

Abstract
The function of the C5a receptors, C5ar (encoded by C5ar) and C5l2 (encoded by Gpr77), especially of C5l2, which was originally termed a ‘default receptor’, remains a controversial topic. Here we investigated the role of each receptor in the setting of cecal ligation and puncture-induced sepsis by using antibody-induced blockade of C5a receptors and knockout mice. In ‘mid-grade’ sepsis (30-40% survival), blockade or absence of either C5ar or C5l2 greatly improved survival and attenuated the buildup of proinflammatory mediators in plasma. In vivo appearance or in vitro release of high mobility group box 1 protein (HMGB1) required C5l2 but not C5ar. In ‘high-grade’ sepsis (100% lethality), the only protective condition was the combined blockade of C5l2 and C5ar. These data suggest that C5ar and C5l2 contribute synergistically to the harmful consequences in sepsis and that C5l2 is required for the release of HMGB1. Thus, contrary to earlier speculation, C5l2 is a functional receptor rather than merely a default receptor.
The complement anaphylatoxin, C5a, is generated during experimental sepsis and has been shown to play adverse roles in survival after cecal ligation and puncture (CLP)1. C5a is known to mediate its proinflammatory effects via interaction with its rhodopsin-type receptor, C5ar2,3. C5l2 represents a second receptor that binds C5a and its degradation product C5adesArg with high affinity4. Because C5l2 is uncoupled from G proteins and because no clear-cut cellular responses developed after binding of C5a to C5l2 in initial studies, this receptor was postulated to act as a default receptor for C5a and C5adesArg4-6. There are unresolved disagreements as to whether C3a and C3adesarg bind to C5l2 (refs. 5,7,8). Therefore, it also remains unclear whether C3a and C3adesarg might exert their anti-inflammatory effects via interaction with C5l2, which has been thought to have anti-inflammatory properties by nonproductively binding C5a9-12. Recent studies suggest that C5l2 can mediate the biological activities of the complement anaphylatoxins C5a and C3a via mitogen-activated protein kinase (MAPK) activation and that C5l2, as a receptor for C3adesArg, contributes to protein acylation and synthesis of triglycerides in adipocytes7,13. Like C5ar, C5l2 is abundantly expressed on both myeloid and nonmyeloid cells14. Loss of C5l2 on blood neutrophils during sepsis correlates with lethality15. Ina mouse model of acute lung injury, the use of Gpr77-/- mice resulted in enhanced tissue injury, supporting the hypothesis that C5l2 may function as a modulating receptor for C5a and may therefore be anti-inflammatory11. As expected, the genetic deletion of C5ar resulted in protection from acute lung injury, indicating its proinflammatory function16. In the current work, we describe evidence for the combined roles of C5ar and C5l2 in the harmful outcomes of CLP-induced sepsis, including lethality and the surge of proinflammatory mediators in plasma. These data suggest that both C5ar and C5l2 cooperatively play functional parts in the setting of sepsis and that the role of C5l2 is specifically linked to the release of HMGB1, a known key mediator in CLP-induced lethality.
RESULTS
Specificity of antibodies to C5a receptors
Using flow cytometry, we evaluated rabbit polyclonal antibodies to the N-terminal peptide regions of C5ar and C5l2. Antibody to C5ar bound to surfaces of blood neutrophils (PMNs) from wild-type mice (Fig. 1a). When the immunogenic peptide used to raise the antibody to C5ar was added, binding of IgG to PMNs was completely blocked (Fig. 1a). Addition of the C5l2 immunogenic peptide to the C5ar-specific antiserum did not alter the binding of IgG to C5ar (Fig. 1a). Likewise, C5l2-specific antiserum showed binding of IgG to blood PMNs (Fig. 1b). Addition of the immunogenic peptide for C5l2 abolished the IgG binding (Fig. 1b), whereas addition of irrelevant peptide (immunogenic peptide for C5ar) did not affect binding (Fig. 1b). These data define the specificities of the antibodies to C5ar and C5l2.
Figure 1.
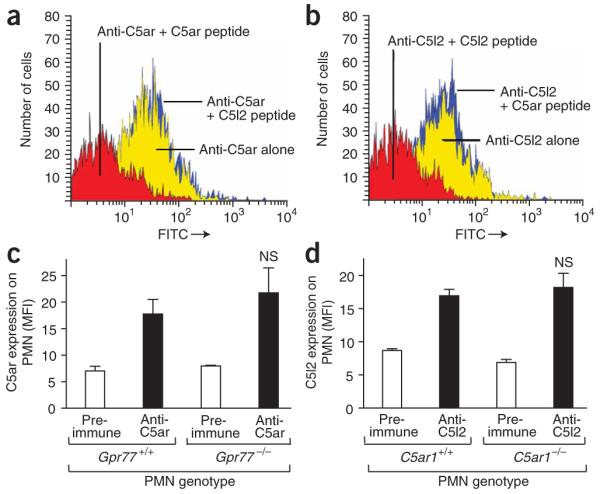
Characterization of antibodies to C5a receptors. (a,b) Binding of rabbit serum IgG to C5ar (a) or C5l2 (b) on mouse blood PMNs, as assessed by flow cytometry. Antisera were pre-incubated with a relevant (red curve) or irrelevant (blue curve) peptide immunogen (100 μg/ml) used to raise the antibodies. (c) C5ar protein expression on blood PMNs from wild-type (Gpr77+/+) mice or Gpr77-/- mice, as assessed by flow cytometry. (d) Expression of C5l2 on PMNs from wild-type (C5ar1+/+) or C5ar1-/- mice. NS, not significant when compared to receptor expression on wild-type PMNs. MFI, mean fluorescence intensity. Studies were done in three separate experiments, with each sample run in duplicate.
