

Abstract
Methamphetamine (MA) is currently the most widespread illegally used stimulant in the United States. Use of MA by smoking is the fastest growing mode of administration, which increases concerns about potential pulmonary and other medical complications. A murine exposure system was developed to study the pulmonary affects of inhaled MA. Mice were exposed to 25–100 mg vaporized MA and assessments were made 3 h following initiation of exposure to model acute lung injury. Inhalation of MA vapor resulted in dose-dependent increases in MA plasma levels that were in the range of those experienced by MA users. At the highest MA dose, histological changes were observed in the lung and small but significant increases in lung wet weight to body weight ratios (5.656 ± 0.176 mg/g for the controls vs. 6.706± 0.135 mg/g for the 100 mg MA-exposed mice) were found. In addition, there was 53% increase in total protein in bronchoalveolar lavage (BAL) fluid, greater than 20% increase in albumin levels in the BAL fluid, greater than 2.5-fold increase in lactate dehydrogenase levels in the BAL fluid, and reduced total BAL cell numbers (approximately 77% of controls). Levels of the early response cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-6 were dose-dependently increased in BAL fluid of MA-exposed mice. Exposure to 100 mg MA significantly increased free radical generation in the BAL cells to 107–146% of controls and to approximately 135% of the controls in lung tissue in situ. Together, these data show that acute inhalation exposure to relevant doses of volatilized MA is associated with elevated free radical formation and significant lung injury.
Methamphetamine hydrochloride (MA) is a substituted amphetamine with potent central nervous system stimulant effects, and is currently the most widespread illegally used stimulant in the United States (NIDA, 2006). Like cocaine, MA can be snorted, injected, ingested, or smoked. According to the 2007 National Drug Threat Assessment Report by the National Drug Intelligence Center, smoking is the fastest growing primary mode of administration. From 1993 to 2004, use of MA by smoking grew from 15% to nearly 60% (NDIC, 2006). Smoking MA may result in more rapid addiction to the drug than snorting or injection because smoking causes a nearly instantaneous, intense, and longer lasting high (NIDA, 2002). These factors may lead to increased rates of addiction that will further strain the resources of public health agencies.
Studies of other inhaled drugs including marijuana and cocaine have shown that these substances can cause a variety of pulmonary complications. Chronic marijuana use is associated with increased prevalence of symptoms of chronic bronchitis, including chronic cough, sputum production, wheeze, and increased outpatient visits for respiratory illness (Bloom et al., 1987; Tashkin et al., 1987; Taylor et al., 2000; Taylor et al., 2002). Marijuana use also affects airway pathology by causing lower-airway injury, inflammation, and impairment in the antimicrobial and tumoricidal function of alveolar macrophages (Fligiel et al., 1997; Roth et al., 1998). Alkaloidal cocaine (free-base or crack cocaine) smoking is associated with a number of acute pulmonary complications, including acute respiratory symptoms, (Tashkin et al., 1992), severe exacerbations of asthma (Haim et al., 1995), and an acute lung injury syndrome (Forrester et al., 1990).
Increasing evidence suggests that MA-induced neurotoxicity involves the production of reactive oxygen and reactive nitrogen species (ROS and RNS), resulting in induction of oxidative stress (Stephans & Yamamoto, 1994). Furthermore, there is evidence that free radicals may play a role in amphetamine-related lung injury. In an isolated rat lung model, Huang et al. demonstrated that the injury observed following amphetamine administration was associated with overproduction of free radicals (Huang et al., 2002). Although most of the reported side effects of smoking MA have been systemic, case reports of pulmonary complications including acute noncardiogenic pulmonary edema (Nestor et al., 1989a, 1989b) and fatal pulmonary hypertension (Schaiberger et al., 1993) have been reported. However, to date there are no published studies on the pulmonary effects of vaporized MA. Based on the published results with other drugs and the case studies reported for MA, we hypothesized that vaporized MA may cause lung injury accompanied with oxidant stress. The present study was designed to test this hypothesis.
MATERIALS AND METHODS
Animals
BALB/c mice used in these studies were bred and maintained in microisolator units in the University of Montana specific pathogen-free animal facility. The mice were given water ad libitum. Mice, 7–10 wk of age, were used for all animal experiments. All animal procedures were approved by the University of Montana Institutional Animal Care and Use Committee (IACUC).
Methamphetamine Exposure
Mice were exposed to vapor from 25–100 mg heated MA utilizing the exposure system described in Figure 1. The exposure chamber consisted of a 100-ml heating vessel above a propane heat source connected to the animal chamber. The animal chamber was designed and built by Dr. Lung-Chi Chen at New York University School of Medicine and holds up to 32 mice in individual compartments. Air was pumped into the heating vessel at a rate of 1.5 L/min using a small pump connected to the top of the heating vessel. The air moved from the heating vessel into the stainless-steel chamber and was distributed evenly throughout the chamber. Air flow through the chamber was kept continuous with the use of a constant vacuum of approximately 1.5 L/min. Temperature and humidity were both monitored inside the chamber using a remote temperature/humidity probe and remained unchanged throughout the exposures.
FIG. 1.

Heated methamphetamine exposure system. MA was heated over a propane heat source in the heating vessel. The resultant MA vapor was moved through the animal chamber by constant air flow of 1.5 L/min generated by an air pump and a vacuum source.
Mice were placed in the chamber and were allowed to acclimate for 5 min. The heat source was ignited and the indicated amount of MA (Sigma Chemical, St. Louis, MO) heated for 20 min. Following the 20-min exposure, the heat source was turned off and the mice remained in the chamber for an additional 5 min. Due to risk of low levels of MA exposures, control mice were subjected to the same conditions as the exposed mice (e.g., transfer to and from the exposure chamber), except for being placed inside the exposure chamber. All mice were sacrificed approximately 3 h following initiation of exposure. Mice receiving the 100 mg dose displayed self-injurious behavior. Therefore, the 3-h time point was chosen to allow for maximal time between the exposure and assessments before significant injury occurred.
