

Abstract
Prophylactic agents acutely administered in response to anticholinesterases intoxication can prevent toxic symptoms, including fasciculations, seizures, convulsions and death. However, anticholinesterases also have long-term unknown pathophysiological effects, making rational prophylaxis/treatment problematic. Increasing evidence suggests that in addition to excessive cholinergic stimulation, organophosphate compounds such as diisopropylphosphorofluoridate (DFP) induce activation of glutamatergic neurons, generation of reactive oxygen (ROS) and nitrogen species (RNS), leading to neurodegeneration. The present study investigated multiple affectors of DFP exposure critical to cerebral oxidative damage and whether antioxidants and NMDA receptor antagonist memantine provide neuroprotection by preventing DFP-induced biochemical and morphometric changes in rat brain. Rats treated acutely with DFP (1.25 mg/kg, s.c.) developed onset of toxicity signs within 7-15 min that progressed to maximal severity of seizures and fasciculations within 60 min. At this time point, DFP caused significant (p<0.01) increases in biomarkers of ROS (F2-isoprostanes, F2-IsoPs; and F4-neuroprostanes, F4-NeuroPs), RNS (citrulline), and declines in high-energy phosphates (HEP) in rat cerebrum. At the same time, quantitative morphometric analysis of pyramidal neurons of the hippocampal CA1 region revealed significant (p<0.01) reductions in dendritic lengths and spine density. When rats were pretreated with the antioxidants N-tert-butyl-α-phenylnitrone (PBN, 200 mg/kg, i.p.), or vitamin E (100 mg/kg, i.p./day for 3 days), or memantine (18 mg/kg, i.p.), significant attenuations in DFP-induced increases in F2-IsoPs, F4-NeuroPs, citrulline, and depletion of HEP were noted. Furthermore, attenuation in oxidative damage following antioxidants or memantine pretreatment was accompanied by rescue from dendritic degeneration of pyramidal neurons in the CA1 hippocampal area. These findings closely associated DFP-induced lipid peroxidation with dendritic degeneration of pyramidal neurons in the CA1 hippocampal area and point to possible interventions to limit oxidative injury and dendritic degeneration induced by anticholinesterase neurotoxicity.
Introduction
Pesticide residues are now among the most ubiquitous synthetic chemicals in our environment, as they are detectable in the tissues of humans, animals, aquatic and wildlife worldwide. Of the wide variety of pesticides available, organophosphate (OP) and carbamate (CM) insecticides are the most commonly used and encountered in accidental, suicidal, and occupational poisonings. Presently, more than 100 different OPs are used as insecticides worldwide (Kwong, 2002; Gupta, 2006). The widespread use and easy accessibility to these compounds result in a significant number of intoxications and several hundred thousand fatalities annually (Gunnell and Eddleston, 2003). Other derivatives of phosphoric or phosphonic acid, such as chemical warfare nerve agents, are considered to be the most toxic compounds among all chemical weapons (Watson et al., 2006, 2009). Therefore, OP compounds (industrial chemicals or weapons of mass destruction) pose a potential threat to civilians as well as military personnel.
Pharmacologically, OPs and CMs are acetylcholinesterase (AChE) inhibitors and their acute symptoms are attributed to accumulation of acetylcholine (ACh), thus exhibiting the signs of cholinergic hyperactivity. Depending upon the degree of AChE inhibition, the severity of poisoning can vary from mild (mild dyspnea, blurred vision and glandular hypersecretion) to severe (severe dyspnea, skeletal muscle fasciculations, convulsions and unconsciousness) cases, and eventually death ensues from respiratory failure (Goldfrank et al., 1982; Weinbroum, 2005).
However, anticholinesterases have long-term pathophysiological effects that are not yet well characterized, making rational prophylaxis and treatment for these effects problematic. Long-term neurological impairments following anticholinesterase exposure including: (a) an intermediate syndrome (IMS) affecting muscles, which can occur within 24 to 96 hours following recovery from severe acute affects (De Bleecker, 2006); (b) a delayed peripheral polyneuropathy associated with some anticholinesterases, that usually occurs within weeks following an acute exposure (Lotti, 1992: Lotti and Moretto, 2005); and (c) subtle, long-term neurological effects which may last months or even years (Behan, 1996; Jamal, 1997). Anticholinesterase initiation of adverse health effects is also associated with potential involvement of glial cells in the neurotoxicity of OPs (Aschner, 2000). Neuronal injury caused by seizures is accompanied by an inflammatory reaction involving gliosis, and induction of inflammatory mediators including prostaglandins, cytokines, cell adhesion proteins and matrix metalloproteinases (Jorgensen et al., 1993; Vezzani et al., 2002; Jourquin et al., 2003; Lehtimaki et al., 2003; Borges et al., 2003). Animals exposed to soman at doses producing convulsions exhibit a rapid increase in active astrocytes and the accumulation of glial fibrillatory acidic protein (GFAP) (Zimmer et al., 1997). In addition to oxidative damage and interference with adenylyl cyclase cell signaling, chlorpyrifos inhibits DNA synthesis to a greater extent in glioma cell lines (C6 cells) than neuronal cell lines (PC12) (Garcia et al., 2001). These effects are independent of cholinergic receptors as the cholinergic antagonist fails to block chlorpyrifos-induced inhibition of DNA synthesis. The findings are also consistent with exposures to another OP compound, diazinon, suggesting that anticholinesterase compounds target glial cells by additional mechanisms of cholinergic toxicity (Walker and Nidiry, 2002).
Involvement of non-cholinergic mechanisms in OP toxicity is also supported by evidence suggesting that anticholinesterases induce activation of glutamatergic neurons. For example, soman-induced seizures increased extracellular glutamate in the pyriform cortex (Wade et al., 1987) and cornu ammonis (CA) region of the hippocampus (Lallement et al., 1992) followed by activation of N-methyl-D-aspartate (NMDA) receptors in the CA1 region. Overstimulation of glutamate receptors causes synaptic and cellular degeneration in the hippocampus (Siman et al., 1989; Bahr et al., 2002, Munirathinam and Bahr, 2004). Excitotoxicity in hippocampal neurons is also associated with enhanced vulnerability to other types of neuropathogenesis (Bahr et al., 1994). Moreover, glutamate stimulates ACh release (Anderson et al., 1994), further contributing to excitatory stimulation and prolongation of the seizures, and thus like a brushfire, it propagates excitotoxic neurodegeneration in vulnerable brain regions. Microdialysis studies revealed an immediate increase in extracellular glutamate concentrations in the septum, pyriform cortex, hippocampal regions and amygdala following soman-triggered seizures (Lallement et al., 1991a, b; Wade et al., 1987). Furthermore, blockage of specific glutamate receptors reduces neuropathogenic responses, including nerve agents' toxicity (Sheardown et al., 1990; Sparenborg et al., 1992). In addition to the activation of NMDA receptors, glutamate release also leads to massive Ca2+ fluxes into the post-synaptic cells, generation of reactive oxygen (ROS) and nitrogen species (RNS), ensuing in neurodegeneration. Elevation of cytosolic free Ca2+ leads to derangement of many intracellular processes that normally regulate Ca2+ sequestration and energy metabolism (Siesjo, 1988). Modulations of Ca2+, glutamate and NMDA receptors also induce some other biochemical mechanisms such as oxidative stress which further compromise cell viability.
