

Abstract
Eticlopride is a substituted benzamide analog with high affinity and selectivity for dopamine (DA) D2‐like receptors that was initially developed as a potential antipsychotic agent. A great deal of research has utilized this drug to better understand central DA receptor function, the role of D2‐like receptors in behavior, and the influence of blockade of these receptors on several preclinical animal models. This review highlights research utilizing this drug and compares it to typical and atypical antipsychotics used clinically. First, we describe structure–activity relationships as it relates to binding at DA receptors and the consequences on behavior. This is followed by a discussion of several imaging strategies including the use of eticlopride for in vivo, in vitro, and ex vivo examination of DA D2‐like receptor densities and function. Finally, we discuss the use of eticlopride in several behavioral models predictive of antipsychotic activity, extrapyramidal side effects (EPS), and learning and memory. While eticlopride is not used clinically, it remains a viable research tool for understanding DA receptor function and behavior.
Keywords: Behavioral pharmacology, Dopamine D2‐like receptors, Eticlopride, Extrapyramidal side effects, Imaging
Introduction
Schizophrenia is a chronic brain disease that affects approximately 1% of the population worldwide [1, 2, 3]. There are three broad categories of symptoms related to schizophrenia: positive, negative, and cognitive [2]. Distortions of thought and perception, including hallucinations, are part of the constellation of symptoms described as “positive.” In contrast, reductions in activity, affect, and motivation are “negative” symptoms of schizophrenia. Finally, cognitive symptoms include poor executive function, lack of ability to sustain attention, and poor working memory. Alleviation of these symptoms is the goal of pharmacotherapies.
The primary target for antipsychotic medication has been the dopamine (DA) neurotransmitter system. Two superfamilies of DA receptors have been identified, the D1‐ and D2‐like receptors. DA D1‐like receptors are located postsynaptically, with the highest abundance in the striatum, a midbrain structure that includes the caudate nucleus, putamen, and nucleus accumbens [4, 5, 6, 7] Conversely, D2‐like receptors are located pre‐ and postsynaptically in striatal brain regions [8]. Additionally, D2‐like receptor binding sites are also present throughout the cerebral cortex, although the densities are less than in the striatum [9]. Within the DA system, several lines of research have linked antipsychotic activity to the DA D2‐like superfamily. For example, there is a positive correlation between blockade of D2‐like receptors (as determined by IC50 values) and average clinical dose for controlling symptoms associated with schizophrenia [10]. The D2‐like receptor has been further implicated in schizophrenia by brain imaging studies. Wong et al. [11] used positron emission tomography (PET) to compare D2‐like receptor availability in drug‐naïve (i.e., unmedicated) schizophrenic and control subjects and found significantly higher levels of D2‐like receptors in the schizophrenic subjects. The elevation in striatal D2‐like receptor densities has been confirmed in postmortem studies and shown to be selective for D2‐ versus D1‐like receptors [12].
The history of antipsychotic medication has been reviewed elsewhere, including the significant role these drugs played in the establishment of animal models of affective disorders [13]. This review will focus specifically on eticlopride, a substituted benzamide initially developed as a potential antipsychotic agent with selectivity at blocking DA D2‐like receptors. However, the primary use of eticlopride is not in the treatment of schizophrenia, but rather as a tool for understanding D2‐like receptor function in schizophrenia and other brain diseases. In this review, we will describe the pharmacology of eticlopride, as determined with in vitro screens, and then describe some of the uses of this drug with several in vivo models related to antipsychotic drug activity. In many cases, we will directly compare eticlopride to clinical antipsychotics. These direct comparisons allow researchers to ask questions related to mechanisms of action. For example, eticlopride is selective for D2‐like receptors compared to D1‐like receptors with Ki values of 0.09 versus 10,000 nM, respectively [14]. Spiperone and sulpiride, two compounds frequently described in the literature, are also D2‐like selective relative to D1‐like receptors, although they have lower affinities for D2‐like receptors compared to eticlopride (0.26 and 18.2 nM, respectively [Ref. 14]). In contrast, chlorpromazine (3.0 vs. 94 nM) and haloperidol (1.2 vs. 55 nM) have lower selectivity for D2‐like relative to D1‐like receptors [14]. In vitro receptor autoradiography measures of D2‐like receptor densities frequently use [3H]raclopride, which has high selectivity for D2‐ versus D1‐like receptors (3.4 vs. 18,000 nM [Ref. 14]). Finally, the affinity of DA for D2‐like receptors has been estimated to be 7.5 nM, which is twofold lower than the affinity of eticlopride for D2‐like receptors [14]. With regard to the pharmacokinetic, chemical, physical, and toxicological properties of eticlopride, many in‐depth analyses are absent from the literature; however, to the best of our knowledge, the available data are included in this review.
Development
Because of the indications that antipsychotics exert their effect through blockade of central DA receptors, and that clinical efficacy correlates with affinity for D2‐ but not D1‐like receptors, several DA D2‐like receptor antagonists have been studied for their possible utility as antipsychotics. Traditionally, antipsychotic drugs have been categorized based on their specificity and can be divided into two basic families, typical and atypical. The typical antipsychotic category includes the phenothiazines (e.g., chlorpromazine), the thioxanthenes (e.g., flupenthixol), and the butyrophenones (e.g., haloperidol) while the atypical antipsychotic category includes the tricyclic antipsychotics (e.g., clozapine), and the benzamides (e.g., amisulpride). All antipsychotics tend to block DA D2‐like receptors in the mesolimbic DA pathway; however, while atypical antipsychotics affect DA turnover in the mesolimbic system preferentially [15, 16], typical antipsychotics less selectively inhibit cholinergic, histaminergic, and adrenergic systems in addition to the broad sweeping inhibition of DA neurotransmission in both mesocorticolimbic and nigrostiatal pathways [1]. As such, atypical antipsychotics have been found to be associated with fewer extrapyramidal side effects (EPS; 15, 16, 17, 18), and there have been great efforts allocated to the development of novel atypical antipsychotics associated with reduced incidence of EPS [19].
Targeted Structure Activity Relationship (SAR) Development
Of the antipsychotics, the substituted‐benzamides are a group of compounds that has proven useful for the design of selective DA D2‐like receptor antagonists [15, 20, 21, 22]. Sulpiride, the prototypical benzamide, was noted for its neuroleptic effects as early as the mid‐ to late‐1970s [17, 18]. However, while sulpiride produced minimal EPS, large doses were required for clinical efficacy, and significant increases in prolactin levels were observed [18, 23]. Using sulpiride as a starting model, chemists at Astra Pharmaceuticals manipulated the aromatic substituents to develop compounds that were more potent in animal models of psychosis and were more selective for DA D2‐like receptors [19, 21, 24]. One of the first major successes in this focused SAR development of ligands was remoxipride, an effective antipsychotic agent with high affinity for DA D2‐like receptors that was introduced into the market in 1992 [25, 26, 27, 28, 29]; however, despite low occurrence of EPS, it was quickly withdrawn from clinical use due to an association with a rare blood dyscrasia [24, 29, 30]. Soon after remoxipride, the binding affinity for DA D2‐like receptors was again increased with the development of raclopride, a 3,5‐dichloro analogue of sulpiride with halogens in the meta position of the aromatic ring [21]. Further evaluation of these two meta positions revealed that each position had a distinct SAR, and affinity for DA D2‐like receptors could be increased further by optimizing the substituent at either position independently [21, 24]. Through a series of substituted 6‐hydroxy‐2‐methoxybenzamides, de Paulis et al. [21, 31] demonstrated that placing a large electron‐donating substituent, such as a propyl group, at the aromatic 5‐position increased the potency to block [3H]spiperone binding in vitro and apomorphine‐induced stereotypy (SAI) and apomorphine‐induced hyperactivity (HAI) in vivo, all assays of D2‐like receptor selectivity and affinity. However, further analysis determined that it was actually the lipophilic nature of the substituent and not necessarily the electronic nature that conferred enhanced potency [21]. Conversely, the 3‐position ortho to the methoxy group required a small substituent to optimize in vivo activity [21, 31]. Eticlopride (Fig. 1) was created by combining the most favorable substituents at either position, an ethyl group at the aromatic 5‐position and a chlorine at the aromatic 3‐position [24, 32]; the result was a compound that was 27 times more potent than remoxipride and nearly 35‐times more potent than raclopride at displacing [3H]spiperone binding [31, 33].
Figure 1.
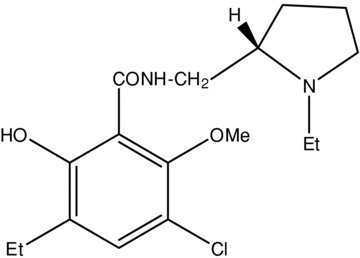
The simple chemical structure of eticlopride.
Synthesis, Structure, and Stability
Eticlopride {2S(–)‐3‐chloro‐5‐ethyl‐N‐[(1‐ethyl‐2‐pyrrolidinyl)methyl]‐6‐hydroxy‐2‐methoxybenzamide} was synthesized from ethyl‐2,4‐dimethoxybenzene through a series of intermediate reactions (including lithiation, carboxylation, demethylation, and chlorination) that produced 3‐chloro‐5‐ethyl‐6‐hydroxy‐2‐methoxybenzoic acid; subsequent treatment of the salicylic acid precursor with S(–)‐2‐aminomethyl‐N‐ethyl‐pyrrolidine resulted in the final salicylamide product (33). The crystal structure of eticlopride consists of two planes: (i) benzene ring and substituents; and (ii) pyrrolidine ring and amide bonds, with a dihedral angle of 38° between the two planes, and the asymmetric carbon atom has the S‐configuration [32, 33]. The resulting structure is a flat, almost planar conformation that is comparable to the conformation of the original benzamide antipsychotic sulpiride [32, 34].