In order to address the concern that the absence of C5l2 might be associated with reduced expression of C5ar, we assessed the amount of C5ar on PMNs from either wild-type (Gpr77+/+) or Gpr77-/- mice (Fig. 1c). No quantitative difference in C5ar content was noted on the surface of PMNs from the two groups of mice. Accordingly, when PMNs from C5ar1-/- and wild-type (C5ar1+/+) mice were stained with the antibody to C5l2, C5ar1-/- cells had similar expression of C5l2 on their surfaces as compared to cells from wild-type mice (Fig. 1d). These results suggest that genetic deletion of either C5a receptor does not measurably affect the content of the other receptor on blood PMNs, despite the fact that the genes of both receptors are in close proximity on chromosome 19 (ref. 11).
Roles of C5a receptors in CLP-induced sepsis
In order to define the roles of C5ar and C5l2 in sepsis, we induced CLP in wild-type, C5ar1-/- and Gpr77-/- mice or blocked the C5a receptors by using polyclonal antibodies to the N-terminal peptides of each receptor. In mid-grade CLP (Fig. 2), survival by day 7 was 31% in wild-type mice, 80% in C5ar1-/- mice and 100% in Gpr77-/- mice (Fig. 2a), indicating harmful roles for both C5a receptors in this sepsis model. All wild-type mice survived after antibody-induced blockade of C5ar (Fig. 2b). When we treated wild-type mice with antiserum to C5l2, 80% survived, whereas only 40% of the mice treated with preimmune serum survived. Collectively, these data indicate that both C5ar and C5l2 contribute to the harmful outcomes of CLP-induced sepsis.
Figure 2.
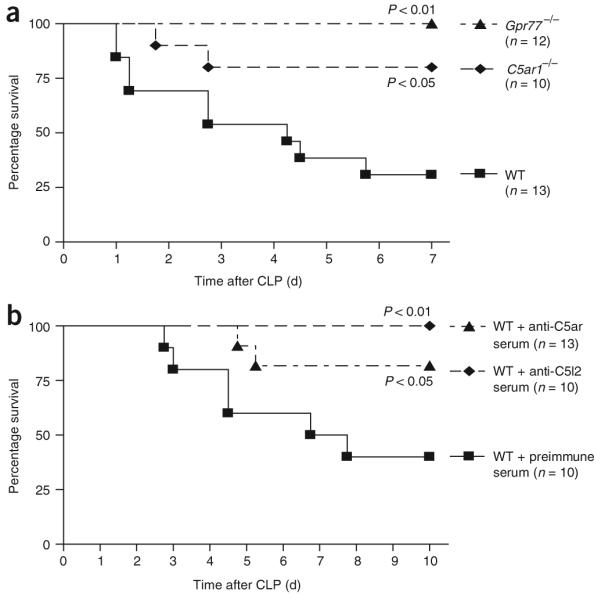
Survival curves for mice after mid-grade CLP. (a) Survival of wild-type (WT) mice, C5ar1-/- mice and Gpr77-/- mice. (b) Survival of WT mice pretreated with preimmune serum or antiserum to C5ar or C5l2 (500 μl each, given intraperitoneally) 12 h before CLP. For each curve, the n value is given, as are the corresponding P values.
In the high-grade form of CLP (Fig. 3), none of the wild-type mice treated with preimmune serum survived by the third day (Fig. 3a). The same outcome occurred in Gpr77-/- mice, whereas in wild-type mice pretreated with antiserum to C5ar, only a single mouse survived beyond day 4 (Fig. 3a). In accordance with these results, C5ar1-/- mice also did not show improved survival after high-grade CLP (Supplementary Fig. 1a online). In striking contrast, the survival rate was 80% in Gpr77-/- mice treated with antiserum to C5ar (Fig. 3a). Likewise, treatment of C5ar1-/- mice with C5l2-specific antiserum greatly improved the outcome after high-grade CLP (survival rate of 87%, as compared to 17% for C5ar1-/- mice treated with preimmune serum or 0% for wild-type mice treated with antiserum to C5l2; Fig. 3a and Supplementary Fig. 1b). Dual blockade of C5a receptors by combined polyclonal antibody pretreatment was also highly protective (Fig. 3b and Supplementary Fig. 1c). However, these protective effects of dual receptor blockade were lost when the antibodies were administered after the CLP procedure (Fig. 3b and Supplementary Fig. 1d-f). Whereas pretreatment with antiserum to both C5l2 and C5ar resulted in a survival rate of 60%, only 20% of mice survived when the antiserum was injected immediately after CLP (Supplementary Fig. 1d). Delayed treatment with antiserum to C5ar and C5l2 at either 12 h or 24 h after CLP did not yield any beneficial effect in terms of improved survival; at most, lethality was modestly delayed (Fig. 3b and Supplementary Fig. 1e,f). These data suggest that the combined blockade of C5a receptors during sepsis is most effective when administered before the onset of sepsis.