There are many methods to quantify oxidative damage to tissues, but in the CNS no method distinguishes oxidative damage between neurons and glia. This is potentially a serious limitation because glia outnumbers neurons with a further increase in this ratio in neurodegenerative diseases. We have shown previously that free radical damage to the brain can be sensitively and accurately quantified by measuring chemically stable oxidative damage products of arachidonic acid (AA) and docosahexaenoic acid (DHA); F2-IsoPs and neuroprostanes (F4-NeuroPs), respectively (Morrow et al., 1990; Milatovic and Aschner, 2009). AA is relatively evenly distributed in brain with similar concentrations in gray matter and white matter, and within glia and neurons. Thus, F2-IsoPs quantification is a reflection of oxidative damage to the brain in general and F4-NeuroPs in particular. Unlike AA, DHA is highly concentrated in neuronal membranes to the exclusion of other cell types. Moreover, F4-NeuroPs are by far the most abundant products of this pathway in the brain (Reich et al., 2000; 2001). Thus, quantification of F4-NeuroPs provides a highly selective quantitative window for neuronal oxidative damage in vivo.
In this study, we have used diisopropylphosphorofluoridate (DFP) as a model compound for OP insecticides or nerve agents, and investigated non-cholinergic mediated activities in rat cerebrum. Novel biomarkers of neuronal oxidative (F4-NeuroPs) and nitrosative (citrulline) damage and Neurolucida-assisted neuronal tracings were employed to explore the mechanisms involved in OP-induced neurotoxicity. Different pharmacological tactics were utilized to attenuate oxidative/nitrosative damage induced by anticholinesterase exposure and investigate extent to which such attenuation is accompanied by protection of dendritic damage in the CA1 sector of hippocampal neurons.
Materials and Methods
Animals
Male Sprague-Dawley rats weighing about 200 g (7-8 weeks old), purchased from Harlan Laboratories (Indianapolis, IN, USA), were used in this investigation. They were housed five per cage in a room with controlled conditions: temperature 21±1 °C, humidity 50±10 %, and 12-h/12-h light/dark cycle. Animals had free access to pelleted food and tap water. Rats were acclimatized to these conditions for 7-10 days before being used. During the treatment, rats were placed in individual cages. The animal facility is approved by the Institutional Animal Care and Use Committee (IACUC), and is under the supervision of a veterinarian. All experiments were conducted in accordance with the guidelines from the National Institutes of Health (USA), with adequate measures taken to minimize any discomfort to the rats.
Drugs and Chemicals
Diisopropylphosphorofluoridate (DFP), vitamin E (α-tocopherol, 97%), N-tert-butyl-α-phenylnitrone (PBN), acetylthiocholine iodide, adenosine 5′-triphosphate (ATP), phosphocreatine (PCr) and L-citrulline were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO., USA). Memantine HC1 (MEM) was purchased from Panorama Research, Inc (Mountain View, CA). All other chemicals used were of highest purity and were purchased from Cayman Chemical Company (Ann Arbor, MI), VWR (West Chester, PA) or Fisher Scientific (Fair Lawn, NJ, USA).
Experimental Design and Animal Treatment
In time-course experiments, the levels of F2-isoprostanes (F2-IsoPs) and F4-neuroprostanes (F4-NeuroPs) were assayed in brain at 30 min, 1 h, 2 h or 6 h post-DFP (1.25 mg/kg, s.c.) injection in rats. The rationale for selecting the DFP dose was based on our previous reports (Gupta et al., 2001a, b; Zivin et al., 1999; Milatovic et al., 2006) showing that 1.25 mg/kg of this anticholinesterase provide sings of maximal severity without causing the fatality. Following DFP administration, rats showed the signs of onset within 7-15 min and signs of maximal severity within 30 to 60 min that lasted for about 2 to 3 h. Rats in the control group received normal saline (1 ml/kg, sc). In protection experiments, pretreatment with memantine HCl (MEM, 18 mg/kg, i.p.) or N-tert-butyl-α-phenylnitrone (PBN, 200 mg/kg, i.p.) was given as a single injection 60 min (time of maximum distribution of MEM, Wesemann et al., 1983) or 30 min before DFP, respectively. Supplemental vitamin E (100 mg α-tocopherol/kg body wt., i.p.) was given daily for 3 days (48 hours, 24 hours, and 30 min before DFP exposure). To test protective effects of these drugs, rats were sacrificed 1 hour after the last treatment.
Biochemical assays
Quantitation of F2-IsoPs and F4- NeuroPs as a marker of ROS
At a predetermined time, rats were sacrificed and brains were rapidly harvested, the cerebral hemispheres flash frozen in liquid nitrogen, and stored at −80 °C until analysis. F2-IsoPs and F4-NeuroPs were determined with a stable isotope dilution method using a gas chromatograph coupled with a mass spectrometer (negative ion chemical ionization and selective ion mode) as previously described (Morrow et al., 1990; Milatovic et al., 2003; Milatovic and Aschner, 2009).
Quantitation of nitric oxide (NO) as a marker of RNS
The levels of citrulline (the co-product of NO synthesis) were determined by the reversed-phase HPLC method of Bagetta et al. (1995) with minor modifications (Gupta et al., 2001b; Gupta et al., 2007). Tissues were homogenized in a aliquot of 0.4 M perchloric acid using a Brinkman homogenizer with a PT-10 probe, followed by sonication for 10 s with a Biosonic Cell Disruptor equipped with a microprobe. The homogenates were allowed to extract for 30 min at ice-cold temperature, followed by centrifugation (10,000 rpm for 20 min at 4°C) using a Sorvall centrifuge (RC 26 plus). The supernatants were aspirated and neutralized to pH 7 with 1 M KOH before being centrifuged again to remove fine precipitate (KClO4). The supernatants were derivatized with OPA and assayed for citrulline concentrations using an HPLC system coupled with a fluorescence detector (excitation at 334 nm and emission at 440 nm). The data are expressed as nmol citrulline/g wet tissue weight.
Measurement of ATP and Phosphocreatine
Levels of ATP and phosphocreatine (PCr) were measured in the perchloric acid extracts (as described above for citrulline), using an HPLC method (Gupta et al., 2001a; 2007). The Waters HPLC system was coupled with a UV detector (Model 2487), which allowed simultaneous determination of ATP and PCr at a wavelength 206 nm.
Quantitative morphology of pyramidal neurons
Quantitative neuronal analysis was conducted on a tissue stained with Golgi impregnation that was uniform throughout the section. Length of dendrites and spine density counts of pyramidal neurons were evaluated in Golgi impregnated 50 microns thick hippocampal sections from paraffin-embedded blocks prepared according to the manufacturer's specifications (FD Rapid GolgiStain Kit). Six or more pyramidal neurons with no breaks in staining along the dendrites from the CA1 area of the hippocampus were selected and spines counted according to the published methods (Leuner et al., 2003; Milatovic et al., 2003). Tracing and counting were performed with a Neurolucida system at 100× under oil immersion (MicroBrightField, VT). Dendritic systems were also quantified to a centrifugal nomenclature by Sholl-method (Scholl, 1953) where spine density and length of dendrites arising from the soma are in the first- (50 μm), second- (50 μm -100 μm) and third-order segments (100 μm -150 μm) from the center of the soma.
Statistical analysis
The data presented are means ± SEM of 4-6 rats in each group. Statistical significance between groups was determined by analysis of variance (ANOVA) followed by Bonferroni's multiple comparison test with statistical significance set at p<0.05. All analyses were carried out with GraphPad Prism 4.02 for Windows (GraphPad Software, San Diego, CA, USA).
Results
A single injection of DFP with an acute dose of 1.25 mg/kg, s.c. produced toxic signs in rats, including salivation, tremors, wet dog shakes, fasciculations with rearing and rolling over within 15- 20 min. Signs of maximal intensity, such as severe muscle fasciculations, seizures, and convulsions developed within 30 min and lasted for about 2 – 3 h before tapering off. By 24 h, animals were free of toxic signs. DFP- induced signs were of typical hypercholinergic preponderance involving both the central and peripheral nervous systems as confirmed by markedly depressed AChE activity. Following DFP exposure, brain AChE activity was reduced to 10.12 ± 1.12 %, 10.05 ± 1.96 %, 14.30 ± 1.41 % and 16.51 ± 1.27% compared to control (569.12 ± 26.80 μmol/hr/g of tissue) at 30 min, 1 h, 2h and 6h, respectively.
