]
- Schwartz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 1985;314:111–114. doi: 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- Stoler MH, Rhodes CR, Whitbeck A, Wolinsky SM, Chow LT, Broker TR. Human Papillomavirus Type 16 and 18 Gene Expression in Cervical Neoplasias. Human Pathology. 1992;23(2):117–128. doi: 10.1016/0046-8177(92)90232-r. [DOI] [PubMed] [Google Scholar]
- Stoppler MC, Straight SW, Tsao G, Schlegel R, McCance DJ. The E5 Gene of HPV-16 Enhances Keratinocyte Immortalization by Full-Length DNA. Virol. 1996;223:251–254. doi: 10.1006/viro.1996.0475. [DOI] [PubMed] [Google Scholar]
- Straight SW, Herman B, McCance DJ. The E5 Oncoprotien of Human Paillomavirus Type 16 Inhibits the Acidification of Endosomes in Human Keratinocytes. J Virol. 1995;69(5):3185–3192. doi: 10.1128/jvi.69.5.3185-3192.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight SW, Hinkle PM, Jewers RJ, McCance DJ. The E5 Oncoprotein of Human Papillomavirus Type 16 Transforms Fibroblasts and Effects the Downregulation of the Epidermal Growth Factor Receptor in Keratinocytes. J Virol. 1993;67(8):4521–4532. doi: 10.1128/jvi.67.8.4521-4532.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprynowicz FA, Disbrow GL, Krawczyk E, Simic V, Lantzky K, Schlegel R. HPV-16 E5 oncoprotein upregulates lipid raft components caveolin-1 and ganglioside GM1 at the plasma membrane of cervical cells. Oncogene. 2008;27(8):1071–1078. doi: 10.1038/sj.onc.1210725. [DOI] [PubMed] [Google Scholar]
- Thomsen P, van Deurs B, Norrild B, Kayser L. The HPV 16 E5 oncogene inhibits endocytic trafficking. Oncogene. 2000;19:6023–6032. doi: 10.1038/sj.onc.1204010. [DOI] [PubMed] [Google Scholar]
- Tomakidi P, Cheng H, Kohl A, Komposch G, Alonso A. Modulation of the epidermal growth factor receptor by the human papillomavirus type 16 E5 protein in raft cultures of human keratinocytes. Eur J of Cell Biol. 2000:407–412. doi: 10.1078/0171-9335-00060. [DOI] [PubMed] [Google Scholar]
- Valle GF, Banks L. The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J Gen Virol. 1995;76:1239–1245. doi: 10.1099/0022-1317-76-5-1239. [DOI] [PubMed] [Google Scholar]
- Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- Williams SMG, Disbrow GL, Schlegel R, Lee D, Threadgill DW, Lambert PF. Requirement of Epidermal Growth Factor Receptor for Hyperplasia Induced by E5, a High-Risk Human Papillomavirus Oncogene. Cancer Res. 2005;65(15):6534–6542. doi: 10.1158/0008-5472.CAN-05-0083. [DOI] [PubMed] [Google Scholar]
- Zhang B, Li P, Wang E, Brahmi Z, Dunn KW, Blum JS, Roman A. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virol. 2003;310:100–108. doi: 10.1016/s0042-6822(03)00103-x. [DOI] [PubMed] [Google Scholar]
- Zhang B, Spandau DF, Roman A. E5 Protein of Human Papillomavirus Type 16 Protects Human Foreskin Keratinocytes from UV B-Irradiation-Induced Apoptosis. J Virol. 2002;76(1):220–231. doi: 10.1128/JVI.76.1.220-231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of HPV16 E5 causes an increase in cell fusion as measured by the fluorescence reporter assay.
Shown are representative data from one of three experiments used to generate Figure 1C. Separate populations of tTA-HaCaT cells were transfected with either pRK-T7 RNA polymerase or pT7-YFP, mixed in a 1:1 ratio, and infected with either adenovirus encoding HPV16 HA-E5 or HPV16 E5-HA in the present or absence of tetracycline. After 72 hours, cells were collected and sorted by FACS to assess the number of YFP-positive cells. Shown are representative FACS images from three experiments. Typically, 0.1 - 0.5% of cells that were not infected with any HPV16 E5 had a low level of autofluorescence.