The amide has a restricted rotational freedom due to some degree of double bond character as indicated by the length of the C–N bond [33]. Side‐chain conformations are stabilized through the formation of two additional six‐membered pseudo‐rings facilitated by intramolecular hydrogen bonds between the phenol hydrogen and carbonyl oxygen atoms, ring B, and the methoxy oxygen and amide nitrogen atoms, ring C [32, 33]. The importance of the hydrogen bond‐induced pseudo‐rings was further investigated by the Astra laboratories, and it was concluded that only ring C was required for biological activity because removal of ring B had no effect on DA antagonism but removal of ring C resulted in a 160‐fold decrease in potency for the blockade of [3H]spiperone binding [33]. Moreover, these data highlight that DA antagonism at the receptor level is not limited only to those compounds that share structural features with DA [33, 35].
Eticlopride has an uncorrected melting point of 144–146°C, and the optical rotation in acetone is –69°[31]. The hydrochloride salt is an off‐white solid with a molecular weight of 377.31 g, is soluble in both aqueous and organic solvents, and produces an optical rotation of –4.7° in methanol. In studying the effect of solvation on the ionization and conformation of 6‐methoxysalicylamides, Tsai et al. [36] demonstrated that in an aqueous phase at physiological pH, 74% of the eticlopride species were in the zwitterionic form. Under an aqueous potentiometric titration, the pKa of the phenolic group was 6.93 ± 0.01 and the pKa of the amino group was 9.67 ± 0.02, creating an isoelectric point of 8.30 [36]. Furthermore, Tsai et al. [36] suggested that at pH around the isoelectric point, 6‐methoxysalicylamides exist as extended or half‐folded zwitterionic conformers in highly polar media (e.g., water) and as neutral molecules in solvents of low polarity (e.g., organic phases), and that the capacity of eticlopride to readily cross the blood–brain barrier is likely a function of strong internal hydrogen bonds that prevent eticlopride from acting as a hydrogen donor in biological lipophilic media. However, in an interesting study exploring the potential influence of the lipophilicity of zwitterionic drugs at an aqueous/organic interface, it was demonstrated that at isoelectric pH in an aqueous solution with varying percentages of a dioxane organic phase, the unionized species and not the zwitterionic form of eticlopride always predominated regardless of the dioxane percentage [37]. Regardless, as the phenolic group must be neutral upon binding to the DA receptor [36, 38, 39, 40], the predominance of neutral eticlopride species should not interfere with receptor binding.
Affinity and Specificity
In general in vitro receptor binding assays comparing the displacement of [3H]spiperone from rat, pig, or calf brain striatum membranes by various DA D2‐like receptor antagonists have shown consistent results. At D2‐like receptors, the Ki for eticlopride was ∼0.92 nM; this is 13 times more potent than haloperidol with a Ki= 1.2 nM, over 200 times more potent than sulpiride with a Ki= 18.2 nM, and over 7000 times more potent than remoxipride with a Ki= 703 nM [14]. While the magnitude of difference in potencies is reduced under in vivo conditions, eticlopride was still far more potent than most compounds tested. For example, in the blockade of DA‐mediated HAI in the rat, eticlopride was nine times more potent than haloperidol, ∼25 times more potent than remoxipride, and over 800 times more potent than sulpiride (ED50: 32, 290, 860, and 28,200 nM/kg ip, respectively; Ref. [31]). Importantly, an interesting in vivo finding was that while apomorphine induced both stereotypy and hyperactivity in the rat, both behaviors are thought to be DA mediated [41, 42, 43], HAI is thought to occur through activation of mesolimbic dopaminergic pathways, and SAI through stimulation of nigrostriatal dopaminergic pathways [43, 44]; and the ability of DA D2‐like receptor antagonists to inhibit these behaviors can often be described in terms of separation of potency. As stated above, eticlopride was nine times more potent than haloperidol at inhibiting HAI; however, eticlopride and haloperidol were nearly equipotent at inhibiting SAI, and haloperidol was equipotent in both assays [31, 33]. This same phenomenon of separation between inhibition potencies was true for the earlier generation benzamides [45, 46] as well as for the more potent 3‐alkyl‐substituted salicylamides [21]; each inhibited HAI at lower doses than required to inhibit SAI. Interestingly, a binding displacement study showed that high doses of sulpiride, which inhibited both HAI and SAI, displaced [3H]spiperone to the same extent in all brain regions studied [47]. However, with low doses of sulpiride, which preferentially inhibited HAI, [3H]spiperone displacement was limited to limbic regions and the nucleus accumbens [47]. This finding suggests that spiperone and, by extension, later generation salicylamides, may affect subpopulations of DA receptors differently [47] or that there is a single effect that occurs preferentially in different regions of the brain [31]. Regardless of the exact mechanisms, a preferential inhibition of HAI over SAI and a limbic binding profile are desirable attributes of potential antipsychotics [16, 19, 48].
In comparing in vitro potency (‐log IC50[3H]spiperone binding displacement) to in vivo potency (log ED50 stereotypy/ED50 hyperactivity), the substituted benzamides typically exhibit a linear, predictive correlation with few outliers [21, 31]; that is, in vitro potency predicts in vivo potency. Eticlopride, for example, had an ED50 value on the hyperactivity parameter that was only six times lower than the stereotypy parameter, and several other potent benzamides studied had even lower stereotypy/hyperactivity ratios [21, 31]. Of the outliers mentioned above, three that deviate most from the normal distribution include sulpiride, remoxipride, and raclopride [20, 31]. The deviation of sulpiride and remoxipride can be explained by poor bioavailability and reduced brain penetration [23] and the production of active metabolites [20, 49], respectively. On the other hand, raclopride, which was 13 times more potent at inhibiting HAI than SAI[31], was selected for further clinical investigation as a potential antipsychotic agent due to its ideal behavioral profile suggestive of low EPS‐liability [31, 50].
While there is strong evidence that eticlopride and many of the substituted benzamides are antagonists at DA D2‐like receptors and not D1‐like receptors [51, 52, 53, 54], several of these studies were conducted prior to the ability to distinguish definitively between D2‐like receptor subtypes, that is, DA D2 and DA D3 receptors. Before the cloning and direct localization of the DA D3 receptor in the early 1990s [55, 56, 57, 58], D2 receptors were defined [59] as those binding sites from which [3H]ADTN (2‐amino‐6,7‐dihydroxy‐1,2,3,4‐tetrahydronaphthalene) binding was displaced by very low concentrations of neuroleptics (IC50 values of 0.15–40 nM), while D3 receptors were those binding sites from which [3H]ADTN was displaced by very high concentrations of neuroleptics (IC50 values of 10–50,000 nM). Nevertheless, differentiation of ligand specificity for D2 receptors versus D3 receptors was challenging (for review see Refs [60, 61]). Some ligands used in the early studies of DA D3 receptor binding included [3H]ADTN [62, 63], [3H]apomorphine [63, 64], and [3H]NPA (N‐propyl‐norapomorphine [65, 66]. Initial affinity studies with eticlopride demonstrated an IC50 value of 113 nM in the displacement of [3H]ADTN from DA D3 receptors and values of 1.0 and 1.6 nM for the displacement of [3H]spiperone and [3H]domperidone, respectively, from DA D2 receptors [67], suggesting that eticlopride was highly selective for the D2 receptor over the D3 receptor. However, more recent studies have suggested that all atypical antipsychotics are potent D3 receptor antagonists and that eticlopride has considerable affinity for the D3 receptor and may even be an inverse agonist at the D3 receptor [55, 68, 69, 70, 71]. In another study using cloned DA receptors in CCL1.3 and MN9D cells, Tang et al. [72] reported that eticlopride had only three‐ to six‐fold greater affinity for D2 receptors (Ki= 0.26 ± 0.12 nM, CCL1.3 cells; 0.23 ± 0.05 nM, MN9D cells) over D3 receptors (Ki= 1.5 ± 0.6 nM, CCL1.3 cells; 0.78 ± 0.36 nM, MN9D cells). While the debate regarding activity at D2 versus D3 receptors has not been settled, it is clear that the effects of eticlopride are not mediated via interaction with D1‐like receptors. Hall et al. [67] demonstrated that eticlopride had a low affinity for DA D1 receptors based on both negligible displacement of [3H]flupenthixol binding (IC50 value = >100,000 nM) and lack of inhibition of DA‐stimulated adenylate cyclase at concentrations below 100 μM. Moreover, eticlopride lacked any significant specificity for all other central receptors tested, including various adrenergic, serotonergic, histaminergic, and muscarinic subtypes, with an overall binding profile similar to that reported for atypical antipsychotics (Table 1; Refs [67, 73]). However, while eticlopride has greatest affinity for DA D2 receptors, it is not possible to rule out the contribution of activity at DA D3, DA D4, or α1‐adrenergic receptors to both the in vitro and in vivo effects of eticlopride.
Table 1.