Determination of MA Plasma Levels
Mice were euthanized by a lethal injection of Euthasol. Blood was collected by cardiac bleeds into 2.0-ml Vacutainer tubes containing sodium fluoride and potassium oxalate (Becton Dickenson, Franklin Lakes, NJ). A basic, liquid—liquid extraction procedure was used to extract MA and amphetamine from the mouse blood. Stock standards (10 ng/μl, 1 ng/μl, 0.1 ng/μl) of MA were used as positive controls. An internal standard (1 ng/μl) MA d-14 was also prepared and 7.5 μl was added to all samples to aid in the quantification of MA. One half milliliter of mouse blood was diluted to 1.0 ml by adding 0.5 ml deionized water. The standards were prepared for quantification in 0.5 ml of negative control blood, and were also diluted to 1.0 ml using 0.5 ml deionized water. Extraction was done by addition of 1.0 ml of 0.5 N NaOH, 4.0 ml n-butyl chloride, mixing for 2 min, and centrifugation for 3 min at 3400 rpm. The supernatant was transferred to new tubes and dried at 37°C for 10 min following the addition of 100 μl 1% methonolic hydrochloride. The dried samples were reconstituted with 100 μl 1% formic acid in water, centrifuged at 14,000 rpm for 1 min, and supernatants were transferred to vials for liquid chromatography—tandem mass spectrometric (LC/MS) analysis. Chromatographic separation was achieved on a 4.6 mm × 150 mm, 3.5 μm Eclipse Plus C18 column (Agilent Technologies, Palo Alto, CA) at a column temperature of 55°C. The mobile phase consisted of 1% formic acid in water and methanol and the flow rate was 1.00 ml/min with a 20-min stop time and a 5-min post time. Signals were produced using selected ion monitoring (SIM) parameters and an atmospheric pressure ionization—electrospray ionization mode with positive polarity. SIM parameters were set to scan for ions at 136, 147, 150, and 164. The spray chamber parameters were 300°C gas temperature, 13.0 L/min drying gas, a capillary voltage of 3.0 kV.
Collection of Bronchoalveolar Lavage Cells and Fluid
Lungs were removed with the heart and then lavaged with five 1.0-ml aliquots of cold phosphate-buffered saline (PBS). The first 1.0 ml was centrifuged at 400 × g for 5 min and the lavage fluid was saved for protein, cytokine, and lactate dehydrogenase (LDH) assessments. The remaining 4 aliquots were centrifuged at 400 × g for 5 min and the cells from all 5 aliquots were pooled. The cell pellet was resuspended in 1 ml PBS and a 40-μl sample was counted using a Z1 Coulter particle counter (Beckman Coulter, Fullerton, CA).
Determination of Protein Levels
Total bronchoalveolar lavage (BAL) fluid protein levels were analyzed for total protein in triplicate using the Bio-Rad Laboratories protein assay (Hercules, CA) according to the manufacturer’s instructions. Bovine serum albumin was used as the standard. Samples were measured undiluted. Colormetric analysis was performed with the Spectra Max 340 plate reader (GE Healthcare, United Kingdom) at 595 nm. Data are expressed as micrograms total protein per milliliter of BAL fluid.
Mouse albumin levels in BAL fluid were measured in triplicate using a mouse albumin enzyme-linked immunosorbent assay (ELISA) kit purchased from Bethyl Laboratories (Mont-gomery, TX) according to the manufacturer’s instructions. Samples were diluted 1:200. Colormetric analysis was performed with the Spectra Max 340 plate reader (GE Healthcare) at 450 nm. Data are expressed as micrograms albumin per milliliter of BAL fluid. The detection limit for this ELISA was 7 ng/ml.
Determination of Lung Weight to Body Weight (LW/BW) Ratios
Lung weight to body weight (LW/BW) ratios were determined by weighing whole mice and whole excised lung. Only nonlavaged lungs were utilized for the determination of LW/BW ratio. The LW/BW ratio is expressed in milligrams per gram.
Determination of LDH in BAL Fluid
LDH activity in the BAL fluid was determined by measuring the oxidation of lactate to pyruvate, which then reacts with tetrazolium salt to form formazan (Decker & Lohmann-Matthes, 1988), using an LDH-cytotoxicity assay kit by BioVision Research Products (Mountain View, CA) according to the manufacture’s instructions. Absorbance was measured by spectrophotometric analysis at 500 nm with the Spectra Max 340 plate reader (GE Healthcare). Data are expressed as optical density at 500 nm.
Histology
Lungs from each mouse were inflation fixed with 3% paraformaldehyde and immersed in the same fixative. Tissue was fixed overnight, washed with PBS, dehydrated, and embedded in paraffin. Tissue sections (7 μm) were stained with hematoxylin—eosin (H&E) for histology.
Determination of BAL Fluid Cytokine Levels
BAL fluid from the first 1.0-ml aliquot was assayed for cytokines with commercially available kits according to the manufacturer’s protocol. Interleukin (IL)-6 and interferon (IFN)-α measurements were determined by using Duo-set kits (R & D Systems). Samples were diluted 1:2. Colormetric analysis was performed with the Spectra Max 340 plate reader (GE Healthcare) at 450 nm. Data are expressed as picograms per milliliter of retrieved culture supernatant.