Figure 3.

Survival curves for high-grade CLP under various conditions. (a) Survival from severe sepsis in WT mice and Gpr77-/- mice pretreated with either preimmune serum or C5ar-specific antiserum or C5ar1-/- mice treated with antiserum to C5l2 (500 μl each by intraperitoneal injection) 12 h before CLP. (b) Survival curves of WT mice with dual blockade of C5a receptors 12 h prior to or 12 h after sepsis induction by CLP. One milliliter combined antiserum to C5ar and C5l2 or 1 ml of preimmune serum was given by subcutaneous injection at the time points indicated. (c) Combined blockade of C5ar and C5l2 by receptor antagonist. WT mice were injected with 200 μl of 10 μM A8Δ71-73 at the time of CLP followed by subcutaneous injection of A8Δ71-73 at 12 h and 24 h after CLP. Mice in the control group received 200 μl PBS at the same time points. (d) Neutralization of C5a in severe sepsis. C5a was blocked by neutralizing polyclonal antibody to mouse C5a, nonspecific IgG (nsIgG) or C5a-specific antibody given intravenously immediately after the CLP procedure. For each condition, the n numbers and P values are shown in the corresponding panel.
To exclude the possibility that the protective effects shown in Figure 3a,b were a specific phenomenon of polyclonal antibodies, we used two additional protocols. C5ar and C5l2 were blocked in wild-type mice by the C5a mutant A8Δ71-73, which functions as a competitive receptor antagonist for C5ar and C5l2 (ref. 17). Under these conditions, survival in severe sepsis was also greatly improved in comparison to mice treated with PBS only (70%; Fig. 3c). In accordance with these results, injection of Gpr77-/- mice with a monoclonal antibody to mouse C5ar also resulted in an improved outcome (Supplementary Fig. 1g). Because C5ar and C5l2 both have been described to bind C5a with high affinity, we evaluated the effect of neutralization of C5a in our severe model of sepsis (high-grade CLP). When C5a was neutralized by polyclonal antibody to mouse C5a, 60% of mice survived, in contrast to 0% survival in the nonspecific IgG-treated group (Fig. 3d), indicating that C5a acts as the key ligand for both C5ar and C5l2 during sepsis. In summary, in this more severe model of sepsis, both C5ar and C5l2 contribute cooperatively to lethal outcomes.
Using the mid-grade form of sepsis, we screened plasma from wild-type control mice and wild-type, C5ar1-/- and Gpr77-/- mice 24 h after CLP for various proinflammatory cytokines and chemokines (Table 1). In wild-type CLP mice, the abundance of all measured mediators was significantly elevated 24 h after CLP in comparison to control mice (P < 0.01). In the absence of C5ar, the abundance of interleukin-1β (IL-1β), IL-6, macrophage inflammatory protein-1α (MIP-1α) and MIP-2 was significantly lower (P < 0.05). Gpr77-/- mice after CLP showed a similar cytokine pattern, with decreased plasma levels of IL-1β, MIP-1α and MIP-2. Notably, the absence of either C5ar or C5l2 did not affect the amount of TNF-α. In accordance with a previous report18, the plasma concentration of IL-6 was significantly increased in Gpr77-/- CLP mice in comparison to wild-type mice (P < 0.05). In contrast to septic Gpr77-/- mice, plasma IL-6 was greatly suppressed in C5ar1-/- CLP mice when compared to wild-type CLP mice. Taken together, these results indicate that both C5a receptors contribute to the regulation of the inflammatory mediator response in sepsis.
Table 1. Plasma cytokine and chemokine concentrations after CLP (pg/ml).
| CLPa |
||||||
|---|---|---|---|---|---|---|
| Cytokine or chemokine | WT control | WT | C5ar1-/- | Change (%) | Gpr77-/- | Change (%) |
| IL-1β | 34.5 ± 1.22 | 881 ± 199 | 143 ± 8.19* | 84 ↓ | 155 ± 38.3* | 82 ↓ |
| IL-6 | 13.2 ± 1.58 | 2,499 ± 329 | 224 ± 43.9* | 91 ↓ | 3,509 ± 185* | 41 ↑ |
| TNF-α | 19.3 ± 1.62 | 146 ± 36.5 | 155 ± 7.40 | 6 ↑ | 147 ± 10.3 | 0 - |
| MIP-1α | 13.8 ± 0.37 | 243 ± 53.7 | 29.5 ± 3.58* | 88 ↓ | 35.0 ± 8.10* | 86 ↓ |
| MIP-2 | 46.2 ± 6.65 | 18,881 ± 4153 | 5,991 ± 697* | 68 ↓ | 645 ± 137* | 97 ↓ |
n ≥ 5 for each group.
P < 0.05; all P values are based on comparison to WT CLP data.