Relative affinity of eticlopride, sulpiride, remoxipride, and raclopride for various receptors in the rat brain determined by in vitro receptor binding displacement assays
| Receptor | Radioligand | Relative affinity (Ki nM)a | |||
|---|---|---|---|---|---|
| Eticlopride | Sulpiride | Remoxipride | Raclopride | ||
| Dopamine D1 | 3H‐flupenthixol | >10,000 | >10,000 | >10,000 | >10,000 |
| Dopamine D2 | 3H‐spiperone | 0.09 | 18.2 | 703 | 3.4 |
| Relative affinity (IC50 nM)b | |||||
| α1‐adrenergic | 3H‐WB4104 | 112 | 54,400 | 42,000 | 32,300 |
| α2‐adrenergic | 3H‐PAC | 699 | 7880 | 15,900 | 38,200 |
| β‐adrenergic | 3H‐DHA | 95,900 | >10,000 | >10,000 | >10,000 |
| 5HT1 | 3H‐5HT | 6220 | >10,000 | >10,000 | 48,800 |
| 5HT2 | 3H‐spiperone | 830 | >10,000 | 32,300 | 12,900 |
| Histamine‐H1 | 3H‐mepyramine | 14,800 | >10,000 | >10,000 | 8430 |
| Muscarinic | 3H‐QNB | 14,200 | >10,000 | 93,600 | >10,000 |
Isotope labeling
Given its potency and high affinity for DA D2‐like receptors, many attempts have been made to label eticlopride with various radioisotopes for in vivo, in vitro, and ex vivo visualization of DA receptor binding. Initial binding studies with [3H]eticlopride showed much promise for this potential radioligand; unlike its predecessor sulpiride, eticlopride readily crossed the blood–brain barrier where it bound with a very low proportion of nonspecific binding, accumulating most densely in the striatum, olfactory tubercles, and nucleus accumbens [67, 74]. While the striatum has a high density of DA D2‐like receptors and thus high accumulation of eticlopride, the cerebellum DA D2‐like receptor density and eticlopride accumulation are negligible. As such, the striatum‐to‐cerebellum distribution volume ratio (DVR) of [3H]eticlopride accumulation reached approximately 10:1 in ex vivo autoradiography studies [74]. Such a high DVR made eticlopride an excellent candidate to pursue as a potential PET radiotracer. However, the utility of eticlopride for use in PET has been limited by the reduced in vivo striatal uptake of labeled eticlopride. While Halldin et al. [75] successfully labeled eticlopride at both the N‐ethyl group and the O‐methyl group with 11C, the best striatum‐to‐cerebellum DVR achieved during PET studies was only 2.5 for [N‐ethyl‐11C]eticlopride, binding nearly half as specific as the DVR of 4–5 achieved with [11C]raclopride [76]. In another disappointing study, using an eticlopride analogue shown to be highly potent at displacing [3H]spiperone (IC50= 3 nM; Refs [33, 76]), labeled methyl‐eticlopride (FLB 524) with 11C for PET analyses in cynomolgus monkeys demonstrated a peak striatum‐to‐cerebellum DVR of only 1.65 at 70 min postinjection.
The 18F isotope has a half‐life of 110 min, making it a more accommodating radionuclide than 11C; however, [18F]eticlopride PET ligands have not shown much promise [19]. Kiesewetter et al. [77] initially attempted to label eticlopride with 18F at the N‐ethyl position; this produced a radioligand with relatively high nonspecific binding and poor specificity for the caudate nucleus over the cerebellum. In another study, the β‐ and γ‐fluoro analogues of eticlopride, [18F]NCQ 134 and [18F]NCQ 135, respectively, each had poor uptake into the striatum compared to the cerebellum, a surprising finding considering that unlabeled NCQ 134 and NCQ 135 both demonstrated high DA D2‐like receptor affinity under in vitro assays [78]. It has been suggested that protein binding, nonspecific binding, temperature effects, or ligand metabolism could all play a role in limiting the ability to predict in vivo PET efficacy from in vitro binding data [78]. Regardless of the exact mechanism, two things are clear from attempts at N‐fluoroalkyl substitution: (i) introduction of a fluorine atom results in a 4‐ to 10‐fold decrease in affinity for the DA D2‐like receptors [24]; and (ii) the electronegative fluorine atom lowers the pKa value of the amine, thus increasing lipophilicity and nonspecific binding [79, 80]; for review see de Paulis [24]. Since these initial studies with [18F]eticlopride, further attempts have been made to label eticlopride analogues with 18F in alternate positions. To date, the highest striatum‐to‐cerebellum DVR obtained from 18F‐labeled eticlopride analogues has been 5.22 for 4‐[18F]fluoropyrroldinyl eticlopride [81]. As a whole, attempts to create a [18F] fluoroalkyl eticlopride analogue with sufficient striatal uptake have been unsuccessful [81], an outcome similar to that reported for raclopride [77, 78].
While there has been limited success in creating eticlopride radiotracers for use in PET imaging, the resultant in‐depth analysis of the chemistry that dictates selection criteria for imaging agents has not been without gain. Since Astra Pharmaceuticals' introduction of the substituted benzamides as potential antipsychotics and subsequent appreciation for their high affinity for DA D2‐like receptors, considerable competition within the field has led to the development of several generations of compounds with ever increasing affinity, specificity, and PET applicability. Studies with a series of substituted benzamides, including raclopride and eticlopride, led to the elucidation of the importance of size and lipophilicity of aromatic substituents [21, 31] as well as the importance of isotope choice and position [79, 80]; for complete review see de Paulis [24]. While future generations of substituted benzamides continue to improve, [11C]raclopride remains the gold‐standard PET tracer for striatal D2‐like receptors [82].
Preclinical Applications
In Vitro and Ex Vivo Antagonism—Cellular Mechanisms
Much of this review so far has focused on eticlopride and other substituted benzamides insofar as their utility as antipsychotics or PET radiotracers; however, their usefulness in cellular studies of dopaminergic regulatory processes, effects of DA signaling, and receptor physiology and pharmacology should not go unmentioned. While presentation of the complete range of these studies is well beyond the scope of this review, the goal of this section is to present findings from some of the more relevant applications of D2‐like receptor antagonism by eticlopride in cellular‐level assays so as to emphasize its preclinical utility. To that end, a few specific examples of in vitro and ex vivo studies will be discussed so as to demonstrate how eticlopride has been used to elucidate receptor function and second messenger signaling.
Many of the substituted benzamides express potent, highly specific receptor binding patterns, which is a feature that makes their use ideal when studying regional differences in the various brain dopaminergic systems. Soon after eticlopride was developed, Magnusson et al. [83] compared sulpiride, raclopride, and eticlopride to haloperidol and chlorpromazine to examine the relationship between the behavioral and the neurochemical effects of D2‐like receptor antagonism with regard to potential antipsychotic efficacy and mesolimbic versus nigrostriatal activity. Before this study, it had been demonstrated that DA receptor antagonists increased DA turnover [84] through activation of tyrosine hydroxylase, resulting in an increase in the concentration of DA metabolites dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) but not DA itself (for reviews, see Refs [85, 86]). However, the antagonists previously evaluated did not exhibit the separation between potencies for inhibiting HAI versus SAI that is characteristic of the substituted benzamides. Comparing the changes in DA metabolite concentration induced by various doses of each antagonist [83] showed that, unlike with haloperidol and chlorpromazine, administration of the ED50 dose of eticlopride for HAI inhibition did not change the concentration of DA metabolites in the striatum. On the other hand, for all antagonists tested, the SAI inhibition ED50 doses resulted in a near threefold increase in striatal DA metabolite concentrations. As discussed above, HAI and SAI are likely mediated by dopaminergic stimulation of different brain regions, and displacement of [3H]spiperone binding by low doses of the substituted benzamide sulpiride, which only inhibited HAI, was limited to limbic brain regions. With that in mind, the findings of Magnusson et al. [83] might suggest that the activation of nigrostriatal and mesolimbic DA receptors differentially alter DA turnover. However, as Magnusson et al. [83] pointed out, it should be considered that HAI may result from dopaminergic activation of several closely interacting brain regions, and increases in DA turnover by eticlopride might have occurred at the HAI ED50 had the entire network been analyzed together rather than each brain region in isolation. Undoubtedly, more studies are needed to parse out the true differences between the nigrostriatal and mesolimbic dopaminergic systems—differences that deserve thorough investigation because understanding them and exploiting them may ultimately lead to more efficacious antipsychotic drugs with fewer side effects. As already demonstrated, the substituted benzamides have been very useful tools in this endeavor, and their application in future studies is sure to provide further insight.
Another example highlighting the usefulness of eticlopride in preclinical assays comes from a study examining the properties of D2‐like receptors in clonal cell lines transfected with D2, D3, or D4 receptors. In this study, Tang et al. [72] were interested in investigating the potential autoreceptor role of each of the D2‐like receptor subtypes; however, their experimental methodology can be applied to investigation of any D2‐like receptor second messenger system or G‐protein coupling. In brief, cells from a mesencephalic cell line, MN9D, that synthesize and release DA, were transfected with one of the three D2‐like receptor subtypes, and the level of DA release was assessed. The transfected cells were then stimulated with the D2‐like agonist quinpirole alone, or with eticlopride, and the effects on baseline DA release were determined under each condition. What this study demonstrated was that the stimulation of both D2 and D3 receptors inhibited DA release, the first demonstration of a functional role for human D3 receptors [72]. While this finding suggests a conservation of function between the D2 and D3 receptor homologs, previous work by this group and others have shown subtle differences. For example, D2 receptors expressed in a multitude of different cell lines couple to Gi proteins and inhibit adenylyl cyclase [87], but D3 receptors only couple to Gi proteins and inhibit adenylyl cyclase in a select few cell lines [88]. Since the identification of unique D2‐like receptor subtypes by molecular cloning techniques (D2: [89]; D3: [55]; D4: [90]), the use of cellular expression systems and simple stimulation/antagonism assays to study receptor function has steadily provided valuable insight into dopaminergic system physiology, pathology, and pharmacology, as briefly highlighted above.