Time-course analysis of cerebral biomarkers of oxidative damage revealed significant increases in F2-IsoPs (142%) and F4-NeuroPs (225%) as early as 30 min after a single acute exposure to DFP (1.25 mg/kg, sc). Both F2-IsoPs and F4-NeuroPs showed transient increase reaching maximum levels at 1h with a return to basal levels by 6 h (Fig. 1). The selective increase in F4-NeuroPs indicates that neurons are specifically targeted by this mechanism (Fig. 1b). Thus, a one-time challenge with DFP produced a transient increase in both F2-IsoPs and F4-NeuroPs, which are sensitive and specific in vivo biomarkers of oxidative damage to AA and DHA, respectively.
Figure 1.

Cerebral concentrations of F2-IsoPs (a) and F4-NeuroPs (b) following DFP (1.25 mg/kg, s.c.) exposure in rats. Values represent mean±SEM (n=4-6). *One-way ANOVA had p<0.001 with Bonferroni's multiple comparison tests showed significant difference (p<0.05) for vehicle injected control vs. DFP treatment.
In an attempt to discern additional mediators of oxidative damage in the rat model of DFP-induced neurotoxicity, the levels of citrulline and high- energy phosphates (HEPs: ATP and PCr) were determined in the cerebral hemispheres of rats 1 h after DFP exposure. NOS catalyzes the conversion of arginine to NO and citrulline, and quantification of citrulline is widely employed as an in vivo marker of NOS/NO activity. While citrulline levels were increased > 3-fold compared to control (139.2 ± 8.4 nmol/g tissue), significant depletion of ATP and PCr (47% and 38 % compared to control values (2.23 ± 0.05 and 8.90 ± 0.33 μmol/g tissue, respectively) was observed in brain of rats 1 h post-DFP injection. Alterations in HEPs are considered as indicators of mitochondrial dysfunction that increases neuronal vulnerability to injury.
We next investigated if neuronal oxidative/nitrosative damage and depletion of energy metabolites is accompanied by altered integrity of dendritic system. Data revealed that high intensity of seizures and peak of oxidative damage to neuronal membranes at 1 h coincided with dendritic degeneration of pyramidal neurons in the CA1 hippocampal area. Representative images of Golgi impregnated hippocampal sections with their traced pyramidal neurons from control and DFP-exposed animal are presented in Figure 2. Images of neurons with Neurolucida-assisted morphometry show that DFP-induced brain hyperactivity targeted the dendritic system with profound dendrite regression of hippocampal neurons. Dendritic morphology of randomly selected pyramidal neurons from CA1 hippocampal area from control and DFP-exposed animals was also evaluated by the Sholl method of concentric circles (Fig. 3). Sholl analysis represents a quantitative method for morphometric neuronal studies with consecutive-circles (50, 51-100, 101-150 μm from the center of soma) that analysis specifies dendritic geometry. Results of the present investigation demonstrate that DFP treatment caused a significant decrease in total dendrite length in the proximal (0-50 μm) and intermediate (51-100 μm) Sholl compartment of CA1 pyramidal neurons (Fig. 3a). Results also revealed that DFP treatment induced a significant decrease in spine density (number of spines per 100 μm of dendrites) in all three (proximal, intermediate and distal) Sholl compartments of CA1 pyramidal neurons (Fig. 3b).
Figure 2.
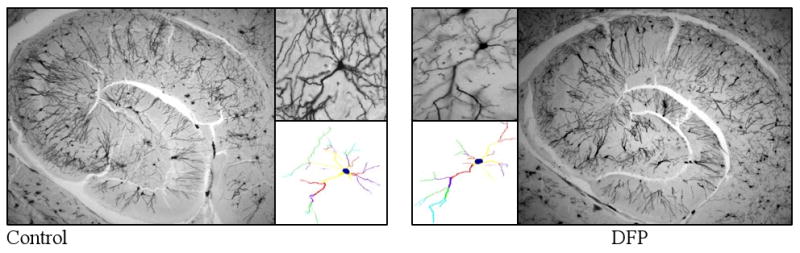
Photomicrographs of rat hippocampi (2.5×) with pyramidal neurons (10×) from CA1 hippocampal area of rat brains 1 h after saline (control) and DFP (1.25 mg/kg, s.c) injections. Treatment with DFP induced degeneration of hippocampal dendritic system, decrease in total length of dendrite and spine density of hippocampal pyramidal neurons. Tracing and counting were done using a Neurolucida system at 100 × under oil immersion (MicroBrightField, VT). Colors indicate the degree of dendritic branching (yellow=1°, red=2°, purple=3°, green=4°, turquoise=5°, gray=6°).
Figure 3.

Dendritic length (a) and spine density (b) in each Sholl compartment of pyramidal neurons from CA1 hippocampal area of rats following DFP (1.25 mg/kg, s.c.) exposure. Brains from rats exposed to DFP were collected 1 h post injections (n≥4). *One-way ANOVA had p<0.001 with Bonferroni's multiple comparison tests showed significant difference for Sholl compartment (p<0.05) from control vs. DFP treatment.
The last part of this investigation determined the effectiveness of two antioxidants (vitamin E and PBN) and NMDA receptor antagonist memantine to suppress DFP-induced oxidative/nitrosative damage and neurodegeneration. Vitamin E, PBN and memantine treatment in non-DFP exposed rats did not alter basal levels of F2-IsoPs, F4-NeuroPs, citrulline, ATP and PCr in brain. But when given as pretreatment E, PBN or memantine completely suppressed DFP-induced alterations in markers of neuronal oxidative damage (Fig. 4), nitrosative damage (Fig. 5), and depletion of HEPs (Fig. 6). Vitamin E, PBN or memantine pretreatment also completely protected against DFP- induced declines in dendrite length (Fig. 7a) and dendritic spine density (Fig. 7b) when measured 1 h after DFP exposure.
Figure 4.

Cerebral F2-IsoPs (a) and F4-NeuroPs (b) concentrations following saline (control) or DFP (1.25 mg/kg, s.c.) exposed rats with or without pretreatment with memantine (MEM, 18 mg/kg, s.c.), N-tert-butyl-α-phenylnitrone (PBN, 200 mg/kg, i.p.) or Vitamin E (Vit E, 100 mg/kg, i.p./day for 3 days). Brains from rats exposed to DFP were collected 1 hr post injections. Values of F2-IsoPs and F4-NeuroPs represent mean±SEM (n=4-6). *One way ANOVA had p<0.001 with Bonferroni's multiple comparison tests significant (p<0.05) for DFP vs. control, Mem+DFP, PBN+DFP or Vit E+DFP treatment.
Figure 5.

Cerebral citrulline concentrations following DFP exposure with or without pretreatment with memantine (MEM, 18 mg/kg, s.c.), alpha-phenyl-N-tert-butylnitrone (PBN, 200 mg/kg, i.p.) or Vitamin E (Vit E, 100 mg/kg, i.p./day for 3 days). Brains from rats exposed to DFP were collected 1 hr post injections. Values of citrulline represent mean±SEM (n=4-6). *One way ANOVA had p<0.001 with Bonferroni's multiple comparison tests significant (p<0.01) for DFP vs. control, Mem+DFP, PBN+DFP or Vit E+DFP treatment.
Figure 6.