Link between C5l2 and release of HMGB1
Because HMGB1 is key as a late mediator in sepsis, we included it in our cytokine screening. We detected HMGB1 in mouse plasma by western blotting (Fig. 4a,b). In the blot of control plasma, the bands corresponding to HMGB1 were faint, whereas, 24 h after CLP, intense bands were present from plasma from wild-type mice (Fig. 4a,b). Blockade of C5ar in wild-type CLP mice had no discernible effect on plasma HMGB1 banding patterns, and the same was observed for septic C5ar1-deficient mice (Fig. 4a,b). In contrast, for Gpr77-/- CLP mice, the HMGB1 bands were much weaker (Fig. 4a,b). Densitometry measurements of the HMGB1 bands are shown in Figure 4c,d. ELISA analysis showed that the HMGB1 abundance in plasma from septic Gpr77-/- mice was barely above that in plasma from control mice, whereas HMGB1 abundance in C5ar1-/- CLP mice was comparable to that in septic wild-type mice (Fig. 4e), confirming the patterns found by western blotting (Fig. 4a,b). In order to evaluate whether C5a is the ligand for C5l2 that triggers In vivo HMGB1 release, we assessed the effect of absence of the complement components C3 or C5 and of neutralization of C5a on plasma HMGB1 levels during sepsis. When we induced sepsis in mice lacking the ability to produce C3 (C3-/-; Fig. 4f) or C5 (Hc-/-; Fig. 4g), the plasma HMGB1 concentrations did not differ from those in C3+/+ or Hc+/+ CLP mice. In striking contrast, in wild-type CLP mice treated with an antibody to C5a, HMGB1 abundance was reduced to baseline, whereas septic mice treated with nonspecific IgG showed full HMGB1 expression (Fig. 4h). Although the interpretation of the data using C3-/- or Hc-/- mice is uncertain, because unknown compensatory mechanisms due to genetic deletion might be involved in causing full expression of HMGB1 in these knockout mice, the findings with the neutralizing antibody clearly suggest that C5a acts as the key ligand for C5l2 in sepsis.
Figure 4.

Build-up of HMGB1 in plasma during experimental sepsis. (a,b) Western blots for plasma HMGB1 obtained 24 h after mid-grade CLP from WT mice, WT mice treated with antiserum to C5ar, C5ar1-/- mice or Gpr77-/- mice. (c,d) Densitometry for western blots in a (c) and b (d). (e) ELISA for plasma HMGB1 in CLP-treated WT, C5ar1-/-or Gpr77-/- mice. (f) Plasma HMGB1 levels in C3+/+ or C3-/- mice after CLP. (g) HMGB1 concentration in plasma from CLP-treated Hc+/+ or Hc-/- mice. (h) Plasma HMGB1 concentrations in septic WT mice treated with C5a-specific IgG in comparison to nonspecific IgG-treated mice. Ctrl, healthy (non-septic) wild-type mice. For each condition, n ≥ 5. * P < 0.05 in comparison to WT positive control.
In additional experiments, we incubated peritoneal macrophages from healthy wild-type, C5ar1-/- or Gpr77-/- mice in vitro with culture medium, lipopolysaccharide (LPS) or recombinant mouse C5a. Unstimulated macrophages released little HMGB1 into supernatant fluids, as detected by western blotting (Fig. 5a). Addition of LPS or C5a caused release of HMGB1 from wild-type macrophages, whereas both stimuli induced very little HMGB1 release from Gpr77-/- macrophages (Fig. 5a). As confirmed by ELISA, wild-type and C5ar1-/- macrophages each produced similar amounts of HMGB1 in the presence of C5a, LPS or the combination of LPS and C5a, whereas the HMGB1 release from Gpr77-/- cells was clearly attenuated (Fig. 5b). To investigate the underlying signaling mechanisms, we used various inhibitors of several members of the MAPK cascade, namely MEK1/2, JNK1/2 and p38. Inhibitors for the phosphatidylinositol 3-kinase (PI3K) and Akt pathways were also used in vitro. Macrophages from C5ar1-/- mice, which only express C5l2 as a C5a-binding receptor, showed a robust release of HMGB1 when incubated with C5a and this release was significantly attenuated in the presence of inhibitors of MEK1/2, JNK1/2 and PI3K/Akt as a function of dose (P < 0.05; Fig. 5c and Supplementary Table 1 online). However, inhibition of p38 did not result in reduced HMGB1 secretion (Fig. 5c). These findings suggest that C5l2 functions as a receptor for C5a, with binding resulting in activation of intracellular MAPK (MEK1/2 and JNK1/2) and Akt pathways. Human peripheral blood monocytes (PBMCs) also released HMGB1 in a manner independent of C5AR but sensitive to antibody-induced blockade of C5L2 and the antagonist A8Δ71-73, which blocks both C5AR and C5L217 (Fig. 5d). Of note, not only human PBMCs, but also isolated human PMNs, released HMGB1 in response to recombinant human C5A (Fig. 5e). in vitro HMGB1 secretion from PMNs and PBMCs was suppressed in the presence of a monoclonal antibody to human C5L2 or A8Δ71-73, but was not inhibited by a monoclonal antibody that only blocks C5AR (Fig. 5d,e)19. Collectively, these data suggest that release of HMGB1 by phagocytes requires the participation of C5l2.
Figure 5.