Lastly, eticlopride has proven useful in in vitro studies examining the role of DA and D2‐like receptors in developmental neurobiology. In one of the early studies in this area, Swarzenski et al. [91] demonstrated that the D2‐like receptor subtypes differentially affected the outgrowth and morphology of neurites in cultures of fetal cortical neurons. This study showed that the stimulation of D2 receptors with quinpirole led to an increased number of neurites and branches per neuron while stimulation of D3 and D4 receptors resulted in an increase in the number of branches per neuron as well as an increase in primary neurite length. In addition, application of eticlopride to cultures of D3 expressing cells was capable of blocking these quinpirole‐induced morphological changes. While it is unlikely that in vivo morphology is dictated by activation of a single D2‐like receptor subtype, it is not difficult to appreciate how normal physiology and function of dopaminergic pathways could be affected by abnormal stimulation of these receptors. Importantly, the implications of studies like this are relevant to several disciplines ranging from disease pathophysiology and substance abuse to prenatal care and developmental neurobiology. Whether the changes induced by dopaminergic stimulation or antagonism are deleterious or advantageous remains to be seen and requires much more in depth analysis of questions such as the effect of developmental stage on susceptibility to dopaminergic control of morphology and the translation to in vivo models. Nevertheless, the necessity of high affinity, high specificity D2, D3, and D4 receptor antagonists for use in this type of model remains.
In Vitro Antagonism—Autoradiography
In order to better understand how the brain works, it is necessary to correlate spatial organization and structure with function. To that end, the topographical maps created through autoradiography have proven to be useful histochemical tools (for review see Pilgrim and Stumpf [92]). While autoradiography has been employed in the study of a vast array of topics ranging from cellular proliferation (incorporation of [3H]thymidine) to neuronal connectivity (utilizing axoplasmic transport) or regional glucose utilization (incorporation of [14C]‐2‐deoxyglucose), the most relevant use of autoradiography with regard to this review is for the study of brain receptor visualization.
As discussed above, early binding displacement studies demonstrated that eticlopride has high affinity and specificity for the D2‐like receptors, making it an ideal candidate for use in autoradiographical studies attempting to localize DA D2‐like receptors. Nonetheless, it remains possible that antagonism of other receptors influenced the findings reported below. In initial studies with rat brain homogenates, [3H]eticlopride binding was predominantly recorded in the striatum with significant binding also occurring in the nucleus accumbens and olfactory tubercles [67]. Subsequent autoradiographical studies using [3H]eticlopride (rodent brain: [74]) and [3H]raclopride (monkey brain: [93]) also demonstrated high binding densities, interpreted as D2‐like receptors, in the striatum, nucleus accumbens, and olfactory tubercles. In addition, both rodent and monkey brains demonstrated moderate tracer binding in the entorhinal cortex. With the advent of newer substituted benzamides, such as the highly efficacious epidepride, the localization of extra‐striatal D2‐like receptors has also been possible [94]. Clearly, as the sophistication of autoradiographical tracers continues to improve, so does the ability to precisely visualize and quantify specific receptors throughout the brain.
One of the positive assets of autoradiography is that the technique has few inherent confounds and can be applied under both normal and pathological states. Relevant to the discussion of eticlopride as a valuable preclinical tool is the example of autoradiography use in the study of schizophrenia. While the exact etiology of schizophrenia remains unclear, there is strong evidence in the literature to support dopaminergic pathology, especially among D2‐like receptors, as one of the key features of the disease [15, 51, 95]. Based on this evidence, several studies have attempted to identify key neurochemical differences between schizophrenic and nonschizophrenic control populations such as in DA receptor localization and density (for reviews see [96, 97, 98]). Unfortunately, these studies have yet to identify any pathognomonic lesions in schizophrenia, and several of the results have proven difficult to replicate. Regardless, eticlopride and several other substituted benzamides have repeatedly demonstrated their utility as radiotracers for autoradiographical studies of schizophrenia pathophysiology. Moreover, nearly any disease or drug treatment thought to affect DA D2‐like receptors can be evaluated with histochemical techniques employing selective DA D2‐like antagonists such as eticlopride.
In Vivo Antagonism—Behavioral Pharmacology
Animal models have provided a wealth of information related to the ability of eticlopride to block the behavioral effects of other drugs, thereby implicating D2‐like receptors in the actions of these drugs. For example, eticlopride has been shown to robustly block the reinforcing effects of cocaine under several conditions and in different species [99, 100, 101]. A PubMed search with the key words “Eticlopride” and “Behavior” yielded 158 hits (http://www.ncbi.nlm.nih.gov/sites/entrez?db=PubMed, Oct 2007). For the sake of brevity, in this section, we will only highlight research involving the behavioral effects of eticlopride, rather than the antagonism of the behavioral effects of other drugs. Thus, issues related to amphetamine‐induced stereotypy, apomorphine‐induced climbing, or PCP‐induced social behavior as models of neuroleptic activity will not be discussed in this section (see reviews in Refs [102, 103]). When possible, we will compare the behavioral effects of eticlopride to those of other neuroleptics and/or atypical antipsychotics. For example, the D2‐selective antagonist raclopride is equipotent to eticlopride at decreasing food intake in free‐feeding rats, suggesting a role of D2‐like receptors in feeding behavior [104]. Interestingly, when these investigators gave eticlopride daily for 2 weeks, eticlopride continued to decrease food intake (i.e., no tolerance developed to this effect), despite the likely compensatory increase in D2‐like receptor densities [105]; also see Czoty et al. [106] for chronic raclopride effects on D2‐like receptor availability.
The original animal model that differentiated chlorpromazine from other drugs was a signaled shock avoidance paradigm first established by Macht [107]; see Pickens [13] for a description. The avoidance model, including modifications to the paradigm [108], has proven to be one of the most reliable and valid animal models of neuroleptic activity [109]. The behavioral profile for neuroleptic drugs is characterized by a drug‐induced reduction in avoidance responding, but no effect on escape latency. This is different from other drugs that sedate the animals, in which case avoidance and escape responding are equally affected. This paradigm is highly predictive of clinical efficacy, with a high correlation between clinical dose and dose that affects avoidance responding [110]. Surprisingly, there is only one publication that we are aware of involving the behavioral effects of eticlopride on avoidance responding [111]. The procedure was a modification of the classic conditioned‐avoidance response paradigm such that rats held down a lever until an auditory stimulus was presented at which time they had 500 ms to release the lever [112]. Under these conditions, eticlopride disrupted avoidance responding by increasing the latency to hold down the lever; there was no effect on latency to escape once the shock was presented [111]. The effective doses of eticlopride were 0.01 and 0.05 mg/kg (s.c.). Consistent with D2‐like receptor affinity, haloperidol was less potent than eticlopride under these conditions [112].
Of particular significance in evaluating neuroleptics are EPS, including blockade of drug‐induced stereotypy [71, 113]. High doses of DA antagonists in rodents can produce profound immobility, referred to as catalepsy [114]. Using doses that did not induce such profound effects, Fowler et al. have developed a model that assesses the ability of drugs to induce “microcatalepsy”[115]. For these experiments, lever‐press responding by water‐restricted rats was maintained by water reinforcement; photobeams measured the duration of time the rats' muzzles were in the dipper well. Immobility was assessed by increases in muzzle entry duration and was sensitive to neuroleptic drugs [115]. As it relates to eticlopride, Fowler and Liou [116] found that eticlopride as well as haloperidol and raclopride, all decreased operant responding and increased maximum muzzle entry duration following 3 weeks of chronic treatment. However, the drugs had different behavioral profiles over the course of the treatment. Haloperidol and raclopride showed increased sensitivity in both disruption of operant responding and in induction of microcatalepsy over the 3 weeks of treatment. In contrast, eticlopride had no effects on either measure on day 1, up to doses of 0.16 mg/kg (i.p.). However, at this high dose, sensitization to the behavioral effects of eticlopride occurred over the 3 weeks of treatment. This finding suggested that the tendency of eticlopride to affect movement occurred only with repeated dosing. Unlike the first generation neuroleptics, clozapine administration in this study resulted in decreases in operant responding without producing microcatalepsy, suggesting that this atypical antipsychotic has a low EPS profile.