Cerebral high-energy phosphates concentartion, ATP (a) and PCr (b) following DFP exposure with or without pretreatment with memantine (Mem, 18 mg/kg, s.c.), alpha-phenyl-N-tert-butylnitrone (PBN, 200 mg/kg, i.p.) or Vitamine E (Vit E, 100 mg/kg, i.p./day for 3 days). Brains from rats exposed to DFP were collected 1 hr post injections. Values of ATp and PCr represent mean±SEM (n=4-6). *One way ANOVA had p<0.001 with Bonferroni's multiple comparison tests significant (p<0.01) for DFP vs. control, Mem+DFP, PBN+DFP or Vit E+DFP treatment.
Figire 7.

Dendritic length (a) and spine density (b) of pyramidal neurons from CA1 hippocampal area of rats following DFP exposure with or without pretreatment with memantine (Mem, 18 mg/kg, s.c.), alpha-phenyl-N-tert-butylnitrone (PBN, 200 mg/kg, i.p.) or Vitamine E (Vit E, 100 mg/kg, i.p./day for 3 days). Brains from rats exposed to DFP were collected 1 hr post injections. Values of dendritic length and spine density represent mean±SEM (n ≥ 12 neurons from 4-6 animals). *One way ANOVA had p<0.001 with Bonferroni's multiple comparison tests significant (p<0.01) for DFP vs. control, Mem+DFP, PBN+DFP or Vit E+DFP treatment.
Discussion
Previous reports have highlighted the involvement of non-cholinergic mechanisms involved in OP induced toxicity (McDonough and Shih, 1997; De Groot et al., 2001). Several lines of evidence have suggested that excessive cholinergic stimulation following anticholinestrase exposure is associated with activation of glutamatergic neurons, NMDA receptors, Ca2+ fluxes into the post-synaptic cells and generation of ROS/RNS, ensuing in neurodegeneration. The present study explores the mechanisms associated with OP-induced neurotoxicity by probing their effects on oxidative/nitrosative stress and associated dendritic degeneration. Findings revealed that DFP induced reversible oxidative damage to cerebral neuronal membranes, alterations in NOS/NO and impairment of mitochondrial function as evidenced by significant depletion of ATP and PCr. Furthermore, data demonstrated that DFP-induced increases in biomarkers of global free radical damage (F2-IsoPs) and the selective peroxidation biomarkers of neuronal membranes (F4-NeuroPs) were accompanied by dendritic degeneration of pyramidal neurons in the CA1 hippocampal area. Importantly, results demonstrated that both neuronal oxidative damage and dendritic degeneration of CA1 pyramidal neurons induced by DFP were completely suppressed by the antioxidants (vitamin E or PBN) or the NMDA receptor antagonist (memantine).
The present results corroborate the findings strongly suggesting that the non-cholinergic system(s) is recruited at an early stage of the OP poisoning (Lallement et al., 1991a, b; Wade et al., 1987). Excessive amounts of glutamate are associated with intense transient influx of Ca2+, leading to mitochondrial structural and/or functional impairments characterized by activation of the permeability transition pores in the inner mitochondrial membrane, cytochrome c release, depletion of ATP and simultaneous formation of ROS (Heinemann et al., 2002; Cadenas and Davies, 2000; Patel, 2002; Nicholls and Ward, 2000; Nicholls et al., 2003). In addition, increase in cytoplasmic Ca2+ triggers intracellular cascades through stimulation of enzymes, including proteases, phospholipase A2, and NOS, which also lead to increased levels of ROS and oxidative stress (Lafon-Cazal et al., 1993; Farooqui et al., 2001). The present data demonstrated that DFP - induced reversible oxidative damage to cerebral neuronal membranes accompanied with depletion of HEP. Oxidative injury was quantified by measuring F2-IsoPs and lipid peroxidation of AA by free radicals. In a recent multi-investigator study, i.e., Biomarkers of Oxidative Stress Study (BOSS), sponsored by the National Institutes of Health, it was suggested that the quantification of F2-IsoPs represents the most accurate method to assess oxidative stress status in vivo (Kadiiska et al., 2005). Furthermore, quantification of F4-NeuroPs (oxidative damage to DHA – highly concentrated in neuronal membranes) provided unique insight into oxidative damage occurring in neurons. Both biomarkers of oxidative damage showed transient increase that reached maximum at the time of the most intensive seizure activity (i.e., 1 h after DFP) and returned toward basal levels by 6 h (Fig. 1). Activation of nNOS and generation of NO, as evidenced by elevation of citrulline at 1 h post-DFP exposure (Fig. 5), may be stimulated by elevation of intracellular Ca2+. Increased NO in the presence of superoxide anion radical (O2-) generates the peroxynitrite radical (OONO-), a powerful oxidant exhibiting a wide array of tissue damaging effects ranging from lipid peroxidation, inactivation of enzymes and ion channels via protein oxidation and nitration to inhibition of mitochondrial respiration (Montine et al., 2002; Milatovic et al., 2002; Virag et al., 2003). NO is involved in glutamate receptor-mediated neurotoxicity by decreasing intracellular ATP levels. There are two possible mechanisms responsible for energy depletion caused by NO in neuronal cells. One is the prolonged activation of poly-(ADP ribose) polymerase (Zhang et al., 1994), leading to depletion of ATP. The other mechanism is the inhibition of the mitochondrial complexes, leading to diminished ATP production. NO impairs mitochondrial/cellular respiration and other functions by inhibiting the activities of several key enzymes, particularly cytochrome c oxidase, and thereby causing ATP depletion (Yang and Dettbarn, 1998; Milatovic et al., 2001; Gupta et al., 2001a, b; Dettbarn et al., 2001). NO was also reported to inhibit complexes II and III (Bolanos et al., 1994), as well as complex IV (Lizasoain et al., 1996) in neuron-derived mitochondria and neuronal energy production in cultured hippocampal neurons (Brorson et al., 1999), leading to rapid ATP depletion. Results of the present study confirmed that an increase in the levels of cerebral citrulline (Fig. 5) was accompanied by depletion of ATP and PCr (Fig. 6) in rats 1 h after DFP administration.
Excitotoxic levels of glutamate with influx of Ca2+ leading to mitochondrial dysfunction are also involved in anticholinesterase-induced synaptic architectural changes (Munirathinam and Bahr, 2004). Although seizures can induce neuronal death, they may also have “nonlethal” pathophysiological effects on neuronal structure and function. Dendritic spines represent the structural sites of contact for the majority of excitatory, glutamatergic synaptic inputs onto neurons and are strongly implicated in mechanisms of synaptic plasticity and learning (Rao and Craig, 1997; O'Brien et al., 1998). NMDA and other glutamate receptor subtypes are clustered in dendritic spines (Yuste and Denk, 1995; Rao and Craig, 1997; O'Brien et al., 1998) suggesting that receptor localization at synapses might be critical to excitotoxicity. Findings of the present study with the Sholl method of concentric circles confirmed an early neuronal damage and showed that anticholinesterase-induced brain hyperactivity targeted the dendritic system with profound dendrite regression of hippocampal neurons. The data also demonstrated that DFP-induced oxidative damage to neuronal membranes is associated with degeneration of the pyramidal dendritic system in the CA1 hippocampal area.
An additional goal of this study was to determine whether suppression of lipid peroxidation prevents neurodegeneration of pyramidal neurons in the CA1 hippocampal area in the model of DFP-induced neurotoxicity. Therefore, efficacy of the antioxidants (vitamin E and PBN) was evaluated. Antioxidants play an important role in preventing many human diseases, including but not limited to cancer, atherosclerosis, stroke, rheumatoid arthritis and neurodegeneration (Fang et al., 2002). Vitamin E has been recognized as one of the most potent and important antioxidants. It acts as a chain- breaking antioxidant and radical scavenger and thus protects cells from peroxidation of PUFA in phospholipids and consequent membrane degeneration (Topinka et al., 1989; VanAcker et al., 1993). Decreased level of vitamin E in response to hyperoxia or treatment with a convulsant (Mori et al., 2004; Onodera et al., 2003; Rauca et al., 2004) suggested that it is consumed to prevent oxidative damage. In addition, vitamin E maintains oxidative phosphorylation in mitochondria, and accelerates restitution of high-energy metabolites (Punz et al., 1998; Kotegawa et al., 1993; Milatovic et al., 2005b).