Requirement of C5l2 for the release of HMGB1 in vitro. (a) Western blots for HMGB1 in supernatant fluids from peritoneal macrophages obtained from WT or Gpr77-/- mice. Cells were stimulated with LPS (100 ng/ml) or recombinant mouse C5a (50 nM) for 12 h. (b) ELISA for HMGB1 in supernatant fluids after stimulation of WT, C5ar1-/- or Gpr77-/- peritoneal macrophages with LPS (100 ng/ml), C5a (50 nM) or the combination of LPS and C5a. (c) HMGB1 release by peritoneal macrophages from C5ar1-/- mice in the presence of inhibitors of MEK1/2 (U0126; 100 μM), JNK1/2 (SP600125; 10 μM), p38 (SB203580; 100 μM) or PI3K/Akt (wortmannin; 500 nM). (d,e) HMGB1 ELISA of supernatant fluids after incubation of 5 × 106 human PBMCs with recombinant human C5A for 12 h (d) or after incubation of 5 × 106 human PMNs with recombinant human C5A for 5 h (e). Human C5AR and C5L2 were blocked by monoclonal antibodies (1 μg/ml) or inhibited by the receptor antagonist A8Δ71-73 (350 nM). For each condition, n ≥ 3. * P < 0.05 in comparison to WT positive control.
DISCUSSION
Sepsis is associated with complement activation together with the release of numerous proinflammatory cytokines and chemokines20. In addition to C5ar, C5l2 represents a second high affinity receptor for C5a and C5adesArg. There is still controversy about the expression of C5l2, its ligands and its functional role in biological responses4-8,13. One way in which C5l2 differs from C5ar is in the so-called DRY region of the third intracellular loop (replacement of arginine by leucine), resulting in an uncoupling of C5l2 from G protein interactions6,7. Binding of C5a (and C5adesArg) to C5l2 does not result in increased intracellular calcium, although recent evidence suggests that there may be activation of MAPKs in phagocytes and that interaction of C3adesArg with C5l2 might be associated with protein acylation and triglyceride synthesis in adipocytes6-8,13. It was hypothesized that C5l2 may function as a default or modulating receptor for C5a, competing with C5ar for binding of C5a11,18,21. Because C3a and C3adesarg have been described to be putative ligands for C5l2 and show anti-inflammatory or immunomodulatory properties, it is also conceivable that these effects are mediated through C5l2 (refs. 9-12). Furthermore, in human sepsis, C5L2 is downregulated on the surfaces of PMNs, the extent of which correlates with the harmful outcomes of sepsis15.
In the current study, both C5l2 and C5ar contributed to lethality during sepsis. Their interception or absence of both receptors greatly improved survival (Figs. 2 and 3). Each receptor also contributed to the appearance of proinflammatory mediators in plasma after CLP (Table 1). However, the patterns of change in mediator abundance suggest a complex system regulating the release of these mediators when one of the two C5a receptors is unavailable (for example, divergent effects on IL-6). Plasma IL-6 levels were elevated in septic Gpr77-/- mice, consistently with a previous report in which over-expression of C5l2 resulted in reduced LPS-induced IL-6 production in vitro13. The fact that unavailability of either receptor caused a >60% reduction in plasma levels of mediators suggests that signaling cascades for C5l2 and C5ar might be interlinked in a sequential manner.
Another finding that may explain the protective effects of C5l2 deficiency in CLP mice is linked to reduced plasma abundance of HMGB1 during CLP (Fig. 4). HMGB1, which has previously been described as a transcriptional factor that binds to cruciform DNA, has recently been discovered to function as a proinflammatory cytokine that mediates the immune response22-24. HMGB1 is released from macrophages incubated with LPS, resulting in release of proinflammatory cytokines from these cells22. As a late mediator of sepsis and endotoxin lethality, HMGB1 is increased in the serum of humans with sepsis22. Blockade of HMGB1 has resulted in improved survival after endotoxemia or CLP-induced sepsis in rodents22,25. Unlike other therapeutic approaches in sepsis, inhibition of HMGB1 activity, even if delayed, significantly increases survival, with rodents being protected from development of organ damage and failure26,27. Our data suggest that C5l2 participates in the release of HMGB1 based on In vivo findings in septic Gpr77-/- mice as well as on in vitro stimulation of macrophages with LPS and C5a (Fig. 5). Notably, the HMGB1 responses of Gpr77-/- macrophages to LPS alone were suppressed, indicating that LPS signaling via Toll-like receptor 4 (Tlr4) depends on the integrity of C5l2. It is tempting to speculate that there is cross-talk between C5l2 and Tlr4, as it is known that C5a negatively regulates Tlr4-induced immune responses, and secretion of HMGB1 partially depends on LPS-mediated signaling through the Tlr4-CD14 complex24,28. In a recent report, it was suggested that Tlr4 signaling is mediated by C5ar and C3ar and involves MAPK-nuclear factor-κB activation29. The effect of complement on Tlr4-mediated cytokine release seems to correlate with the degree of complement activation29, and, in turn, Tlr4-induced cytokines upregulate the expression of C5ar and C3ar30. These observations suggest that there is substantial crosstalk between signaling pathways downstream of complement receptors and other receptors of the innate immune system31,32, which may have ramifications for other inflammatory responses and diseases.