A final set of studies relates the effects of eticlopride and other typical and atypical antipsychotics on learning, memory, and cognition. While these studies did not directly assess the “antipsychotic” potential of drugs, they are of clear relevance in understanding the role of D2‐like receptors in cognitive impairments, including the poor decision making that is characteristic of schizophrenia. Guarraci et al. [117] used a Pavlovian fear conditioning paradigm and central administration of eticlopride to better understand the role of D2‐like receptors within the amygdala on acquisition and expression of this conditioned fear response. Rats received three Pavlovian conditioning trials. Each trial began with the presentation of a tone (a conditioned stimulus; CS); the offset of the tone was followed by a footshock (an unconditioned stimulus; US). A test session was conducted on the fourth trial—the conditioned response (CR) was freezing in response to the tone. Four groups were studied: (i) eticlopride administered directly into the amygdala before each acquisition trial and retention test; (ii) saline before both conditions; (iii) eticlopride before each acquisition trial and saline before the test trial; and (iv) saline before each acquisition trial and eticlopride before the test trial. Guarraci et al. [117] found no effect of eticlopride on the acquisition of the CR. That is, irrespective of whether rats received eticlopride or saline before the CS presentation, both groups showed increases in freezing (i.e., CR) across training trials. However, during retention testing, rats from groups 1 and 3 (i.e., those that received eticlopride before the CS presentation during acquisition) demonstrated less freezing compared to rats from group 2 (saline‐saline). These findings, using eticlopride as a tool to assess D2‐like receptor function, indicate that D2‐like receptors within the amygdala may contribute to associative learning during Pavlovian fear conditioning.
Another behavioral paradigm that is sensitive to pharmacological manipulations of the striatum is the conditioned reaction‐time paradigm [118]. In this paradigm, rats are trained to hold down a lever until a visual stimulus is illuminated (a CS); rats have to release the lever within 700 ms to receive a food pellet reinforcer. Reaction time was defined as the elapsed time between the presentation of the CS and the release of the lever. Eticlopride (0.005–0.02 mg/kg, s.c.) produced dose‐dependent increases in the number of incorrect responses (i.e., incorrect because the rat either released the lever before the CS or held it longer than 700 ms and then released it). There are several possible explanations to account for the findings, including motor impairment induced by eticlopride or attentional deficits leading to poor performance. The investigators concluded that D2‐like receptors appear to be important for accurate processing of sensorimotor information [118].
Clinical Implications and Future Directions
The selective DA D2‐like receptor antagonist eticlopride currently has no clinical indications and has never been tested in humans as a potential antipsychotic medication. However, the very closely related compound raclopride has been examined in clinical trials for treatment of schizophrenia and psychosis and was found to be well tolerated and to have similar efficacy to haloperidol with reduced occurrence of EPS [50, 119, 120, 121, 122, 123]. The discovery of “atypical” or “second‐generation” antipsychotics, such as clozapine, risperidone, olanzapine, quietiapine, sertindole, and ziprasidone beginning in the 1990s has led to clinical efficacy with rare occurrence of EPS. However, because the causes of schizophrenia remain unknown, these medications are only attempting to eliminate the symptoms of schizophrenia (see [2]). Thus, drugs such as eticlopride will remain in use as tools to assess the DA system. Despite the lack of testing in the clinical realm, eticlopride has been a valuable tool in the preclinical evaluation of DA receptor physiology and pharmacology. Our furthered understanding of the physiological underpinnings of DA D2‐like receptors depends on continued advancement of the tools available—technological, pharmacological, and otherwise. As such, the development of high‐affinity ligands for D2‐like receptors, including eticlopride and newer generation substituted benzamides, has greatly aided our capacity to study these receptors. Since its introduction, eticlopride has been used to elucidate several of the numerous contributions of DA D2‐like receptors ranging from the role in the neural circuitry of reward to the role in supraspinal analgesia, and it is findings such as these that allow the outcomes of preclinical studies to guide pharmacotherapy design and the direction of clinical pursuits. For example, results from imaging studies conducted throughout a course of treatment with known high‐efficacy antipsychotics associated with a low occurrence of EPS could provide valuable information about specific binding of the drug and fluctuations in DA receptor function that could guide future drug development.
Inevitably, there are clear gaps in our knowledge that can only be addressed with additional research. For example, all of the animal models described in this review used adult subjects. However, a recent study of US trends noted that the individuals aged 20 years or younger had increased frequency of office visits, including prescription of antipsychotic medication, from approximately 201,000 in 1993–1995 to approximately 1,224,000 in 2002 [124]. Second‐generation antipsychotics are most often prescribed to young people despite not being FDA approved for use in pediatric patients [124]. What are the long‐term consequences of such treatment? Do adolescent DA receptors respond in a manner similar to those of adults treated chronically with antipsychotic medications? In this young population, how do the second‐generation antipsychotics compare to the older neuroleptics? Clearly, these are critically important avenues of future research, which can be addressed through the use of animal models and the application of well‐studied dopamineric antagonists such as eticlopride.
As it relates to animal models, there are several concerns that need continued emphasis. Of particular importance is the issue of species differences. While rodent studies have there advantages, there remain questions about differences in DA receptor distribution and function between rodents and primates [125, 126], which may impact our understanding of neuroleptic activity. Studies using eticlopride under similar conditions in rodents and nonhuman primates would provide valuable information related to these questions. This is particularly true with improved PET and MR imaging capabilities allowing the study of both species in the same instrument.
In closing, it is worth noting how valuable a research tool can be even if it has no clinical applicability. The ability to compare across studies and with compounds that have since been developed with greater specificity and efficacy at particular targets, allows for a better understanding of the role of D2‐like receptor subtypes in the etiology, symptomolgy, and prognosis of many DA‐mediated affective disorders.
Conflict of Interest
The authors have no conflict of interest.
Acknowledgments
We thank Amy Hauck Newman for comments on an earlier version of this manuscript. Preparation of this review was supported by F30 DA021920 (JLM) and R37 DA10584, R01 DA17763, and R01 DA12460 (MAN).
References
- 1. Baldessarini RJ, Tarazi FI. Pharmacotherapy of psychosis and mania In: Brunton LL, Lazo JS, Parker KL, eds. Goodman and Gilman's the pharmacological basis of therapeutics. New York : McGraw‐Hill, 2006; 461–500. [Google Scholar]
- 2. NIMH, National Institute of Mental Health (2008) “What is schizophrenia”, http://www.nimh.nih.gov; accessed 26 June 2008.
- 3. Regier DA, Narrow WE, Rae DS, Manderscheid RW, Locke BZ, Goodwin FK. The de facto US mental and addictive disorders service system. Epidemiologic catchment area prospective 1‐year prevalence rates of disorders and services. Arch Gen Psychiatry 1993;50:85–94. [DOI] [PubMed] [Google Scholar]
- 4. Dearry A, Gingrich JA, Falardeau P, Fremeau RT Jr., Bates MD, Caron MG. Molecular cloning and expression of the gene for a human D1 dopamine receptor. Nature 1990;347:72–76. [DOI] [PubMed] [Google Scholar]