Measurement of ROS and RNS in BAL Cells
For analysis of intracellular production (referred to as ROS) and intracellular ONOO− production (referred to as RNS), dihydroethidium (DHE) and dihydrorhodamine 123 (DHR) levels were determined as previously described (Haugen et al., 1999). Briefly, 1.5 × 105 cells were incubated on ice with DHE (25 μM) or DHR (25 μM) for 20 min and analyzed using a FACSCalibur (Becton Dickinson). Data were processed using the Cell Quest software (Becton Dickinson). The membrane-permeable fluorescent dye DHE is oxidized in the presence of ROS and fluoresces red (absorption/emission 518/605 nm) upon DNA intercalation. The membrane-permeable nonfluorescent dye DHR is oxidized to the fluorescent product rhodamine, which has absorption/emission spectra of 505/534 nm. Increased cellular fluorescence, as measured by flow cytometry, indicates increased production of ROS or RNS as is reported as mean channel fluorescence.
In Situ Detection of ROS
In situ production of ROS can be evaluated microscopically in frozen sections using DHE (Miller et al., 1998; Paravicini et al., 2002; Grobe et al., 2006). Briefly, lungs were inflated with 0.5 ml of a 1:1 ratio of Tissue-Tek OCT compound (Sakura Finetek USA, Torrance, CA) and water. They were then frozen by placing immediately above liquid nitrogen until rigid and then submerging in liquid nitrogen for 10 min, and they were stored at −80°C. Tissues were cryosectioned at 7 μm, collected onto Superfrost plus slides (VWR Scientific, West Chester, PA), allowed to air-dry at room temperature, and stored at −80°C until used. Slides were placed into PBS for 30 min at room temperature and then stained with DHE (10 μM) in PS for 30 min in a moist chamber in the dark. The slides were rinsed extensively with PBS, coverslipped, and imaged with a Kodak DC120 digital camera. DHE fluorescence was quantified with a CompuCyte LSC laser scanning cytometer (CompuCyte, Cambridge, MA). The desired area of analysis was located visually using epifluorescence visual microscopy on the instrument. Scan areas were set and the scan initiated using the 20× objective and the argon laser operating at 10 mW. Phantom contours (Megyeri et al., 2005) were used to obtain fluorescence measurements of both airways and interstitial tissue. Long red fluorescence was collected with a 650–700 nm bandpass filter/PMT set. Four different measurements were taken and averaged for each animal. Maximum pixel median values and percent positive over background statistics were used. Data were acquired and analyzed with WinCyte acquisition software (CompuCyte).
To confirm specificity of DHE for , slides were also pretreated with polyethylene glycol superoxide dismutase (PEG-SOD) (50 U/slide) before being stained with DHE as described earlier. PEG-SOD was obtained from Sigma Chemical.
Statistical Analysis
The mean ± SEM was calculated for all samples and p values were calculated using an unpaired t-test or a one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison to a single control group. For calculation of correlation between MA dose and plasma levels, a Pearson correlation test was utilized. All data are expressed as mean × SEM. Less than 5% probability of type I error was accepted as statistically significant.
RESULTS
Mouse Plasma MA Levels
To determine the relative exposure of the mice to the volatized MA, we first established whether there was a correlation between the amount of MA that was heated and the levels of MA in plasma of the exposed mice. We exposed mice to 25 mg, 50 mg, 75 mg, or 100 mg heated MA. Plasma levels of MA for the exposed mice and the unexposed controls are shown in Figure 2. We observed a relative linear increase in plasma MA levels following increasing doses of heated MA. A significant ( p < .001) correlation between MA dose and plasma levels was determined using a Pearson correlation test (r = .7450).
FIG. 2.
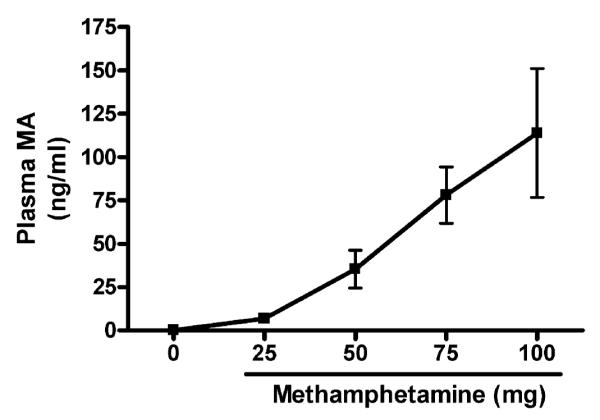
MA plasma levels with increasing doses of MA. Plasma MA levels were determined by LC/MS. Following exposure to the vapors from increasing amounts of heated MA, MA plasma levels were increased in the exposed mice. Values are means ± SEM from four mice in each group. Correlation between MA dose and plasma levels was calculated using a Pearson correlation test (r = .7450,p <.001).
Lung Injury Following Exposure to Vaporized MA
To explore the effects of vaporized MA in the lungs of mice following inhalation we assessed several parameters of lung injury and inflammation following exposure to increasing MA doses. Protein leak across the alveolar—capillary barrier can be determined by measurement of total protein concentration in the BAL fluid (Kenyon et al., 2002). We measured total protein levels in the BAL fluid of control and exposed mice (Figure 3A) and found a significant (53%) increase in total protein in BAL fluid of animals exposed to the 100-mg dose of MA. In addition, BAL fluid albumin levels were significantly increased in 100 mg MA-exposed animals (Figure 3B; 143.4 ± 8.738 μg/ml in controls vs. 173.0 ± 8.053 μg/ml in MA-exposed mice; p < .05). Finally, the lung wet weight/body weight ratio (LW/W) was assessed and was significantly higher in the MA-exposed animals compared to controls (5.656 ± 0.176 mg/g for the controls vs. 6.706 ± 0.135 mg/g for the MA-exposed mice; n = 4, p < .05).