As outlined above, the ligands for C5l2 remain a topic of debate. The data in the present study strongly suggest that C5a functions as the key ligand for C5ar and C5l2 in the setting of sepsis, as blockade of C5a, as well as the combined blockade of C5a receptors, greatly improved survival in severe sepsis (Fig. 3). In line with this, neutralization of C5a resulted in suppressed HMGB1 release, indicating that C5a is the main ligand of C5l2 for the regulation of HMGB1 release (Fig. 4h). The fact that HMGB1 abundance in C3-/- and Hc-/- mice did not differ from wild-type CLP mice may mean that in C3-/- and Hc-/- mice, unknown compensatory mechanisms are engaged to maintain the full inflammatory response despite an impairment of complement activation. In binding studies of wild-type and Gpr77-/- PMNs and macrophages, we excluded HMGB1 from being a specific ligand for C5l2 in terms of a positive autocrine feedback mechanism (data not shown). Nevertheless, C5l2 appears to be required for optimal release of HMGB1 in vitro and In vivo. Of note, not only did murine macrophages and human PBMCs secrete HMGB1 in a C5l2-dependent manner, but also PMNs seemed to be an abundant source of HMGB1, in accordance with a previous report33. HMGB1 release by monocytes is known to be regulated by nuclear factor-κB through a nontranscriptional mechanism34. In another report, inhibition of MEK1/2, PI3K or Akt resulted in suppressed HMGB1 secretion by monocytes35. Recently, it has been suggested that C5l2 engages ERK1/2 and Akt in PMNs as an intracellular signaling event13. In accordance with these results, C5a-induced HMGB1 release was dependent on activation of the MAPK and the Akt pathways in our in vitro intervention studies (Fig. 5c). However, at this point it is unclear whether MAPKs are directly activated by C5l2 or whether their activation occurs indirectly as a downstream event of Tlr4 engagement, the signaling pathways of which may crosstalk with the C5a pathway.
In summary, it is unlikely that the sole function of C5l2 is to act as a default receptor for C5a and C5adesArg. Instead, C5l2 seems to contribute to mediator release in the inflammatory response in the setting of experimental sepsis. C5ar and C5l2 both contribute synergistically to the harmful events during sepsis. A maximal beneficial effect can be achieved by the blockade or absence of both receptors, which might have implications on future complement-blocking strategies in the clinical setting of sepsis. These findings indicate a new functional role for C5l2.
METHODS
Induction of sepsis by cecal ligation and puncture
In these studies, we used adult male C57BL/6 (Jackson Laboratories), C5ar1-/-,Gpr77-/-and C3-/- mice (all on a C57BL/6 background11,16) and Hc+/+ and Hc-/- mice (congenic strains B10.D2/oSn and B10.D2/nSn). All studies were performed in accordance with the University of Michigan Committee on Use and Care of Animals. After abdominal midline incision, we ligated the cecum below the ileocecal valve, followed by a single ‘through and through’ perforation (21-gauge needle). Midgrade CLP entailed ligation of half of the cecum (30-40% survival), whereas high-grade CLP involved ligation of three-quarters of the cecum (100% lethality). For blockade of C5ar or C5l2, we gave mice an intraperitoneal injection of 500 μl rabbit antiserum to mouse C5ar or mouse C5l2 (Lampire Biological Laboratories) or preimmune rabbit serum (control; Jackson Immunoresearch) 12 h before the CLP procedure. For dual blockade of C5ar and C5l2, we subcutaneously administered a single injection (1 ml) of mixed (1:1) antiserum to C5ar and C5l2 or 1 ml of preimmune serum either 12 h before CLP or at 0 h, 12 h or 24 h after the CLP procedure. In additional experiments, we injected wild-type mice with 200 μl of a C5a receptor antagonist that blocks C5ar and C5l2 (10 μM; (A8Δ71-73)17 at 0 h, 12 h and 24 h after CLP. Alternatively, we injected Gpr77-/- mice with 100 μg of a monoclonal rat antibody to mouse C5ar (clone 20/70; AbD Serotec)36,37 or monoclonal control rat IgG2b (AbD Serotec). For neutralization of C5a, we injected wild-type mice intravenously with polyclonal rabbit antibody to mouse C5a (40 μg; R&D Systems) or nonspecific rabbit IgG (Jackson Immunoresearch).
Preparation and characterization of antibody to mouse C5ar and C5l2
For the immunization of rabbits, we used mouse C5ar or C5l2 peptides (with the published sequences18) conjugated to keyhole limpet hemocyanin (Lampire Biological Laboratories). Western blot analysis of PMN lysates with the rabbit serum containing polyclonal antibody to mouse C5ar or C5l2 yielded specific bands for each receptor at 45 kDa.
Measurements of plasma mediator concentration
For detection of cytokines and chemokines, we drew blood 24 h after CLP into syringes containing Anticoagulant Citrate Dextrose Solution (Baxter). After centrifugation, we collected plasma for ELISA analysis (R&D Systems).