- 5. Mansour A, Meador‐Woodruff JH, Zhou QY, Civelli O, Akil H, Watson SJ. A comparison of D1 receptor binding and mRNA in rat brain using receptor autoradiographic and in situ hybridization techniques. Neuroscience 1991;45:359–371. [DOI] [PubMed] [Google Scholar]
- 6. Meador‐Woodruff JH, Mansour A, Civelli O, Watson SJ. Distribution of D2 dopamine receptor mRNA in the primate brain. Prog Neuropsychopharmacol Biol Psychiatry 1991;15:885–893. [DOI] [PubMed] [Google Scholar]
- 7. Monsma FJ Jr., Mahan LC, McVittie LD, Gerfen CR, Sibley DR. Molecular cloning and expression of a D1 dopamine receptor linked to adenylyl cyclase activation. Proc Natl Acad Sci 1990;87:6723–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sibley DR, Monsma FJ Jr. Molecular biology of dopamine receptors. Trends Pharmacol Sci 1992;13:61–69. [DOI] [PubMed] [Google Scholar]
- 9. Lidow MS, Goldman‐Rakic PS, Rakic P, Innis RB. Dopamine D2 receptors in the cerebral cortex: Distribution and pharmacological characterization with [3H]raclopride. Proc Natl Acad Sci 1989;86:6412–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seeman P, Lee T, Chau‐Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature 1976;261:717–719. [DOI] [PubMed] [Google Scholar]
- 11. Wong DF, Wagner HN Jr., Tune LE, Dannals RF, Pearlson GD, Links JM, Tamminga CA, Broussolle EP, Ravert HT, Wilson AA, et al Positron emission tomography reveals elevated D2 dopamine receptors in drug‐naive schizophrenics. Science 1986;234:1558–1563. [DOI] [PubMed] [Google Scholar]
- 12. Seeman P, Bzowej NH, Guan HC, Bergeron C, Reynolds GP, Bird Ed, Riederer P, Jellinger K, Tourtellotte WW. Human brain D1 and D2 dopamine receptors in schizophrenia, Alzheimer's, Parkinson's, and Huntington's diseases. Neuropsychopharmacology 1987;1:5–15. [DOI] [PubMed] [Google Scholar]
- 13. Pickens RW. Behavioral pharmacology: A brief history In: Thompson T, Dews PB, eds. Advances in behavioral pharmacology. New York : Academic Press, 1977; 229–257. [Google Scholar]
- 14. Seeman P, Ulpian C. Dopamine D1 and D2 receptor selectivities of agonists and antagonists. Adv Exp Med Biol 1988;235:55–63. [DOI] [PubMed] [Google Scholar]
- 15. Lowe JA III, Seeger TF, Vinick FJ. Atypical antipsychotics—recent findings and new perspectives. Med Res Rev 1988;8:475–497. [DOI] [PubMed] [Google Scholar]
- 16. Ogren SO, Hogberg T. Novel dopamine‐D‐2 antagonists for the treatment of schizophrenia. ISI Atlas Sci Pharmacol 1988;2:141–147. [Google Scholar]
- 17. Jenner P, Marsden CD. The substituted benzamides—a novel class of dopamine antagonists. Life Sci 1979;25:479–485. [DOI] [PubMed] [Google Scholar]
- 18. Rich TD. Sulpiride: Assessment of a pharmacologically and chemically distinct neuroleptic. Med Hypotheses 1984;14:69–81. [DOI] [PubMed] [Google Scholar]
- 19. Hogberg T. The development of dopamine D2‐receptor selective antagonists. Drug Des Discov 1993;9:333–350. [PubMed] [Google Scholar]
- 20. Ogren SO, Hall H, Kohler C, Magnusson O, Lindbom LO, Angeby K, Florvall L. Remoxipride, a new potential antipsychotic compound with selective antidopaminergic actions in the rat brain. Eur J Pharmacol 1984;102:459–474. [DOI] [PubMed] [Google Scholar]
- 21. De Paulis T, Kumar Y, Johansson L, Ramsby S, Florvall L, Hall H, Ngeby‐Moller K, Ogren SO. Potential neuroleptic agents. 3. Chemistry and antidopaminergic properties of substituted 6‐methoxysalicylamides. J Med Chem 1985;28:1263–1269. [DOI] [PubMed] [Google Scholar]
- 22. Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1987;1:133–152. [DOI] [PubMed] [Google Scholar]
- 23. Wiesel FA, Alfredsson G, Ehrnebo M, Sedvall G. The pharmacokinetics of intravenous and oral sulpiride in healthy human subjects. Eur J Clin Pharmacol 1980;17:385–391. [DOI] [PubMed] [Google Scholar]
- 24. De Paulis T. The discovery of epidepride and its analogs as high‐affinity radioligands for imaging extrastriatal dopamine D(2) receptors in human brain. Curr Pharm Des 2003;9:673–696. [DOI] [PubMed] [Google Scholar]
- 25. Casey DE. Extrapyramidal syndromes in nonhuman primates: Typical and atypical neuroleptics. Psychopharmacol Bull 1991b;27:47–50. [PubMed] [Google Scholar]
- 26. Mohell N, Sallemark M, Rosqvist S, Malmberg A, Hogberg T, Jackson DM. Binding characteristics of remoxipride and its metabolites to dopamine D2 and D3 receptors. Eur J Pharmacol 1993;238:121–125. [DOI] [PubMed] [Google Scholar]
- 27. Vartiainen H, Leinonen E, Putkonen A, Lang S, Hagert U, Tolvanen U. A long‐term study of remoxipride in chronic schizophrenic patients. Acta Psychiatr Scand 1993;87:114–117. [DOI] [PubMed] [Google Scholar]
- 28. Eriksson L. Remoxipride in the treatment of psychoses. Prog Neuropsychopharmacol Biol Psychiatry 1994;18:619–623. [DOI] [PubMed] [Google Scholar]
- 29. Keks NA, Burrows GD. Withdrawal of remoxipride. Med J Aust 1994;160:591. [PubMed] [Google Scholar]
- 30. Philpott NJ, Marsh JC, Gordon‐Smith EC, Bolton JS. Aplastic anaemia and remoxipride. Lancet 1993;342:1244–1245. [DOI] [PubMed] [Google Scholar]
- 31. De Paulis T, Kumar Y, Johansson L, Ramsby S, Hall H, Sallemark M, Ngeby‐Moller K, Ogren SO. Potential neuroleptic agents. 4. Chemistry, behavioral pharmacology, and inhibition of [3H]spiperone binding of 3,5‐disubstituted N‐[(1‐ethyl‐2‐pyrrolidinyl)methyl]‐6‐methoxysalicylamides. J Med Chem 1986;29:61–69. [DOI] [PubMed] [Google Scholar]
- 32. Wagner A, Stensland B, Csoregh I, De Paulis T. Molecular structure and absolute configuration of the hydrochloride of a novel dopamine receptor antagonist: 2S(‐)‐5‐chloro‐3‐ethyl‐N‐[(1‐ethyl‐2‐pyrrolidinyl) methyl]‐6‐methoxysalicylamide. Acta Pharm Suec 1985;22:101–110. [PubMed] [Google Scholar]
- 33. De Paulis T, Hall H, Ogren SO, Wagner A. Synthesis, crystal structure and antidopaminergic properties of eticlopride (FLB 131). Eur J Med Chem 1985;29:273–276. [Google Scholar]
- 34. Blaton NM, Peeters OM, De Ranter CJ. Cryst Struct Comm 1981;10:833. [Google Scholar]
- 35. Carnmalm B, Johansson L, Ramsby S, Stjernstrom NE, Wagner A. Absolute configuration of potentially neuroleptic rigid spiro amines. A study on the topography of the neuroleptic receptor. Acta Pharm Suec 1979;16:239–246. [PubMed] [Google Scholar]
- 36. Tsai RS, Carrupt P‐A, Testa B, Gaillard P, El Tayar N. Effects of solvation on the ionization and conformation of raclopride and other antidopaminergic 6‐methoxysalicylamides: Insight into pharmacophore. J Med Chem 1993;36:196–204. [DOI] [PubMed] [Google Scholar]
- 37. Bouchard G, Pagliara A, Carrupt P‐A, Testa B, Gobry V, Girault HH. Theoreical and experimental exploration of the lipophilicity of zwitterionic drugs in the 1,2‐dichloroethane/water system. Pharmaceut Res 2002;19:1150–1159. [DOI] [PubMed] [Google Scholar]
- 38. Collin S, El tayar N, Van De Waterbeemd H, Moureau F, Vercauteren DP, Durant F, Langlois M, Testa B. QSAR of nortropane‐substituted benzamides: Use of lipophilic (RP‐HPLC) and electronic (1H‐NMR) parameters. Eur J Med Chem 1989;24:163–169. [Google Scholar]
- 39. Norinder U, Hogberg T. QSAR on substituted salicylamides In: Fauchere JL, ed. QSAR: Quantitative struture‐activity relationships in drug design. New York : Alan R. Liss, Inc, 1989;369–372. [PubMed] [Google Scholar]
- 40. De Paulis T, El Tayar N, Carrupt PA, Testa B, Vande Waterbeemd H. Quantitative structure‐activity relationships of dopamine D2 receptor antagonists: A comparison between orthopramides and 6‐methoxysalicylamides. Helv Chem Acta 1991;74:241–254. [Google Scholar]
- 41. Costall B, Naylor RJ. The role of telencephalic dopaminergic systems in the mediation of apomorphine‐stereotyped behaviour. Eur J Pharmacol 1973;24:8–24. [DOI] [PubMed] [Google Scholar]
- 42. Costall B, Naylor RJ. The behavioural effects of dopamine applied intracerebrally to areas of the mesolimbic system. Eur J Pharmacol 1975;32:87–92. [DOI] [PubMed] [Google Scholar]
- 43. Cools ER, Honig WMM, Pijneburg AJJ, Van Rossum JM. The nucleus accumbens of rats and dopaminergic mechanisms regulating locomotor behaviour. Neurosci Lett 1976;3:335. [DOI] [PubMed] [Google Scholar]
- 44. Pijnenburg AJ, Honig WM, Van Der Heyden JA, Van Rossum JM. Effects of chemical stimulation of the mesolimbic dopamine system upon locomotor activity. Eur J Pharmacol 1976;35:45–58. [DOI] [PubMed] [Google Scholar]
- 45. Ljungberg T, Ungerstedt U. Classification of neuroleptic drugs according to their ability to inhibit apomorphine‐induced locomotion and gnawing: Evidence for two different mechanisms of action. Psychopharmacology (Berl) 1978;56:239–247. [DOI] [PubMed] [Google Scholar]