FIG. 3.

Assessment of lung injury following acute exposure to vaporized MA. Mice were exposed to vapors from 25 to 100 mg of heated MA. Tissues were collected 3 h following exposure to assess parameters of lung injury. (A) Total protein in the BAL fluid was assessed (controls, n = 15; 25–100 mg MA, n = 5 each). (B) Albumin in the BAL fluid was assessed (n = 4 for all groups). (C) Cell numbers from whole lung lavage was determined (controls, n = 15; 25 mg MA, n = 8; 50 mg MA, n = 11; 100 mg MA, n = 9). (D) LDH in BAL fluid was measured (n = 8 for all groups). Asterisk indicates significant at p < .05 versus controls. Data are shown as means ± SEM.
BAL total cellularity was also determined and found to be mildly but significantly decreased in animals exposed to all doses of MA (Figure 3C). At the 100-mg dose, exposed mice had a total BAL cell number of 2.42 ± 0.151 × 105 cells vs. 3.13 ± 0.204 × 105 cells in the control mice ( p <.05). In both control and MA-exposed mice, the cells present in the BAL were predominantly macrophages and the BAL return was similar. To assess whether this decrease in cell number could be loss through cell death, we assessed lactate dehydrogenase (LDH) levels in BAL fluid. We observed a greater than 2.5-fold increase in BAL fluid LDH levels in mice exposed to the highest dose of MA (Figure 3D). Lower doses of MA also increased BAL fluid LDH; however, these increases were not significant.
MA-Induced Changes in Lung Histopathology
Histopathologic assessment of the lung was performed to assess whether the physiological indices reported earlier in this article correlated with any morphological changes. For this assessment, we used 100 mg dose of MA, since exposure to this level of MA resulted in plasma MA levels below the mean MA plasma level of 730 ng/ml reported for human MA users (Schwilke et al., 2006). Representative H&E sections from control (Figure 3, panels A—B) and MA-exposed (Figure 3, panels C—D) mice indicate that inhalation of vaporized MA appeared to have caused subtle but detectable lung injury (Figure 4). Compared to control mice (panel A), there were fewer small arterioles in the peripheral lung tissue of MA-exposed mice (panel C). In addition, MA exposure appears to result in damage to airway epithelial cells as evidenced by exposed nuclei and exposed basement membrane (panel D). This is in contrast to the epithelial cell layer in control mice, which was uniform and intact (panel B). Taken together, these results indicate that exposure to relevant levels of MA resulted in detectable lung injury.
FIG. 4.
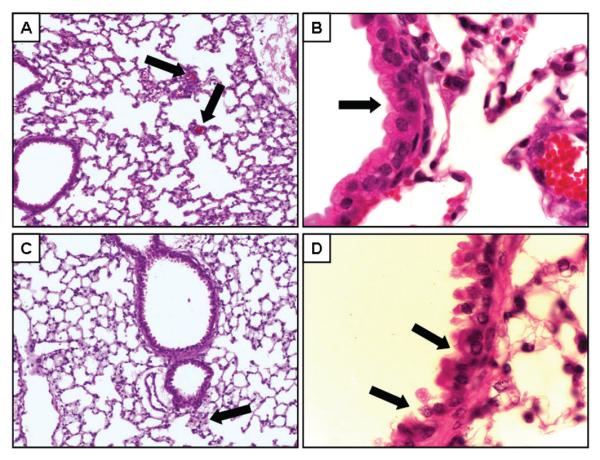
Assessment of lung histopathology following acute exposure to vaporized MA. Lung morphology was evaluated by H&E staining of paraffin sections. Representative sections from control mice (n = 4; panels A—B) and mice 3 h after MA exposure (n = 4; panels C—D). Panels A and B show representative images of lung tissue (original magnification, 20×). In panel A, the capillaries (black arrows) are apparent throughout the tissue. In panel C, no clear capillaries could be identified, but what appears to be the remains of a small arteriole is shown (black arrow). Higher magnification (original magnification 100×) of the airway epithelial cells are shown in panels B and D. The black arrows in panel D point to areas of disruption of normal cellular architecture.
Early-Response Cytokines Following Acute Exposure to Vaporized MA
To determine whether the induction of lung injury correlated with increased expression of early response cytokines, TNF-α and interleukin IL-6 were measured in the BAL fluid of control and MA-exposed mice (Figure 5). TNF-α levels were significantly increased following exposure to the 50 mg doses of MA. TNF-α levels also appeared higher at the 100 mg dose of MA; however, this increase did not reach statistical significance. IL-6 levels were only significantly increased following exposure to 100 mg MA. Consistent with other agents that cause lung injury, these results demonstrate that the MA-induced lung injury was associated with rapid elevation of early response cytokines.
FIG. 5.

Levels of early response cytokines in BAL fluid following acute exposure to vaporized MA. Three hours following 25–100 mg MA exposure, TNF-α production (A) and IL-6 production (B) in BAL fluid was determined (n = 8 for all groups). Asterisk indicates significant at p < .05 versus controls. Data are shown as means ± SEM.