The synthetic spin trapping agent, N-tert-butyl-α-phenylnitrone (PBN) is also an efficient scavenger of free radicals. PBN is widely used to trap ROS in a variety of physical, chemical and biological conditions. PBN concentrates in the mitochondria, where it reacts with ROS and forms stable adducts, and thereby maintains normal levels of energy metabolites. The protective effects of PBN have been described in experimental models of brain ischemia/reperfusion (Phillis and Clough-Helfman, 1990; Carney and Floyd,1991; Gido et al., 1997; Fetcher et al., 1997), excitotoxicity (Cheng and Sun,1994; Lancelot et al., 1997; Milatovic et al., 2002; Zaja-Milatovic et al., 2008), inhibition of NOS induction (Krishna et al., 1996; Miyajima and Kotake, 1997) and in different models of seizures (He et al., 1997; Thomas et al., 1997). Thus, these agents have been proven to rescue neurons in multiple experimental injury models. Results of the present study confirmed protective effects of these antioxidants and showed that both agents fully suppressed DFP-induced increases in cerebral and neuronal markers of oxidative damage, F2-IsoPs and F4-neuroPs, respectively (Fig. 4). In addition to attenuating DFP-induced increased production of citrulline and depletion of ATP and PCr (Fig. 5 and 6), both antioxidants fully protected hippocampal CA1 pyramidal neurons from dendritic degeneration (Fig. 7). Vitamin E pretreatment did not prevent DFP- seizure severity, indicating that its protective effect is most likely mediated by scavenging ROS, thus preventing lipid peroxidation and consequent neuronal damage. However, PBN pretreatment 1 h prior to DFP exposure substantially decreased the intensity of induced seizures. Corroborating with our previous studies, this effect appears to reflect the protective interaction of PBN with AChE, sufficient to protect a critical fraction of AChE against phosphorylation by DFP (Zivin et al., 1999; Milatovic et al., 2000a, b). Despite the protective interaction of PBN with AChE and NOS, the suppression of lipid peroxidation and the parallel reduction in neuronal damage following the increase in vitamin E and PBN, strongly supports oxidative stress mechanisms as causal mediators of DFP-induced seizures and neurodegeneration.
Since excitotoxicity induced neuronal damage in the model of anticholinesterase- seizures is associated with excessive release of glutamate, we tested if the NMDA receptor antagonist, memantine, could afford protection against deleterious effects of DFP. Memantine is an uncompetitive NMDA receptor antagonist, clinically used for the treatment of Alzheimer's disease, Parkinson's disease and spasticity (Ozsuer et al., 2005; Lipton, 2005). Memantine exerts various pharmacological effects by multiple mechanisms: (1) blockage of nicotinic acetylcholine receptor-ion channel complex (Masuo et al., 1986), (2) prevention of neural hyperexcitability (McLean et al., 1992), (3) reduced high-frequency repetitive activation of peripheral nerves (Wesemann et al., 1983) and (4) protection of AChE activity from inhibition by OP and CM insecticides, and OP nerve agents and prototype compound DFP (Gupta and Kadel, 1990; Gupta and Dettbarn, 1992; McLean et al., 1992; Gupta and Dekundy, 2005). Since memantine is able to prevent the pathogenic Ca2+ influx caused by continuous activation by low-level glutamate, it is expected that memantine would also suppress formation ROS/RNS, depletion of high-energy phosphates, and mitochondrial/neuronal damage. Indeed, pretreatment with memantine (18 mg/kg, s.c.) suppressed DFP-induced lipid peroxidation (Fig. 4), increased citrulline production (Fig. 5) and reduced HEP levels (Fig. 6). Memantine, when administered alone, did not induce any alteration in neuronal morphometry; however, when administered as a pretreatment, it provided protection against DFP-induced morphometric changes in hippocampal neurons. Memantine completely suppressed both the reduction in dendrite length and spine density of pyramidal neurons in the CA1 hippocampal area of DFP exposed rats (Figs. 7). These effects are consistent with our recent studies on memantine's efficacy in reversing carbofuran's effects on lipid peroxidation, and alteration in citrulline and HEP levels in muscles and brain (Milatovic et al., 2005a; Gupta et al., 2007). Taken together, memantine can reduce free radical generation and lipid peroxidation, prevent HEPs depletion and attenuate the morphological injury, thus providing further support for the role of ROS and RNS in anticholinesterase-induced neurotoxicity.
In conclusion, data of the present investigation suggest that oxidative and nitrosative stress and alterations in energy metabolism are early responses to DFP exposure. These data closely associated DFP-induced lipid peroxidation with dendritic degeneration of pyramidal neurons in the CA1 hippocampal area. This study also investigated different pathways to attenuate biomarkers of oxidative damage associated with anticholinesterase exposure and the extent to which such attenuation is accompanied by rescue from neurodegeneration. Specifically, vitamin E, PBN and memantine efficiently suppressed oxidative injury. Future studies should investigate not only the prophylactic, but also therapeutic effects of these neuroprotectants. Successful identification of safe and effective neuroprotectants that suppress non-cholinergic activities associated with anticholinesterase exposure will provide new pharmacological modalities to protect and treat both the acute and delayed effects of anticholinesterase agent.