In vitro incubation of phagocytic cells
Four days after intraperitoneal thioglycolate injection of wild-type, C5ar1-/- and Gpr77-/- mice, we harvested macrophages by peritoneal lavage with PBS. After washing, the purity of cell suspension was >95%. We incubated 5 × 106 macrophages per experimental condition in OPTI-MEM I medium (Gibco Invitrogen) for 12 h at 37 °C with LPS (100 ng/ml; Sigma-Aldrich), recombinant mouse C5a (50 nM; R&D Systems) or the combination of both. To investigate the underlying signaling mechanisms, we preincubated (1 h) C5ar1-/- macrophages with inhibitors of MEK1/2 (U0126, 50-200 μM; Cell Signaling), JNK1/2 (SP600125, 1-100 μM; Calbiochem), p38 (SB203580, 10-100 μM; Calbiochem) or PI3K/Akt (Wortmannin, 0.1-1 μM; Cell Signaling) followed by incubation with recombinant mouse C5a (50 nM). Human leukocytes were separated from whole blood by Ficoll-Hypaque (Nycomed Pharma) gradient centrifugation. After washing of the ‘buffy coat’, we incubated PBMCs (3.5 × 106 cells/ml) with recombinant human C5A (50 nM; R&D Systems) for 12 × h at 37 °C in OPTI-MEM I. We isolated human PMNs as described elsewhere15 and incubated them (5 × 106 cells/ml) for 5 h under the same conditions described above. As indicated in Figure 5, we pre-exposed PMNs and PBMCs to monoclonal antibodies to human C5AR19 or C5L2 (1 μg/ml) or to A8Δ71-73 (350 nM) 1 h before C5A exposure (50 nM)17.
Western blotting and ELISA for HMGB1
We separated equal protein amounts of plasma or supernatant fluids by SDS-PAGE and transferred them onto a PVDF membrane (Amersham). After blocking, we incubated the membranes with HMGB1-specific antibody (1 μg/ml; Abcam). As a secondary antibody, we added horseradish peroxidase-conjugated goat antibody to rabbit IgG (1:10,000; Amersham). For quantification of HMGB1, we used a commercially available ELISA assay (Shino-Test)38.
Analysis of C5ar and C5l2 on blood PMNs
For flow cytometric analysis (BD Pharmingen), we incubated 110 μl rabbit antiserum to mouse C5ar or C5l2 or preimmune rabbit serum (Jackson Immunoresearch) with 110 μl of whole blood from untreated wild-type, Gpr77-/- or C5ar1-/- mice. In order to define antibody specificity, we blocked C5ar or C5l2 antisera by adding the appropriate antigen peptide before analysis. After washing, we incubated cells with phycoerythrin-labeled antibody to rabbit IgG (Invitrogen). Sub-sequently, we lysed erythrocytes and resuspended the leukocytes in a 1% paraformaldehyde solution.
Statistical analysis
All values are expressed as mean ± s.e.m. For analysis of survival curves, we used log-rank and -2 log-rank tests. We analyzed data sets by one-way analysis of variance and compared individual group means using the Tukey multiple comparison test. We considered differences significant when P < 0.05.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by US National Institutes of Health grants GM-29507, GM-61656 and HL-31963 (to P.A.W.), AI-057839 (to J.K.) and HL-69511 (to C.G.) and Deutsche Forschungsgemeinschaft grants HU823/2-2 and HU823/2-3 (to M.H.-L.). We thank B. Schumann and S. Scott for secretarial assistance in preparation of the manuscript.
References
- 1.Czermak BJ, et al. Protective effects of C5a blockade in sepsis. Nat. Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 2.Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature. 1991;349:614–617. doi: 10.1038/349614a0. [DOI] [PubMed] [Google Scholar]
- 3.Gerard C, Gerard NP. C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu. Rev. Immunol. 1994;12:775–808. doi: 10.1146/annurev.iy.12.040194.004015. [DOI] [PubMed] [Google Scholar]
- 4.Cain SA, Monk PN. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-Arg74. J. Biol. Chem. 2002;277:7165–7169. doi: 10.1074/jbc.C100714200. [DOI] [PubMed] [Google Scholar]
- 5.Johswich K, et al. Ligand specificity of the anaphylatoxin C5L2 receptor and its regulation on myeloid and epithelial cell lines. J. Biol. Chem. 2006;281:39088–39095. doi: 10.1074/jbc.M609734200. [DOI] [PubMed] [Google Scholar]
- 6.Okinaga S, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry. 2003;42:9406–9415. doi: 10.1021/bi034489v. [DOI] [PubMed] [Google Scholar]
- 7.Kalant D, et al. The chemoattractant receptor-like protein C5L2 binds the C3a des-Arg77/acylation-stimulating protein. J. Biol. Chem. 2003;278:11123–11129. doi: 10.1074/jbc.M206169200. [DOI] [PubMed] [Google Scholar]
- 8.Kalant D, et al. C5L2 is a functional receptor for acylation-stimulating protein. J. Biol. Chem. 2005;280:23936–23944. doi: 10.1074/jbc.M406921200. [DOI] [PubMed] [Google Scholar]
- 9.Kildsgaard J, et al. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J. Immunol. 2000;165:5406–5409. doi: 10.4049/jimmunol.165.10.5406. [DOI] [PubMed] [Google Scholar]
- 10.Takabayashi T, et al. A new biologic role for C3a and C3a desArg: regulation of TNF-alpha and IL-1 beta synthesis. J. Immunol. 1996;156:3455–3460. [PubMed] [Google Scholar]