Oxidative Stress in Lung Cells and Tissue Following Exposure to Vaporized MA
To test for possible mechanistic explanations for MA-induced lung injury, we evaluated whether the observed lung injury following exposure to 100 mg MA was associated with increased oxidative stress. The fluorescent dyes DHE and DHR were utilized to detect increased ROS and RNS, respectively, in the BAL cells and lung tissue from exposed mice. The oxidation of DHE has been reported to be relatively specific for with minimal oxidation from H2O2 or ONOO− and has been used as a marker for production (referred to as ROS) (Benov et al., 1998; Fink et al., 2004). Peroxynitrite efficiently oxidizes the nonfluorescent molecule DHR to the fluorescent product rhodamine. This method has been employed for detection of ONOO− (referred to as RNS) and can react efficiently with ONOO− even at very low concentrations (Kooy et al., 1994). As previously described (Haugen et al., 1999), DHE and DHR fluorescence was measured by flow cytomentry in intact BAL cells to determine the effect of MA exposure on the level of intracellular ROS and RNS levels (Table 1). The mean channel fluorescence for both DHE and DHR was significantly increased in the BAL cells from mice exposed to heated MA, indicating elevated intracellular ROS and RNS levels following exposure.
TABLE 1.
Effect of Inhaled Methamphetamine on Intracellular Reactive Oxygen and Reactive Nitrogen Species
Note: Data are means ± SEM. DHR, dihydrorhodamine-123; DHE, dihydroethidium; MCF, mean channel fluorescence. n = 4–5;
P < 0.05 vs. control.
The relative levels in lungs in situ were also assessed utilizing DHE staining of frozen tissue sections (Figure 6A). The regions of DHE staining correlated with the airway epithelial cells that were injured, as shown in Figure 4D. The intensity of DHE fluorescence was quantified utilizing Laser Scanning Microscopy (Figure 6B). These studies revealed that the DHE fluorescence was significantly higher (1756 ± 91.33 in controls vs. 2339 ± 172.5 in MA-exposed; n = 4, p < .05) and there was an increase in the percent DHE-positive cells in mice exposed to MA compared to unexposed controls (19.70% ± 1.167% in controls vs. 26.70% ± 1.950% in MA-exposed; n =4, p < .05).
FIG. 6.

Determination of oxidative stress in lung cells and tissue following exposure to vaporized MA. Frozen sections were stained with DHE ± PEG-SOD (see Materials and Methods), imaged, and analyzed by laser scanning cytometry (LSC). (A) DHE staining in lung sections. Representative images from control and MA-exposed mice are shown. (B) The maximum pixel values and percentages of DHE-positive cells are graphed (n = 4). Asterisk indicates significant at p < .05 versus controls. Data are shown as means ± SEM.
It has been reported that concentrations higher than 1 μM DHE can result in -independent fluorescence from nonspecific ethidium cation binding to nuclear DNA, resulting in an enhancement of fluorescence (Budd et al., 1997). Furthermore, DHE can be oxidized by routes independent of such as ferricytochrome c oxidation in conditions where mitochondria are the primary source production or cytochrome c release into the cytosol during apoptosis (Green & Reed, 1998). Therefore, DHE oxidation was also assessed in the presence of PEG-SOD (50 U/slide) to confirm that the increased DHE oxidation following exposure to MA was due to generation (Grobe et al., 2006). Pretreatment of tissue sections with PEG-SOD significantly reduced the DHE fluorescence in both control and exposed animals, confirming that this fluorescence is likely due to the presence of (Figure 6A) rather than to nonspecific DHE oxidation.
DISCUSSION
This study employed an MA inhalation exposure system to explore the acute pulmonary effects of MA. To our knowledge, this is the first report of pulmonary effects in mice following inhalation exposure to MA. We show that an acute exposure to MA causes lung injury and increased production of free radicals in the lungs of exposed mice. It has been previously reported that MA is readily vaporized upon heating, and that approximately 81–91% of methamphetamine-HCl is recovered intact when heated at temperatures used to smoke the drug (NIDA, 1991). Furthermore, in humans, MA is rapidly and well absorbed by inhalation of the vapor (Cook et al., 1993). In our study, we observed dose-dependent increases in MA plasma levels following exposure to the drug, suggesting that MA is also rapidly absorbed by inhalation of the vapor in our animal exposure model. We measured a linear increase in MA plasma concentrations following increasing doses of MA. In humans, several studies have demonstrated that the fraction of MA excreted in urine decreases with increasing drug dose, and the amount of MA in urine is not proportional to the dose of the drug absorbed (Cook et al., 1993; Kim et al., 2004). This suggests that detoxification by renal clearance might become saturated with only moderate doses of MA.
Because the lungs are the principal organ exposed to the products of smoked substances, it is expected that pulmonary complications would be prominent among the health consequences of smoking. The present results show that acute inhalation of MA caused increased microvascular permeability as measured by protein and albumin levels in the BAL fluid and lung weight gain. Furthermore, increased LDH levels in the BAL fluid suggest that the decrease is likely through cell death. There was a decrease in lavagable lung cells that may be due to MA-induced cytotoxicity. Alternatively, this could be due to increased cell adherence preventing macrophages from being lavaged or increased trafficking of the cells out of the lung. Further studies will be required to confirm the cause of decreased BAL cellularity. It is important to note that these acute effects on the lungs occurred at MA plasma levels below those seen in MA users.
This study focused on acute response of the lung to MA smoke exposure. Preliminary studies of mice exposed to the 50-mg MA dose at up to 24 h postexposure suggest that any detectable injury resolves (data not shown). The 100-mg MA dose resulted in self-injurious behavior not allowing long-term evaluation beyond the 3-h time point. However, it will be important to evaluate repeated and long-term MA exposure at lower doses to mimic human exposures.