Acknowledgments
This study was partly supported by grants from National Institute of Health: NS057223 (DM) and NIEHS07331 (MA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson JJ, Kuo S, Chase TN. Endogenous excitatory amino acids tonically stimulate striatal acetylcholine release through NMDA but not AMPA receptors. Neurosci Lett. 1994;176:264–268. doi: 10.1016/0304-3940(94)90097-3. [DOI] [PubMed] [Google Scholar]
- Aschner M. Interaction between pesticides and glia: an unexplored experimental field. NeuroToxicology. 2000;21:175–180. [PubMed] [Google Scholar]
- Bagetta G, Rodino P, Paoletti AM, Arabia A, Massoud R, Nistico G. Systemic administration of lithium chloride and tacrine but not kainic acid augments citrulline content of rat brain. Eur J Pharmacol. 1995;294:314–324. doi: 10.1016/0014-2999(95)00689-3. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Abai B, Gall C, Venerklish PW, Hoffman KB, Lynch G. Induction of b-amyloid-containing polypeptides in hippocampus: evidence for a concomitant loss of synaptic proteins and interactions with an excitotoxin. Exp Neurol. 1994;129:81–94. doi: 10.1006/exnr.1994.1149. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Bendiske J, Brown QB, Munirathinam S, Caba E, Rudin M, Urwyler S, Sauter A, Rogers G. Survival signaling and selective neuroprotection through glutamatergic transmission. Exp Neurol. 2002;174:37–47. doi: 10.1006/exnr.2001.7852. [DOI] [PubMed] [Google Scholar]
- Behan PD. Chronic fatigue syndrome as a delayed reaction to low dose organophosphate exposure. J Nutrition Environ Med. 1996;6:341–350. [Google Scholar]
- Bolanos JP, Peuchen S, Heales SJ, Land JM, Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem. 1994;63:910–916. doi: 10.1046/j.1471-4159.1994.63030910.x. [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, MacDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exper Neur. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Brorson JR, Scumacker PT, Zhang H. Nitric oxide acutely inhibits neuronal energy production. The Committees on Neurobiology and Cell Physiology. J Neurosci. 1999;19:147–158. doi: 10.1523/JNEUROSCI.19-01-00147.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Carney JM, Floyd RA. Protection against oxidative damage to CNS by alpha-phenyl-tert-butylnitrone (PBN) and other spin-trapping agents: a novel series of nonlipid free radical scavengers. J Mol Neurosci. 1991;3:47–57. doi: 10.1007/BF02896848. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Sun AY. Oxidative mechanisms involved in kainate-induced cytotoxicity in cortical neurons. Neurochem Res. 1994;19:1557–1564. doi: 10.1007/BF00969006. [DOI] [PubMed] [Google Scholar]
- De Bleecker JL. Intermediate syndrome in organophosphate poisoning. In: Gupta RC, editor. Toxicology of Organophosphate and Carbamate Compounds. Academic Press/Elsevier; Amsterdam: 2006. pp. 371–380. [Google Scholar]
- De Groot DM, Bierman EP, Bruijnzeel PL, Carpentier P, Kulig BM, Lallement G, et al. Beneficial effects of TCP on soman intoxication in guinea pigs: seizures, brain damage and learning behavior. J Appl Toxicol. 2001;21:S57–S65. doi: 10.1002/jat.812. [DOI] [PubMed] [Google Scholar]
- Dettbarn WD, Milatovic D, Zivin M, Gupta RC. Oxidative stress, acetylcholine and excitoxicity. In: Marwah J, Kanthasamy A, editors. International Conference on Antioxidants; Scottsdale, AZ: Prominant Press; 2001. pp. 183–211. [Google Scholar]
- Fang YZ, Yang S, Guoyao W. Free radicals, antioxidant, and nutrition. Nutrition. 2002;18:872–879. doi: 10.1016/s0899-9007(02)00916-4. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Yi Ong W, Lu XR, Halliwell B, Horrocks LA. Neurochemical consequences of kainate-induced toxicity in brain: involvement of arachidonic acid release and prevention of toxicity by phospholipase A(2) inhibitors. Brain Res Rev. 2001;38:61–78. doi: 10.1016/s0169-328x(01)00214-5. [DOI] [PubMed] [Google Scholar]
- Fetcher LD, Liu Y, Pearce TA. Cochlear protection from carbon monoxide exposure by free radical blockers in the guinea pig. Toxicol Appl Pharmacol. 1997;142:47–55. doi: 10.1006/taap.1996.8027. [DOI] [PubMed] [Google Scholar]
- Garcia SJ, Seidler FJ, Crumpton TL, Slotkin TA. Does the developmental neurotoxicity of chlorpyrifos involve glial targets? Macromolecule synthesis, adenyl cyclase signaling, nuclear transcription factors and formation of reactive oxygen in C6 glioma cells. Neurotoxicology. 2001;22:129–154. doi: 10.1016/s0006-8993(00)03189-9. [DOI] [PubMed] [Google Scholar]
- Gido G, Kristian T, Siesjo BK. Extracellular potassium in a neocortical cone area after transient focal ischemia. Stroke. 1997;28:206–210. doi: 10.1161/01.str.28.1.206. [DOI] [PubMed] [Google Scholar]
- Goldfrank L, Flomenbaum N, Lewin N, Weisman R, Howland MA, Kaul B. Anticholinergic poisoning. J Toxicol Clin Toxicol. 1982;19:17–25. doi: 10.3109/15563658208990362. [DOI] [PubMed] [Google Scholar]
- Gunnell D, Eddleston M. Suicide by intentional ingestion of pesticides: a continuing tragedy in developing countries. Int J Epidemiol. 2003;32:902–909. doi: 10.1093/ije/dyg307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RC, Kadel WL. Methyl parathion acute toxicity: prophylaxis and therapy with memantine and atropine. Arch Intl Pharmacod et de Ther. 1990;305:208–221. [PubMed] [Google Scholar]
- Gupta RC, Dettbarn WD. Potential of memantine, d-tubocurarine and atropine in preventing acute toxic myopathy induced by organophosphate nerve agents: soman, sarin, tabun and VX. Neurotoxicol. 1992;13:500–514. [PubMed] [Google Scholar]
- Gupta RC, Milatovic D, Dettbarn WD. Depletion of energy metabolites following acetylcholinesterase inhibitor-induced status epilepticus: protection by antioxidants. Neurotoxicol. 2001a;22:271–282. doi: 10.1016/s0161-813x(01)00013-4. [DOI] [PubMed] [Google Scholar]
- Gupta RC, Milatovic D, Dettbarn WD. Nitric oxide modulates high-energy phosphates in brain regions of rats intoxicated with diisopropylphosphorofluoridate or carbofuran: prevention by N-tert- butyl-α- phenylnitrone or vitamin E. Arch Toxicol. 2001b;75:346–356. doi: 10.1007/s002040100249. [DOI] [PubMed] [Google Scholar]
- Gupta RC, Dekundy A. Memantine does not influence AChE inhibition in rat brain by donepezil or rivastigmine but does with DFP and Metrifonate in in vivo studies. Drug Develop Res. 2005;64:71–81. [Google Scholar]
- Gupta RC. Classification and uses of organophosphates and carbamates. In: Gupta RC, editor. Toxicology of Organophosphate and Carbamate Compounds. Academic Press/Elsevier; Amsterdam: 2006. pp. 5–24. [Google Scholar]
- Gupta RC, Milatovic S, Dettbarn WD, Aschner M, Milatovic D. Neuronal oxidative injury and dendritic damage induced by carbofuran: protection by memantine. Toxicol Appl Pharmacol. 2007;219:97–105. doi: 10.1016/j.taap.2006.10.028. [DOI] [PubMed] [Google Scholar]
- He QP, Smith ML, Li PA, Siesjo BK. Necrosis of the substantia nigra, pars reticulata, influorothyl-induced status epilepticus is ameliorated by the spin trap alpha-phenyl-N-tert-butylnitrone. Free Rad Biol Med. 1997;22:917–922. doi: 10.1016/s0891-5849(96)00478-9. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Buchheim K, Gabriel S. Cell death and metabolic activity during epileptiform discharges and status epilepticus in the hippocampus. Prog Brain Res. 2002;135:197–210. doi: 10.1016/S0079-6123(02)35019-2. [DOI] [PubMed] [Google Scholar]
- Jamal GA. Neurological syndromes of organophosphorous compounds. Adv Drug React Toxic Rev. 1997;16:133–170. [PubMed] [Google Scholar]