- 11.Gerard NP, et al. An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. J. Biol. Chem. 2005;280:39677–39680. doi: 10.1074/jbc.C500287200. [DOI] [PubMed] [Google Scholar]
- 12.Johswich K, Klos A. C5L2—an anti-inflammatory molecule or a receptor for acylation stimulating protein (C3a-desArg)? Adv. Exp. Med. Biol. 2007;598:159–180. doi: 10.1007/978-0-387-71767-8_12. [DOI] [PubMed] [Google Scholar]
- 13.Chen NJ, et al. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature. 2007;446:203–207. doi: 10.1038/nature05559. [DOI] [PubMed] [Google Scholar]
- 14.Ohno M, et al. A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Mol. Immunol. 2000;37:407–412. doi: 10.1016/s0161-5890(00)00067-5. [DOI] [PubMed] [Google Scholar]
- 15.Huber-Lang M, et al. Changes in the novel orphan, C5a receptor (C5L2), during experimental sepsis and sepsis in humans. J. Immunol. 2005;174:1104–1110. doi: 10.4049/jimmunol.174.2.1104. [DOI] [PubMed] [Google Scholar]
- 16.Hopken UE, Lu B, Gerard NP, Gerard C. Impaired inflammatory responses in the reverse arthus reaction through genetic deletion of the C5a receptor. J. Exp. Med. 1997;186:749–756. doi: 10.1084/jem.186.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otto M, et al. C5a mutants are potent antagonists of the C5a receptor (CD88) and of C5L2: position 69 is the locus that determines agonism or antagonism. J. Biol. Chem. 2004;279:142–151. doi: 10.1074/jbc.M310078200. [DOI] [PubMed] [Google Scholar]
- 18.Gao H, et al. Evidence for a functional role of the second C5a receptor C5L2. FASEB J. 2005;19:1003–1005. doi: 10.1096/fj.04-3424fje. [DOI] [PubMed] [Google Scholar]
- 19.Lee H, et al. Human C5aR knock-in mice facilitate the production and assessment of anti-inflammatory monoclonal antibodies. Nat. Biotechnol. 2006;24:1279–1284. doi: 10.1038/nbt1248. [DOI] [PubMed] [Google Scholar]
- 20.Czermak BJ, et al. In vitro and in vivo dependency of chemokine generation on C5a and TNF-α. J. Immunol. 1999;162:2321–2325. [PubMed] [Google Scholar]
- 21.Gavrilyuk V, et al. Identification of complement 5a-like receptor (C5L2) from astrocytes: characterization of anti-inflammatory properties. J. Neurochem. 2005;92:1140–1149. doi: 10.1111/j.1471-4159.2004.02942.x. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 23.Muller S, et al. New EMBO members’ review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J. 2001;20:4337–4340. doi: 10.1093/emboj/20.16.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 25.Suda K, et al. Anti-high-mobility group box chromosomal protein 1 antibodies improve survival of rats with sepsis. World J. Surg. 2006;30:1755–1762. doi: 10.1007/s00268-005-0369-2. [DOI] [PubMed] [Google Scholar]
- 26.Yang H, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin S, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J. Exp. Med. 2006;203:1637–1642. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawlisch H, et al. C5a negatively regulates Toll-like receptor 4-induced immune responses. Immunity. 2005;22:415–426. doi: 10.1016/j.immuni.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, et al. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110:228–236. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koleva M, et al. Induction of anaphylatoxin C5a receptors in rat hepatocytes by lipopolysaccharide in vivo: mediation by interleukin-6 from Kupffer cells. Gastroenterology. 2002;122:697–708. doi: 10.1053/gast.2002.31883. [DOI] [PubMed] [Google Scholar]
- 31.Kohl J. The role of complement in danger sensing and transmission. Immunol. Res. 2006;34:157–176. doi: 10.1385/IR:34:2:157. [DOI] [PubMed] [Google Scholar]
- 32.Hawlisch H, Kohl J. Complement and Toll-like receptors: key regulators of adaptive immune responses. Mol. Immunol. 2006;43:13–21. doi: 10.1016/j.molimm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 33.Kim JY, et al. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;288:L958–L965. doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 35.Bonaldi T, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Godau J, et al. C5a initiates the inflammatory cascade in immune complex peritonitis. J. Immunol. 2004;173:3437–3445. doi: 10.4049/jimmunol.173.5.3437. [DOI] [PubMed] [Google Scholar]
- 37.Baelder R, et al. Pharmacological targeting of anaphylatoxin receptors during the effector phase of allergic asthma suppresses airway hyperresponsiveness and airway inflammation. J. Immunol. 2005;174:783–789. doi: 10.4049/jimmunol.174.2.783. [DOI] [PubMed] [Google Scholar]
- 38.Yamada S, Inoue K, Yakabe K, Imaizumi H, Maruyama I. High mobility group protein 1 (HMGB1) quantified by ELISA with a monoclonal antibody that does not cross-react with HMGB2. Clin. Chem. 2003;49:1535–1537. doi: 10.1373/49.9.1535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.