- 46. Florvall L, Ogren SO. Potential neuroleptic agents. 2,6‐Dialkoxybenzamide derivatives with potent dopamine receptor blocking activities. J Med Chem 1982;25:1280–1286. [DOI] [PubMed] [Google Scholar]
- 47. Kohler C, Ogren SO, Haglund L, Angeby T. Regional displacement by sulpiride of [3H]spiperone binding in vivo. Biochemical and behavioural evidence for a preferential action of limbic and nigral dopamine receptors. Neurosci Lett 1979;13:51–56. [DOI] [PubMed] [Google Scholar]
- 48. Hogberg T, Ramsby S, Ogren SO, Norinder U. New selective dopamine D‐2 antagonists as antipsychotic agents. Pharmacological, chemical, structural and theoretical considerations. Acta Pharm Suec 1987;24:289–328. [PubMed] [Google Scholar]
- 49. Ogren SO, Hall H, Widman M, Angeby‐Moller K. Effects of remoxipride's metabolites on dopamine D2 receptors and receptor functions in the rat. Pharmacol Toxicol 1993;73:325–334. [DOI] [PubMed] [Google Scholar]
- 50. Farde L, Wiesel FA, Jansson P, Uppfeldt G, Wahlen A, Sedvall G An open label trial of raclopride in acute schizophrenia. Confirmation of D2‐dopamine receptor occupancy by PET. Psychopharmacology 1988;94:1–7. [DOI] [PubMed] [Google Scholar]
- 51. Kebabian JW, Petzold GL, Greengard P. Dopamine‐sensitive adenylate cyclase in caudate nucleus of rat brain, and its similarity to the “dopamine receptor”. Proc Natl Acad Sci U S A 1972;69:2145–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clement‐Cormier YC, Kebabian JW, Petzold GL, Greengard P. Dopamine‐sensitive adenylate cyclase in mammalian brain: A possible site of action of antipsychotic drugs. Proc Natl Acad Sci U S A 1974;71:1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karobath M, Leitich H. Antipsychotic drugs and dopamine‐stimulated adenylate cyclase prepared from corpus striatum of rat brain. Proc Natl Acad Sci U S A 1974;71:2915–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Iversen LL. Dopamine receptors in the brain: A dopamine‐sensitive adenylate cyclase models synaptic receptors, illuminating antipsychotic drug action. Science 1975;188:1084–1089. [DOI] [PubMed] [Google Scholar]
- 55. Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990;347:146–151. [DOI] [PubMed] [Google Scholar]
- 56. Giros B, Martres MP, Sokoloff P, Schwartz JC. Gene cloning of human dopaminergic D3 receptor and identification of its chromosome. C R Acad Sci III 1990;311:501–508. [PubMed] [Google Scholar]
- 57. Bouthenet ML, Souil E, Martres MP, Sokoloff P, Giros B, Schwartz JC. Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: Comparison with dopamine D2 receptor mRNA. Brain Res 1991;564:203–219. [DOI] [PubMed] [Google Scholar]
- 58. Murray AM, Ryoo HL, Gurevich E, Joyce JN. Localization of dopamine D3 receptors to mesolimbic and D2 receptors to mesostriatal regions of human forebrain. Proc Natl Acad Sci U S A 1994;91:11271–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. List SJ, Wreggett KA, Seeman P. Striatal binding of 2‐amino‐6,7‐[3H]dihydroxy‐1,2,3,4‐tetrahydronaphthalene to two dopaminergic sites distinguished by their low and high affinity for neuroleptics. J Neurosci 1982;2:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Seeman P. Brain dopamine receptors. Pharmacol Rev 1980;32:229–313. [PubMed] [Google Scholar]
- 61. Creese I, Sibley DR, Hamblin MW, Leff SE. The classification of dopamine receptors: Relationship to radioligand binding. Annu Rev Neurosci 1983;6:43–71. [DOI] [PubMed] [Google Scholar]
- 62. Creese I, Snyder SH. Dopamine receptor binding of 3H‐ADTN (2‐amino‐6,7‐dihydroxy‐1,2,3,4‐tetrahydronaphthalene) regulated by guanyl nucleotides. Eur J Pharmacol 1978;50:459–461. [DOI] [PubMed] [Google Scholar]
- 63. Seeman P, Woodruff GN, Poat JA. Similar binding of 3H‐ADTN and 3H‐apomorphine to calf brain dopamine receptors. Eur J Pharmacol 1979;55:137–142. [DOI] [PubMed] [Google Scholar]
- 64. Thal L, Creese I, Snyder SH. 3H‐Apomorphine interactions with dopamine receptors in calf brain. Eur J Pharmacol 1978;49:295–299. [DOI] [PubMed] [Google Scholar]
- 65. Creese I, Padgett L, Fazzini E, Lopez F. 3H‐N‐n‐propylnorapomorphine: A novel agonist ligand for central dopamine receptors. Eur J Pharmacol 1979;56:411–412. [DOI] [PubMed] [Google Scholar]
- 66. Titeler M, Seeman P. Selective labelling of different dopamine receptors by a new agonist 3H‐ligand: 3H‐N‐propylnorapomorphine. Eur J Pharmacol 1979;56:291–292. [DOI] [PubMed] [Google Scholar]
- 67. Hall H, Kohler C, Gawell L. Some in vitro receptor binding properties of [3H]eticlopride, a novel substituted benzamide, selective for dopamine‐D2 receptors in the rat brain. Eur J Pharmacol 1985;111:191–199. [DOI] [PubMed] [Google Scholar]
- 68. Baldessarini RJ, Kula NS, McGrath CR, Bakthavachalam V, Kebabian JW, Neumeyer JL. Isomeric selectivity at dopamine D3 receptors. Eur J Pharmacol 1993;239:269–270. [DOI] [PubMed] [Google Scholar]
- 69. Damsma G, Bottema T, Westerink BH, Tepper PG, Dijkstra D, Pugsley TA, MacKenzie RG, Heffner TG, Wikstrom H. Pharmacological aspects of R‐(+)‐7‐OH‐DPAT, a putative dopamine D3 receptor ligand. Eur J Pharmacol 1993;249: R9‐10 . [DOI] [PubMed] [Google Scholar]
- 70. Griffon N, Pilon C, Sautel F, Schwartz JC, Sokoloff P. Antipsychotics with inverse agonist activity at the dopamine D3 receptor. J Neural Transm 1996;103:1163–1175. [DOI] [PubMed] [Google Scholar]
- 71. Giuliani D, Ferrari F. Involvement of dopamine receptors in the antipsychotic profile of (‐) eticlopride. Physiol Behav 1997;61:563–567. [DOI] [PubMed] [Google Scholar]
- 72. Tang L, Todd RD, O'Malley KL. Dopamine D2 and D3 receptors inhibit dopamine release. J Pharmacol Exp Ther 1994;270:475–479. [PubMed] [Google Scholar]
- 73. Hall H, Sallemark M, Jerning E. Effects of remoxipride and some related new substituted salicylamides on rat brain receptors. Acta Pharmacol Toxicol (Copenh) 1986;58:61–70. [DOI] [PubMed] [Google Scholar]
- 74. Kohler C, Hall H, Gawell L. Regional in vivo binding of the substituted benzamide [3H]eticlopride in the rat brain: Evidence for selective labelling of dopamine receptors. Eur J Pharmacol 1986;120:217–226. [DOI] [PubMed] [Google Scholar]
- 75. Halldin C, Farde L, Hogberg T, Hall H, Sedvall G. Carbon‐11 labelling of eticlopride in two different positions—a selective high‐affinity ligand for the study of dopamine D‐2 receptors using PET. Int J Rad Appl Instrum [A] 1990;41:669–674. [DOI] [PubMed] [Google Scholar]
- 76. Farde L, Ehrin E, Eriksson L, Greitz T, Hall H, Hedstrom CG, Litton JE, Sedvall G. Substituted benzamides as ligands for visualization of dopamine receptor binding in the human brain by positron emission tomography. Proc Natl Acad Sci U S A 1985;82:3863–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kiesewetter DO, Kawai R, Chelliah M, Owens E, McLellan C, Blasberg RG. Preparation and biological evaluation of 18F‐labeled benzamide analogs as potential dopamine D2 receptor ligands. Int J Rad Appl Instrum B 1990;17:347–356. [DOI] [PubMed] [Google Scholar]
- 78. Halldin C, Farde L, Hogberg T, Hall H, Strom P, Ohlberger A, Solin O. A comparative PET‐study of five carbon‐11 or fluorine‐18 labelled salicylamides. Preparation and in vitro dopamine D‐2 receptor binding. Int J Rad Appl Instrum B 1991;18:871–881. [DOI] [PubMed] [Google Scholar]
- 79. Kessler RM, Ansari MS, De Paulis T, Schmidt DE, Clanton JA, Smith HE, Manning RG, Gillespie D, Ebert MH. High affinity dopamine D2 receptor radioligands. 1. Regional rat brain distribution of iodinated benzamides. J Nucl Med 1991;32:1593–1600. [PubMed] [Google Scholar]
- 80. Schmidt DE, Votaw JR, Kessler RM, De Paulis T. Aromatic and amine substituent effects on the apparent lipophilicities of N‐[(2‐pyrrolidinyl)methyl]‐substituted benzamides. J Pharm Sci 1994;83:305–315. [DOI] [PubMed] [Google Scholar]
- 81. Maeda M, Sasaki S, Fukumura T, Fukuzawa E, Watanabe K, Kojima M, Tahara T, Masuda K, Ichiya Y. Positron‐emitting N‐[18F]fluoroalkyl and [18F]fluoropyrrolidinyl analogues of eticlopride as potential in vivo radioligands for dopamine D2 receptors. Chem Pharm Bull (Tokyo) 1992;40:1793–1798. [DOI] [PubMed] [Google Scholar]
- 82. Heiss WD, Herholz K. Brain receptor imaging. J Nucl Med 2006;47:302–312. [PubMed] [Google Scholar]
- 83. Magnusson O, Fowler CJ, Kohler C, Ogren SO. Dopamine D2 receptors and dopamine metabolism. Relationship between biochemical and behavioural effects of substituted benzamide drugs. Neuropharmacology 1986;25:187–197. [DOI] [PubMed] [Google Scholar]
- 84. Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963;20:140–144. [DOI] [PubMed] [Google Scholar]