- Jorgensen MB, Finsen BR, Jensen MB, Castellano B, Diemer NH, Zimmer J. Microglial and astroglial reactions to ischemic and kainic acid-induced lesions of the adult rat hippocampus. Exp Neurol. 1993;120:70–88. doi: 10.1006/exnr.1993.1041. [DOI] [PubMed] [Google Scholar]
- Jourquin J, Tremblay E, Decanis N, Charton G, Hanessian S, Chollet AM, Le Diguardher T, Khrestchatisky M, Rivera S. Neuronal activity-dependent increase of net matrix metalloproteinase activity is associated with MMP-9 neurotoxicity after kainate. Eur J Neurosci. 2003;18:1507–1517. doi: 10.1046/j.1460-9568.2003.02876.x. [DOI] [PubMed] [Google Scholar]
- Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, Nyska A, Wachsman JT, Ames BN, Basu S, Brot N, Fitzgerald GA, Floyd RA, George M, Heinecke JW, Hatch GE, Hensley K, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, II, Rokach J, Shigenaga MK, Sohal RS, Sun J, Tice RR, Van Thiel DH, Wellner D, Walter PB, Tomer KB, Mason RP, Barrett JC. Biomarkers of Oxidative Stress Study II: Are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radic Biol Med. 2005;38:698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Kotegawa M, Sugiyama M, Shoji T, Haramaki N, Orgura R. Effect of α-tocopherol on high energy phosphate metabolite levels in rat heart by 31 P-NMR using a Langendorff perfusion technique. J Mol Cell Cardiol. 1993;25:1067–1074. doi: 10.1006/jmcc.1993.1119. [DOI] [PubMed] [Google Scholar]
- Krishna MC, Russo A, Mitchell JB. Do nitroxide antioxidants act as scavengers of O2 or as SOD mimics? J Biol Chem. 1996;271:26026–26031. doi: 10.1074/jbc.271.42.26026. [DOI] [PubMed] [Google Scholar]
- Kwong TC. Organophosphate pesticides: biochemistry and clinical toxicology. Ther Drug Monit. 2002;24:144–149. doi: 10.1097/00007691-200202000-00022. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Lallement G, Carpenrier P, Pernot-Marino I, Baubichon D, Blanchet G. Involvement of the different rat hippocampal glutamatergic receptors in development of seizures induced by soman: an autoradiographic study. Neurotoxicol. 1991a;12:655–664. [PubMed] [Google Scholar]
- Lallement G, Carpentier P, Collet A, Pernot-Marino I, Baubichon D, Blanchet G. Effects of soman-induced seizures on different extracellular amino acid levels and on glutamate uptake in rat hippocampus. Brain Res. 1991b;563:234–240. doi: 10.1016/0006-8993(91)91539-d. [DOI] [PubMed] [Google Scholar]
- Lallement G, Denoyer M, Collet A, Pernot-Marino I, Baubichon D, Monmaur P, et al. Changes in hippocampal acetylcholine and glutamate extracellular levels during soman-induced seizures: influence of septal cholinoceptive cells. Neurosci Lett. 1992;139:104–107. doi: 10.1016/0304-3940(92)90868-8. [DOI] [PubMed] [Google Scholar]
- Lancelot E, Revaud ML, Boulee RG. Alpha-N-tert-butylnitone attenuates excitotoxicity in rat striatum by preventing hydroxyl radical accumulation. Free Rad Biol Med. 1997;23:1031–1034. doi: 10.1016/s0891-5849(97)00128-7. [DOI] [PubMed] [Google Scholar]
- Lehtimaki KA, Peltola J, Koskikallio E, Keranen T, Honkaniemi J. Expression of cytokines and cytokine receptors in the rat brain after kainic acid-induced seizures. Brain Res Mol Brain Res. 2003;110:253–260. doi: 10.1016/s0169-328x(02)00654-x. [DOI] [PubMed] [Google Scholar]
- Leuner B, Falduto J, Shors T. Associative memory formation increases the observation of dendritic spines in the hippocampus. J Neurosci. 2003;23:659–665. doi: 10.1523/JNEUROSCI.23-02-00659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. The molecular basis of memantine action in Alzheimer's disease and other neurologic disorders: low-affinity uncompetitive antagonism. Curr Alzheimer Res. 2005;2:155–165. doi: 10.2174/1567205053585846. [DOI] [PubMed] [Google Scholar]
- Lizasoain I, Moro MA, Knowles RG, Darley-Usmar V, Moncada S. Nitric oxide and peroxynitrite exert distinct effects on mitochondrial respiration which are differentially blocked by glutathione or glucose. Biochem J. 1996;314:877–880. doi: 10.1042/bj3140877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti M. The pathogenesis of organophosphate neuropathy. Crit Rev Toxicol. 1992;21:465–487. doi: 10.3109/10408449209089884. [DOI] [PubMed] [Google Scholar]
- Lotti M, Moretto A. Organophosphate-induced delayed polyneuropathy. Toxicol Rev. 2005;24:37–49. doi: 10.2165/00139709-200524010-00003. [DOI] [PubMed] [Google Scholar]
- Masuo K, Enomoto KI, Maine T. Effects of memantine on the frog neuromuscular junction. Eur J Pharmacol. 1986;130:187–195. doi: 10.1016/0014-2999(86)90267-0. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Shih TM. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci Biobehav Rev. 1997;21:559–579. doi: 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- McLean MJ, Gupta RC, Dettbarn WD, Wamil AW. Prophylactic and therapeutic efficacy of memantine against seizures produced by soman in the rat. Toxicol Appl Pharmacol. 1992;112:95–103. doi: 10.1016/0041-008x(92)90284-y. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Radic Z, Zivin M, Dettbarn WD. Atypical effect of some spin trapping agents: reversible inhibition of acetylcholinesterase. Free Rad Biol Med. 2000a;28:597–603. doi: 10.1016/s0891-5849(99)00270-1. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Zivin M, Dettbarn WD. The spin trapping agent phenyl-N-tert-butyl-nitrone (PBN) prevents excitotoxicity in skeletal muscle. Neurosci Lett. 2000b;278:25–28. doi: 10.1016/s0304-3940(99)00904-0. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Zivin M, Gupta RC, Dettbarn WD. Alterations in cytochrome-c-oxidase and energy metabolites in response to kainic acid-induced status epilepticus. Brain Res. 2001;912:67–78. doi: 10.1016/s0006-8993(01)02657-9. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Gupta RC, Dettbarn WD. Involvement of nitric oxide in kainic acid-induced excitotoxicity in rat brain. Brain Res. 2002;957:330–337. doi: 10.1016/s0006-8993(02)03669-7. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Zaja-Milatovic S, Montine KS, Horner PJ, Montine TJ. Pharmacologic suppression of neuronal oxidative damage and dendritic degeneration following direct activation of glial innate immunity in mouse cerebrum. J Neurochem. 2003;87:1518–1526. doi: 10.1046/j.1471-4159.2003.02120.x. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Gupta RC, Dekundy A, Montine TJ, Dettbarn WD. Carbofuran-induced oxidative stress in slow and fast skeletal muscles: prevention by memantine. Toxicology. 2005a;208:13–24. doi: 10.1016/j.tox.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Milatovic D, VanRollins M, Li K, Montine KS, Montine TJ. Suppression of murine cerebral F2-isoprostanes and F4-neuroprostanes from excitotoxicity and innate immune response in vivo by alpha- or gamma-tocopherol. J Chromatogr B Analyt Technol Biomed Life Sci. 2005b;827:88–93. doi: 10.1016/j.jchromb.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Gupta RC, Aschner M. Anticholinesterase toxicity and oxidative stress. The Scientific World J. 2006;6:295–310. doi: 10.1100/tsw.2006.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milatovic D, Aschner M. Measurement of isoprostanes as markers of oxidative stress in neuronal tissue. Current Protocols in Toxicology. 2009:1–12. doi: 10.1002/0471140856.tx1214s39. unit 12.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima T, Kotake Y. Spin trap phenyl N-tert-butylnitrone (PBN) for the inhibition of nitric oxide synthase induction in mice. Free Radic Biol Med. 1997;22:463–470. doi: 10.1016/s0891-5849(96)00391-7. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Milatovic D, Gupta RC, Morrow JD, Breyer R. Neuronal oxidative damage from activated innate immunity in EP2 receptor-dependent. J Neurochem. 2002;83:463–470. doi: 10.1046/j.1471-4159.2002.01157.x. [DOI] [PubMed] [Google Scholar]