These findings are consistent with observed effects of other smoked substances including tobacco, marijuana, and alkaloidal cocaine. Habitual marijuana use in humans is associated with histopathologic evidence of airway injury (Roth et al., 1998) and increased frequency and severity of epithelial inflammation and loss of ciliated surface epithelium (Gong et al., 1987; Fligiel et al., 1997). Similarly, long-term effects of tobacco smoking, with or without other substances, include increases in alveolar epithelial permeability (Susskind et al., 1991; Tashkin et al., 1997). Although there are no data available on the lung patho-physiology of MA inhalation exposures, it has been well documented that MA can cause neurotoxic damage, including degeneration of monoaminergic terminals and apoptosis of non-monoaminergic cells in the brain (Preston et al., 1985; Ricaurte & McCann, 1992; Deng et al., 1999, 2001; Jayanthi et al., 2001). Furthermore, numerous studies indicate that MA-induced neurotoxic damage is due, at least in part, to the formation of ROS (LaVoie & Hastings, 1999; Davidson et al., 2001), and MA-induced neurotoxicity can be attenuated in transgenic mice that overexpress copper-zinc superoxide dismutase (Cadet et al., 1994). Recently, Mori et al. demonstrated that oxidative stress mediated by the activation of neuronal nitric oxide synthase is associated with MA-induced self-injurious behavior following MA administration (Mori et al., 2007). Our data demonstrate that inhalation of MA also results in cellular toxicity in the lung, and this toxicity is associated with increased cellular ROS and RNS. An important target of toxicity may be airway epithelial cells, as evidenced by increased DHE staining (oxidant stress) seen in Figure 6A that correlated with morphological injury shown in Figure 4D.
The production of free radicals may be responsible for tissue injury in various cardiopulmonary diseases (Yu, 1994), and acute lung injury and consequent acute respiratory distress syndrome have been shown to result from free radical formation (Downey et al., 1999). Furthermore, Huang et al. demonstrated that free radicals mediate amphetamine-induced acute pulmonary edema in isolated rat lung (Huang et al., 2002). Therefore, it is reasonable to suggest that in the model reported here, increased free radical formation results in cellular toxicity contributing to pulmomary edema.
In the present study, there was a significant increase in the early response cytokines TNF-α and IL-6 in BAL fluid of mice exposed to MA. In human brain endothelial cells, Lee et al. demonstrated that MA-induced disturbances in cellular redox status and activation of AP-1 and NF-κB can play critical roles in the signaling pathways leading to upregulation of inflammatory genes (Lee et al., 2001). In vitro studies have shown that humoral-mediated and cell-mediated responses are affected by MA (Zule & Desmond, 1999). For example, it has been demonstrated that MA affects immune function with a significant suppression of IL-2, but not IL-4 production by T lymphocytes, as well as a suppression of B lymphocyte proliferation (House et al., 1994). In vivo, chronic intraperitoneal administration of MA in a mouse model significantly decreased the production of IL-2 and interferon (IFN)-γ , while significantly increasing the production of TNF-α by cultured splenocytes (Yu et al., 2002), further suggesting that MA may have immunomodulatory activity (Yu et al., 2002). The results reported here are consistent with MA having an immunomodulatory effect in the lung, although the mechanisms of this remain to be discovered.
Currently, there are no peer-reviewed reports on the relationship between individuals exposed to MA smoke and resultant health consequences. There have been anecdotal reports of increased asthma, pulmonary fibrosis, and upper respiratory complaints (Nestor et al., 1989b; Schaiberger et al., 1993); however, no documented health statistics are available at this time. The data presented here show that a single inhalation exposure to MA can result in lung injury in mice and support the need to conduct further studies to determine whether there may be a link between MA exposures and respiratory effects in humans. Given that most exposures to MA are chronic in nature, the data presented here support the need to conduct additional studies of the effects of chronic, repeated exposure to MA. Demonstrating that inhalation of MA can result in lung injury should prove useful for consideration of improved respiratory treatment of patients removed from situations where MA exposures may have occurred.
Acknowledgments
This work was supported in part by the National Center for Research Resources (NCRR; P20RR017670 to A. H.) and the National Heart, Lung, and Blood Institute (NHLBI; 1F32HL086154 to S. M. W.), both components of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and not necessarily the official views of NCRR, NHLBI, or NIH. The authors thank R. Hamilton for statistical support, L. Hoerner for technical assistance with the laboratory animals, and P. Shaw for technical assistance with the LSC measurements. In addition, we thank A. Harris and B. Klietz at the Montana State Crime Laboratory for their technical assistance with the development of the exposure model.