- 85. Westerink BHC. The effects of drugs on dopamine biosynthesis and metabolism in the brain In: Horn AS, Korf J, Westerink BHC, eds. The neurobiology of dopamine. London : Academic Press, 1979;255–291. [Google Scholar]
- 86. Masserano JM, Weiner N. Tyrosine hydroxylase regulation in the central nervous system. Mol Cell Biochem 1983;5–5:129–152. [DOI] [PubMed] [Google Scholar]
- 87. Tang L, Todd RD, Heller A, O'Malley KL. Pharmacological and functional characterization of D2, D3 and D4 dopamine receptors in fibroblast and dopaminergic cell lines. J Pharmacol Exp Ther 1994;268:495–502. [PubMed] [Google Scholar]
- 88. Chio CL, Lajiness ME, Huff RM. Activation of heterologously expressed D3 dopamine receptors: Comparison with D2 dopamine receptors. Mol Pharmacol 1994;45:51–60. [PubMed] [Google Scholar]
- 89. Bunzow JR, Van Tol HH, Grandy DK, Albert P, Salon J, Christie M, Machida CA, Neve KA, Civelli O. Cloning and expression of a rat D2 dopamine receptor cDNA. Nature 1988;336:783–787. [DOI] [PubMed] [Google Scholar]
- 90. Van Tol HH, Bunzow JR, Guan HC, Sunahara RK, Seeman P, Niznik HB, Civelli O. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 1991;350:610–614. [DOI] [PubMed] [Google Scholar]
- 91. Swarzenski BC, Tang L, Oh YJ, O'Malley KL, Todd RD. Morphogenic potentials of D2, D3, and D4 dopamine receptors revealed in transfected neuronal cell lines. Proc Natl Acad Sci U S A 1994;91:649–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pilgrim C, Stumpf WE. Applications of autoradiography in neurobiological research. J Histochem Cytochem 1987;35:917–928. [DOI] [PubMed] [Google Scholar]
- 93. Kohler C, Radesater AC. Autoradiographic visualization of dopamine D‐2 receptors in the monkey brain using the selective benzamide drug [3H]raclopride. Neurosci Lett 1986;66:85–90. [DOI] [PubMed] [Google Scholar]
- 94. Hall H, Farde L, Halldin C, Hurd YL, Pauli S, Sedvall G. Autoradiographic localization of extrastriatal D2‐dopamine receptors in the human brain using [125I]epidepride. Synapse 1996;23:115–123. [DOI] [PubMed] [Google Scholar]
- 95. Snyder SH, Creese I, Burt DR. The brain's dopamine receptor: Labeling with (3H) dopamine and (3H) haloperidol. Psychopharmacol Commun 1975;1:663–673. [PubMed] [Google Scholar]
- 96. Bracha HS, Kleinman JE. Postmortem neurochemistry in schizophrenia. Psychiatr Clin North Am 1986;9:133–141. [PubMed] [Google Scholar]
- 97. Kleinman JE, Casanova MF, Jaskiw GE. The neuropathology of schizophrenia. Schizophr Bull 1988;14:209–216. [DOI] [PubMed] [Google Scholar]
- 98. Sedvall G, Farde L. Chemical brain anatomy in schizophrenia. Lancet 1995;346:743–749. [DOI] [PubMed] [Google Scholar]
- 99. Bergman J, Kamien JB, Spealman RD. Antagonism of cocaine self‐administration by selective dopamine D1 and D2 antagonists. Behav Pharmacol 1990;1:355–363. [DOI] [PubMed] [Google Scholar]
- 100. Nader MA, Green KL, Luedtke RR, Mach RH. The effects of benzamide analogues on cocaine self‐administration in rhesus monkeys. Psychopharmacology 1999;147:143–152. [DOI] [PubMed] [Google Scholar]
- 101. Winger G. Dopamine antagonist effects on behavior maintained by cocaine and alfentanil in rhesus monkeys. Behav Pharmacol 1994;5:141–152. [DOI] [PubMed] [Google Scholar]
- 102. Ellenbroek BA, Cools AR. Animal models with construct validity for schizophrenia. Behav Pharmacol 1990;1:469–490. [PubMed] [Google Scholar]
- 103. Ellenbroek BA, Cools AR. Animal models for the negative symptoms of schizophrenia. Behav Pharmacol 2000;11:223–233. [DOI] [PubMed] [Google Scholar]
- 104. Pawlowski J, Kmieciak‐Kolada K, Ouchowicz E, Krysiak R, Herman ZS. Effect of substituted benzamides on feeding and hypothalamic neuropeptide Y‐like immunoreactivity (NPY‐LI) in rats. Neuropeptides 2001;35:204–210. [DOI] [PubMed] [Google Scholar]
- 105. LaHoste GJ, Marshall JF. Chronic eticlopride and dopamine denervation induce equal nonadditive increases in striatal D2 receptor density: Autoradiographic evidence against the dual mechanism hypothesis. Neuroscience 1991;41:473–481. [DOI] [PubMed] [Google Scholar]
- 106. Czoty PW, Gage HD, Nader MA. PET imaging of striatal dopamine D2 receptors in nonhuman primates: Increases in availability produced by chronic raclopride treatment. Synapse 2005;58:215–219. [DOI] [PubMed] [Google Scholar]
- 107. Macht DI. Effect of sulfonamides on cerebral and neuromuscular actions. Exp Med Surgery 1943;1:260–272. [Google Scholar]
- 108. Sidman M. Avodiance condioning with brief shock and no exteroceptive warning signal. Science 1953;118:157–158. [DOI] [PubMed] [Google Scholar]
- 109. Cook L, Weidley EF, Morris RW, Mattis PA. Neuropharmacological and behavioral effectgs of “Thorazine” hydrochloride (chlorpromazine). J Pharmacol Exp Ther 1955;113:11–12. [Google Scholar]
- 110. Kuribara H, Tadokoro S. Correlation between antiavoidance activities of antipsychotic drugs in rats and daily clinical doses. Pharmacol Biochem Behav 1981;14:181–192. [DOI] [PubMed] [Google Scholar]
- 111. White IM, Rebec GV. Performance on a lever‐release, conditioned avoidance response task involves both dopamine D1 and D2 receptors. Eur J Pharmacol 1994;253:167–169. [DOI] [PubMed] [Google Scholar]
- 112. White IM, Ciancone MT, Haracz JL, Rebec GV. A lever‐release version of the conditioned avoidance response paradigm: Effects of haloperidol, clozapaine, sulpiride, and BMY‐14802. Pharmacol Biochem Behav 1991;41:29–35. [DOI] [PubMed] [Google Scholar]
- 113. Ferrari F, Giuliani D. Behavioural assessment in rats of the antipsychotic potential of the potent dopamine D2 receptor antagonist, (‐)eticlopride. Pharmacol Res 1995;31:261–267. [DOI] [PubMed] [Google Scholar]
- 114. Sanberg PR, Pisa M, Faulks IJ, Fibiger HC. Experimental influences on catalepsy. Psychopharmacology 1980;69:225–226. [DOI] [PubMed] [Google Scholar]
- 115. Fowler SC, Skjoldageer PD, Liao R‐M, Chase JM, Johnson JS. Distinguishing between haloperidol's and decamethonium's disruptive effects on operant behavior in rats: Use of measurements that complement response rate. J Exp Anal Behav 1991;56:239–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fowler SC, Liou J‐R. Haloperidol, raclopride, and eticlopride induce microcatalepsy during operant performance in rats, but clozapine and SCH 23390 do not. Psychopharmacology 1998;140:81–90. [DOI] [PubMed] [Google Scholar]
- 117. Guarraci FA, Frohardt RJ, Falls WA, Kapp BS. The effects of intra‐amygdaloid infusions of a D2 dopamine receptor antagonist on Pavlovian fear conditioning. Behav Neurosci 2000;114:647–651. [DOI] [PubMed] [Google Scholar]
- 118. Smith AD, Smith DL, Zigmond MJ, Amalric M, Koob GF. Differential effects of dopamine receptor subtype blockade on performance of rats in a reaction‐time paradigm. Psychopharmacology 2000;148:355–360. [DOI] [PubMed] [Google Scholar]
- 119. Cookson JC, Natorf B, Hunt N, Silverstone T, Uppfeldt B. Efficacy, safety and tolerability of raclopride, a specific D2‐receptor blocker, in acute schizophrenia: An open trial. Int Clin Psychopharmacol 1989;4:61–70. [DOI] [PubMed] [Google Scholar]
- 120. Casey DE. A double blind multicentre trial comparing raclopride and haloperidol in schizophrenia. Biol Psychiatry 1991;2 9 (Suppl.):645. [Google Scholar]
- 121. McCreadie RG. A double‐blind comparison of raclopride and haloperidol in the acute phase of schizophrenia. Acta Psychiatr Scand 1992;86:391–398. [DOI] [PubMed] [Google Scholar]
- 122. McEvoy JP, Movin‐Osswald G, Uppfeldt G, Williams T, Dutcher S, Apperson J. An open study of the pharmacokinetics and the tolerability of raclopride extended release capsules in psychiatric patients. Psychopharmacol 1993;112:371–374. [DOI] [PubMed] [Google Scholar]
- 123. Gendron A, Sirois G, Nair NPV, Bloom D, Movin‐Osswald G, Uppfeldt G. An open study of the tolerability and pharmacokinetics of raclopride extended release capsules in psychiatric patients: A Canadian study. J Psychiatry Neurosci 1995;20:287–296. [PMC free article] [PubMed] [Google Scholar]
- 124. Olfson M, Blanco C, Liu L, Moreno C, Laje G. National trends in the outpatient treatment of children and adolescents with antipsychotic drugs. Arch Gen Psychiatry 2006;63:679–685. [DOI] [PubMed] [Google Scholar]
- 125. Berger B, Gaspar P, Verney C. Dopaminergic innervation of the cerebral cortex: Unexpected differences between rodents and primates. Trends Neurosci 1991;14:21–27. [DOI] [PubMed] [Google Scholar]
- 126. Joel D, Weiner I. The connections of dopaminergic system with the striatum in rats and primates: An analysis with respect to the functional and compartmental organization of the striatum. Neuroscience 2000;96:451–474. [DOI] [PubMed] [Google Scholar]