- Mori A, Yokoi I, Noda Y, Willmore LJ. Natural antioxidants may prevent posttraumatic epilepsy: a proposal based on experimental animal studies. Acta Med Okayama. 2004;58:111–118. doi: 10.18926/AMO/32111. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., II A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87(23):9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munirathinam S, Bahr BA. Repeated contact with subtoxic soman leads to synaptic vulnerability in hippocampus. J Neurosci Res. 2004;77:739–746. doi: 10.1002/jnr.20209. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Vesce S, Kirk L, Chalmers S. Interactions between mitochondrial bioenergetics and cytoplasmic calcium in cultured cerebellar granule cells. Cell Calcium. 2003;34:407–424. doi: 10.1016/s0143-4160(03)00144-1. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Onodera K, Omoi NO, Fukui K, Hayasaka T, Shinkai T, Suzuki S, Abe K, Urano S. Oxidative damage of rat cerebral cortex and hippocampus, and changes in antioxidative defense systems caused by hyperoxia. Free Radic Res. 2003;37:367–372. doi: 10.1080/1071576031000090019. [DOI] [PubMed] [Google Scholar]
- Ozsuer H, Gorgulu A, Kiris T. The effect of memantine on lipid peroxidation following closed-head trauma in rats. Neurosurgery Rev. 2005;28:143–147. doi: 10.1007/s10143-004-0374-1. [DOI] [PubMed] [Google Scholar]
- Patel MN. Oxidative stress, mitochondrial dysfunction, and epilepsy. Free Radic Res. 2002;36:1139–1146. doi: 10.1080/1071576021000016391. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Clough-Helfman C. Protection from cerebral ischemic injury in gerbils with the spin trap agent N-tert-butyl-alpha-phenylnitrone (PBN) Neurosci Lett. 1990;116:315–319. doi: 10.1016/0304-3940(90)90093-o. [DOI] [PubMed] [Google Scholar]
- Punz A, Nanobashvili N, Feugl A, et al. Effect of α-tocopherol pretreatment on high energy metabolites in rabbit skeletal muscle after ischemia-reperfusion. Clin Nutr. 1998;17:85–87. doi: 10.1016/s0261-5614(98)80311-7. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Rauca C, Wiswedel I, Zerbe R, Keilhoff G, Krug M. The role of superoxide dismutase and alpha-tocopherol in the development of seizures and kindling induced by pentylenetetrazol—influence of the radical scavenger alpha-phenyl-N-tert-butylnitrone. Brain Res. 2004;1009:203–212. doi: 10.1016/j.brainres.2004.01.082. [DOI] [PubMed] [Google Scholar]
- Reich EE, Markesbery W, Roberts JL, II, Swift L, Morrow J, Montine T. Brain regional quantification of F-ring and D/E-ring isoprostanes and neuroprostanes in Alzheimer's disease. Am J Pathol. 2001;158:293–297. doi: 10.1016/S0002-9440(10)63968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich EE, Zackert WE, Brame CJ, Chen Y, Roberts LJ, II, Hachey DL, Montine TJ, Morrow JD. Formation of novel D-ring and E-ring isoprostane-like compounds (D4/E4-neuroprostanes) in vivo from docosahexaenoic acid. Biochemistry. 2000;39:2376–2383. doi: 10.1021/bi992000l. [DOI] [PubMed] [Google Scholar]
- Scholl D. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science. 1990;247:571–574. doi: 10.1126/science.2154034. [DOI] [PubMed] [Google Scholar]
- Siesjo BK. Historical overview. calcium, ischemia, and death of brain cells. Ann New York Acad Sci. 1988;522:638–661. doi: 10.1111/j.1749-6632.1988.tb33410.x. [DOI] [PubMed] [Google Scholar]
- Siman R, Noszek JC, Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosc. 1989;9:1579–1590. doi: 10.1523/JNEUROSCI.09-05-01579.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparenborg S, Brennecke LH, Jaax NK, Braitman DJ. Dizocilpine (MK-801) arrests Status epilepticus and prevents brain damage induced by soman. Neuropharmacology. 1992;31:357–368. doi: 10.1016/0028-3908(92)90068-z. [DOI] [PubMed] [Google Scholar]
- Thomas CE, Ohlweiler DF, Taylor VL, Schmidt CJ. Radical trapping and inhibition of iron-dependent CNS damage by cyclic nitrone spin traps. J Neurochem. 1997;68:1173–1182. doi: 10.1046/j.1471-4159.1997.68031173.x. [DOI] [PubMed] [Google Scholar]
- Topinka J, Bincova B, Sram RJ, Erin AN. The influence of a-tocopherol and pyritinol on oxidative DNA damage and lipid peroxidation in human lymphocytes. Mutat Res. 1989;225:131–136. doi: 10.1016/0165-7992(89)90130-9. [DOI] [PubMed] [Google Scholar]
- VanAcker SABE, Koymans LMH, Bast A. Molecular pharmacology of vitamin E: structural aspects of antioxidant activity. Free Radic Biol Med. 1993;15:311–328. doi: 10.1016/0891-5849(93)90078-9. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Moneta D, Richichi C, Aliprandi M, Burrows SJ, Ravizza T, Perego C, De Simoni MG. Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia. 2002;43:30–35. doi: 10.1046/j.1528-1157.43.s.5.14.x. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo E, Gergely P, Szabo C. Peroxynitrite-induced cytotoxicity: mechanism and opportunity for intervention. Toxicol Lett. 2003;140-141:113–124. doi: 10.1016/s0378-4274(02)00508-8. [DOI] [PubMed] [Google Scholar]
- Wade JV, Samson FE, Nelson SR, Pazdernik TL. Changes in extracellular amino acids during soman-and kainic acid-induced seizures. J Neurochem. 1987;49:645–650. doi: 10.1111/j.1471-4159.1987.tb02912.x. [DOI] [PubMed] [Google Scholar]
- Walker B, Nidiry J. Current concepts: Organophosphate toxicity. Inhal Toxicol. 2002;14:975–990. doi: 10.1080/08958370290084728. [DOI] [PubMed] [Google Scholar]
- Watson A, Bakshi K, Opresko D, Young R, Hauschild V, King J. Cholinesterase inhibitors as chemical warfare agents: Community preparedness guidelines. In: Gupta RC, editor. Toxicology of Organophosphate and Carbamate Compounds. Academic Press/Elsevier; Amsterdam: 2006. pp. 47–68. [Google Scholar]
- Watson A, Opresko D, Young R, Hauschild V, King J, Bakshi K. Organophosphate nerve agents. In: Gupta RC, editor. Handbook of Toxicology of Chemical Warfare Agents. Academic Press/Elsevier; Amsterdam: 2009. pp. 43–67. [Google Scholar]
- Wesemann W, Sontag KH, Maj J. Zur Pharmacodynamic und Pharmacokinetic des Memantine. Arzneimittel-Forschung. 1983;33:1122–1134. [PubMed] [Google Scholar]
- Weinbroum AA. Pathophysiological and clinical aspects of combat anticholinesterase poisoning. British Med Bull. 2005;72:119–133. doi: 10.1093/bmb/ldh038. [DOI] [PubMed] [Google Scholar]
- Yang ZP, Dettbarn WD. Lipid peroxidation and changes of cytochrome c oxidase and xanthine oxidase in organophosphorus anticholinesterase induced myopathy Xth International Symposium on Cholinergic Mechanisms. J Physiology (Paris) 1998;92:157–162. doi: 10.1016/s0928-4257(98)80002-8. [DOI] [PubMed] [Google Scholar]
- Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Gupta RC, Aschner M, Montine TJ, Milatovic D. Pharmacologic suppression of oxidative damage and dendritic degeneration following kainic acid-induced excitotoxicity in mouse cerebrum. Neurotoxicology. 2008;29(4):621–627. doi: 10.1016/j.neuro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly (ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- Zimmer LA, Ennis M, Shipley MT. Soman-induced seizures rapidly activates astrocytes and microglia in discrete brain regions. J Comp Neurol. 1997;378:482–492. doi: 10.1002/(sici)1096-9861(19970224)378:4<482::aid-cne4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Zivin M, Milatovic D, Dettbarn WD. Nitrone spin trapping compound N-tert-butyl-α-phenylnitrone prevents seizure induced by anticholinesterases. Brain Res. 1999;850:63–72. doi: 10.1016/s0006-8993(99)02101-0. [DOI] [PubMed] [Google Scholar]