REFERENCES
- Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radical Biol. Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- Bloom JW, Kaltenborn WT, Paoletti P, Camilli A, Lebowitz MD. Respiratory effects of non-tobacco cigarettes. Br. Med. J. Clin. Res. Ed. 1987;295:1516–1518. doi: 10.1136/bmj.295.6612.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Sheng P, Ali S, Rothman R, Carlson E, Epstein C. Attenuation of methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. J. Neurochem. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- Cook CE, Jeffcoat AR, Hill JM, Pugh DE, Patetta PK, Sadler BM, White WR, Perez-Reyes M. Pharmacokinetics of methamphetamine self-administered to human subjects by smoking S-(+)-methamphetamine hydrochloride. Drug Metab. Dispos. 1993;21:717–723. [PubMed] [Google Scholar]
- Davidson C, Gow AJ, Lee TH, Ellinwood EH. Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res. Brain Res. Rev. 2001;36:1–22. doi: 10.1016/s0165-0173(01)00054-6. [DOI] [PubMed] [Google Scholar]
- Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods. 1988;115:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- Deng X, Ladenheim B, Tsao LI, Cadet JL. Null mutation of c-fos causes exacerbation of methamphetamine-induced neurotoxicity. J. Neurosci. 1999;19:10107–10115. doi: 10.1523/JNEUROSCI.19-22-10107.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Wang Y, Chou J, Cadet JL. Methamphetamine causes widespread apoptosis in the mouse brain: evidence from using an improved TUNEL histochemical method. Brain Res. Mol. Brain Res. 2001;93:64–69. doi: 10.1016/s0169-328x(01)00184-x. [DOI] [PubMed] [Google Scholar]
- Downey GP, Dong Q, Kruger J, Dedhar S, Cherapanov V. Regulation of neutrophil activation in acute lung injury. Chest. 1999;116:46S–54S. [PubMed] [Google Scholar]
- Fink B, Laude K, McCann L, Doughan A, Harrison DG, Dikalov S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am. J. Physiol. Cell Physiol. 2004;287:C895–902. doi: 10.1152/ajpcell.00028.2004. [DOI] [PubMed] [Google Scholar]
- Fligiel SE, Roth MD, Kleerup EC, Barsky SH, Simmons MS, Tashkin DP. Tracheobronchial histopathology in habitual smokers of cocaine, marijuana, and/or tobacco. Chest. 1997;112:319–326. doi: 10.1378/chest.112.2.319. [DOI] [PubMed] [Google Scholar]
- Forrester JM, Steele AW, Waldron JA, Parsons PE. Crack lung: An acute pulmonary syndrome with a spectrum of clinical and histopathologic findings. Am. Rev. Respir. Dis. 1990;142:462–467. doi: 10.1164/ajrccm/142.2.462. [DOI] [PubMed] [Google Scholar]
- Gong H, Jr., Fligiel S, Tashkin DP, Barbers RG. Tracheobronchial changes in habitual, heavy smokers of marijuana with and without tobacco. Am. Rev. Respir. Dis. 1987;136:142–149. doi: 10.1164/ajrccm/136.1.142. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Grobe AC, Wells SM, Benavidez E, Oishi P, Azakie A, Fineman JR, Black SM. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: Role of NADPH oxidase and endothelial NO synthase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L1069–1077. doi: 10.1152/ajplung.00408.2005. [DOI] [PubMed] [Google Scholar]
- Haim DY, Lippmann ML, Goldberg SK, Walkenstein MD. The pulmonary complications of crack cocaine. A comprehensive review. Chest. 1995;107:233–240. doi: 10.1378/chest.107.1.233. [DOI] [PubMed] [Google Scholar]
- Haugen TS, Skjonsberg OH, Kahler H, Lyberg T. Production of oxidants in alveolar macrophages and blood leukocytes. Eur. Respir. J. 1999;14:1100–1105. doi: 10.1183/09031936.99.14511009. [DOI] [PubMed] [Google Scholar]
- House RV, Thomas PT, Bhargava HN. Comparison of immune functional parameters following in vitro exposure to natural and synthetic amphetamines. Immunopharmacol. Immunotoxicol. 1994;16:1–21. doi: 10.3109/08923979409029897. [DOI] [PubMed] [Google Scholar]
- Huang KL, Shaw KP, Wang D, Hsu K, Huang TS, Chen HI. Free radicals mediate amphetamine-induced acute pulmonary edema in isolated rat lung. Life Sci. 2002;71:1237–1244. doi: 10.1016/s0024-3205(02)01847-7. [DOI] [PubMed] [Google Scholar]
- Jayanthi S, Deng X, Bordelon M, McCoy MT, Cadet JL. Methamphetamine causes differential regulation of pro-death and anti-death Bcl-2 genes in the mouse neocortex. FASEB J. 2001;15:1745–1752. doi: 10.1096/fj.01-0025com. [DOI] [PubMed] [Google Scholar]
- Kenyon NJ, van der Vliet A, Schock BC, Okamoto T, McGrew GM, Last JA. Susceptibility to ozone-induced acute lung injury in iNOS-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;282:L540–545. doi: 10.1152/ajplung.00297.2001. [DOI] [PubMed] [Google Scholar]
- Kim I, Oyler JM, Moolchan ET, Cone EJ, Huestis MA. Urinary pharmacokinetics of methamphetamine and its metabolite, amphetamine following controlled oral administration to humans. Ther. Drug Monit. 2004;26:664–672. doi: 10.1097/00007691-200412000-00013. [DOI] [PubMed] [Google Scholar]
- Kooy NW, Royall JA, Ischiropoulos H, Beckman JS. Peroxynitrite-mediated oxidation of dihydrorhodamine 123. Free Radical Biol. Med. 1994;16:149–156. doi: 10.1016/0891-5849(94)90138-4. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J. Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YW, Hennig B, Yao J, Toborek M. Methamphetamine induces AP-1 and NF-kappaB binding and transactivation in human brain endothelial cells. J. Neurosci. Res. 2001;66:583–591. doi: 10.1002/jnr.1248. [DOI] [PubMed] [Google Scholar]
- Megyeri A, Bacso Z, Shields A, Eliason JF. Development of a stereological method to measure levels of fluoropyrimidine metabolizing enzymes in tumor sections using laser scanning cytometry. Cytometry A. 2005;64:62–71. doi: 10.1002/cyto.a.20121. [DOI] [PubMed] [Google Scholar]
- Miller FJ, Jr., Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ. Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- Mori T, Ito S, Kita T, Narita M, Suzuki T, Matsubayashi K, Sawaguchi T. Oxidative stress in methamphetamine-induced self-injurious behavior in mice. Behav. Pharmacol. 2007;18:239–249. doi: 10.1097/FBP.0b013e328153dae1. [DOI] [PubMed] [Google Scholar]; National Drug Intelligence Center . National drug threat assessment 2007. National Drug Intelligence Center, U.S. Department of Justice; Washington, DC: 2006. [Google Scholar]
- National Institute on Drug Abuse . In: Methamphetamine abuse: Epidemiologic issues and implications. Miller NJKMA, editor. National Institute on Drug Abuse; Washington, DC: 1991. Research Monograph 115. [Google Scholar]
- National Institute on Drug Abuse . Research report: Methamphetamine: Abuse and addiction. National Institute on Drug Abuse, National Institutes of Health; Washington, DC: 2002. [Google Scholar]
- National Institute on Drug Abuse . Methamphetamine abuse and addiction. National Institute on Drug Abuse, National Institutes of Health; Washington, DC: 2006. [Google Scholar]
- Nestor TA, Tamamoto WI, Kam TH, Schultz T. Acute pulmonary oedema caused by crystalline methamphetamine. Lancet. 1989a;2:1277–1278. doi: 10.1016/s0140-6736(89)91883-7. [DOI] [PubMed] [Google Scholar]
- Nestor TA, Tamamoto WI, Kam TH, Schultz T. Crystal methamphetamine-induced acute pulmonary edema: a case report. Hawaii Med. J. 1989b;48:457–458. 460. [PubMed] [Google Scholar]
- Paravicini TM, Gulluyan LM, Dusting GJ, Drummond GR. Increased NADPH oxidase activity, gp91phox expression, and endothelium-dependent vasorelaxation during neointima formation in rabbits. Circ. Res. 2002;91:54–61. doi: 10.1161/01.res.0000024106.81401.95. [DOI] [PubMed] [Google Scholar]
- Preston KL, Wagner GC, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on monoamine neurons in the rhesus monkey brain. Brain Res. 1985;338:243–248. doi: 10.1016/0006-8993(85)90153-2. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, McCann UD. Neurotoxic amphetamine analogues: Effects in monkeys and implications for humans. Ann. NY Acad. Sci. 1992;648:371–382. doi: 10.1111/j.1749-6632.1992.tb24586.x. [DOI] [PubMed] [Google Scholar]
- Roth MD, Arora A, Barsky SH, Kleerup EC, Simmons M, Tashkin DP. Airway inflammation in young marijuana and tobacco smokers. Am. J. Respir. Crit. Care Med. 1998;157:928–937. doi: 10.1164/ajrccm.157.3.9701026. [DOI] [PubMed] [Google Scholar]
- Schaiberger PH, Kennedy TC, Miller FC, Gal J, Petty TL. Pulmonary hypertension associated with long-term inhalation of “crank” methamphetamine. Chest. 1993;104:614–616. doi: 10.1378/chest.104.2.614. [DOI] [PubMed] [Google Scholar]
- Schwilke EW, dos Santos M. I. Sampaio, Logan BK. Changing patterns of drug and alcohol use in fatally injured drivers in Washington State. J. Forens. Sci. 2006;51:1191–1198. doi: 10.1111/j.1556-4029.2006.00239.x. [DOI] [PubMed] [Google Scholar]
- Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: Roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- Susskind H, Weber DA, Volkow ND, Hitzemann R. Increased lung permeability following long-term use of free-base cocaine (crack) Chest. 1991;100:903–909. doi: 10.1378/chest.100.4.903. [DOI] [PubMed] [Google Scholar]
- Tashkin DP, Coulson AH, Clark VA, Simmons M, Bourque LB, Duann S, Spivey GH, Gong H. Respiratory symptoms and lung function in habitual heavy smokers of marijuana alone, smokers of marijuana and tobacco, smokers of tobacco alone, and nonsmokers. Am. Rev. Respir. Dis. 1987;135:209–216. doi: 10.1164/arrd.1987.135.1.209. [DOI] [PubMed] [Google Scholar]
- Tashkin DP, Khalsa ME, Gorelick D, Chang P, Simmons MS, Coulson AH, Gong H., Jr. Pulmonary status of habitual cocaine smokers. Am. Rev. Respir. Dis. 1992;145:92–100. doi: 10.1164/ajrccm/145.1.92. [DOI] [PubMed] [Google Scholar]
- Tashkin DP, Kleerup EC, Hoh CK, Kim KJ, Webber MM, Gil E. Effects of ’crack’ cocaine on pulmonary alveolar permeability. Chest. 1997;112:327–335. doi: 10.1378/chest.112.2.327. [DOI] [PubMed] [Google Scholar]
- Taylor DR, Fergusson DM, Milne BJ, Horwood LJ, Moffitt TE, Sears MR, Poulton R. A longitudinal study of the effects of tobacco and cannabis exposure on lung function in young adults. Addiction. 2002;97:1055–1061. doi: 10.1046/j.1360-0443.2002.00169.x. [DOI] [PubMed] [Google Scholar]
- Taylor DR, Poulton R, Moffitt TE, Ramankutty P, Sears MR. The respiratory effects of cannabis dependence in young adults. Addiction. 2000;95:1669–1677. doi: 10.1046/j.1360-0443.2000.951116697.x. [DOI] [PubMed] [Google Scholar]
- Yu BP. Cellular defenses against damage from reactive oxygen species. Physiol. Rev. 1994;74:139–162. doi: 10.1152/physrev.1994.74.1.139. [DOI] [PubMed] [Google Scholar]
- Yu Q, Zhang D, Walston M, Zhang J, Liu Y, Watson RR. Chronic methamphetamine exposure alters immune function in normal and retrovirus-infected mice. Int. Immunopharmacol. 2002;2:951–962. doi: 10.1016/s1567-5769(02)00047-4. [DOI] [PubMed] [Google Scholar]
- Zule WA, Desmond DP. An ethnographic comparison of HIV risk behaviors among heroin and methamphetamine injectors. Am. J. Drug Alcohol Abuse. 1999;25:1–23. doi: 10.1081/ada-100101843. [DOI] [PubMed] [Google Scholar]