

Abstract
It is known that C3 is required for optimal expansion of T cells during acute viral infections. However, it is not yet determined whether T cell responses to intracellular bacterial infections require C3. Therefore, we have investigated the requirement for C3 to elicit potent T cell responses to Listeria monocytogenes (LM). We show that expansion of antigen-specific CD8 and CD4 T cells during a primary response to LM was markedly reduced in the absence of C3 activity. Further studies indicated that, unlike in an influenza virus infection, the regulation of LM-specific T cell responses by C3 might not involve the downstream effector C5a. Moreover, reduced T cell responses to LM was not linked to defective maturation of DCs or developmental anomalies in the peripheral T cell compartment of C3-deficient mice. Experiments involving adoptive transfer of C3-deficient CD8 T cells into the C3-sufficient environment of wild type mice showed that these T cells do not have intrinsic proliferative defects and paracrine source of C3 will suffice for clonal expansion of CD8 T cells in vivo. However, stimulation of purified C3-deficient CD8 T cells by plastic-immobilized anti-CD3 showed that C3 promotes T cell proliferation directly, independent of its effects on antigen-presenting cells. Based on these findings, we propose that diminished T cell responses to LM in C3-deficient mice might be at least in part due to lack of direct effects of C3 on T cells. These studies have furthered our understanding of C3-mediated regulation of T cell immunity to intracellular pathogens.
Introduction
It is known for a long time that complement components form an integral arm of innate immunity (1, 2). However, in recent years, it has become increasingly evident that complement components are also important in both induction and effector phases of adaptive immunity. Specifically, it is well established that B cell activation, antibody production, and some of the effector functions of antibodies require complement (3–5). Therefore, complement acts as a bidirectional link in both afferent and efferent phases of humoral immunity. In addition, complement plays an important role in the clearance of antigen/antibody complexes, and protect against immune complex diseases (6–8). Although most studies on complement have focused mainly on innate and humoral immunity (3–5, 9), there is emerging evidence that complement component C3 promotes T cell responses to viral infections including influenza virus and lymphocytic choriomeningitis virus (LCMV) (10, 11). However, our understanding of mechanisms underlying the regulation of T cell responses by complement is incomplete. Experimental models of infections and transplantation have indicated that C3 might regulate T cell responses by distinct mechanisms in a context-dependent fashion. For example, during influenza virus infection of mice, full activation of CD8 T cells require C5a, in addition to C3 (10, 12). However, in the murine visceral leishmaniasis model, induction of CD8 T cell responses by vaccination is dependent upon natural antibodies and complement-dependent IL-4 production (13). Additionally, complement has been shown to be important in the genesis of autoimmune myocarditis, and localized production of C3 plays a key role in allogeneic T cell-mediated rejection of renal transplants (14).
Experimental infection of mice with Listeria monocytogenes (LM) has provided seminal insights into the mechanisms of innate and adaptive immunity to facultative intracellular bacteria (15). At the cellular level, early killing of LM is dependent upon innate immunity mediated by neutrophils, macrophages, and NK cells, but complete bacterial clearance requires CD8 T cells (16–19). Cytokines TNFα and IFNγ play non-redundant roles in controlling bacterial growth because both TNFα and IFNγ-deficient mice are highly susceptible to LM infection (20–23). Importantly, some of the protective effects of TNFα and IFNγ against LM might be complement dependent (24, 25). Moreover, it has been reported that LM activates complement, and complement receptor 3 (CR3) is important for phagocytosis and killing of LM by activated macrophages in vitro (24, 26, 27). Therefore, it is possible that CR3-dependent phagocytosis could be an important step in antigen processing and presentation to T cells during an LM infection. However, the role of complement component C3 in the elicitation of T cell responses in the context of an intracellular bacterial infection has not been examined. Here, we have determined the requirement for C3 and C5a in the induction of antigen-specific CD8 and CD4 T cell responses to LM in mice. These studies show that activation and full expansion of CD8 and CD4 T cells during a primary LM infection requires C3 but not C5a. To understand the mechanisms underlying the regulation of T cell responses by C3, we have investigated: 1) the effect of C3 deficiency on the numbers of T cells, B cells, and dendritic cells (DCs) in spleen prior to infection; 2) whether C3 deficiency influences LM-induced maturation of DCs in vitro and in vivo; 3) the importance of autocrine and paracrine sources of C3 in driving clonal expansion of CD8 T cells; 4) whether C3 promotes TCR signaling-induced proliferation of CD8 T cells. These studies further our understanding of the role of C3 in regulating T cell responses to intracellular bacteria, and have significant implications in vaccine development and treatment of T cell-dependent immunopathology.
Materials and Methods
Mice
The C3-deficient (C3−/−) and C5a receptor-deficient (C5aR−/−) mice on the C57BL/6 background were kindly provided by Drs. Rick Wetsel and Craig Gerard respectively (28, 29). Control wild type (+/+) C57BL/6 mice were either littermates or purchased from National Cancer Institute (Bethesda, MD). Congenic C57BL/6 mice (Ly5.1) were purchased from Jackson labs (Bar Harbor, ME). All animal experiments were performed as per institutional animal care guidelines.
LM Infection
The recombinant Listeria monocytogenes that expresses the GP33–41 epitope (rLM/GP33) or nucleoprotein (rLM/NP) of LCMV were generated by Dr. H. Shen (University of Pennsylvania School of Medicine, Philadelphia, PA) (30) . Mice were infected with 5×104 CFU of rLM/GP33 or rLM/NP by intravenous injection. Bacterial load in tissues was quantitated by plating tissue homogenates on brain-heart infusion (BHI) agar plates (30).
DC propagation
Bone marrow-derived DCs were generated as previously described (31). Briefly, bone marrow cells were collected from the tibias and femurs of +/+ or C3−/− mice. After RBC lysis, cells were resuspended at a concentration of 106/ml and plated in 6-well plates in RPMI 1640 media containing 10% FBS and 20 ng/ml of murine GM-CSF (PEPROTECH Inc, NJ). DCs were cultured for 6 days at 37C in 5% CO2 and half of the media containing GM-CSF was replaced on days 2 and 4. On day 6, DCs were harvested for infection with LM and T cell priming assay as described below.
Induction of dendritic cell maturation by LM infection
Bone marrow-derived DCs (105/ml) were plated onto 48-well plates and infected with rLM/GP33 or rLM/NP (multiplicity of infection [MOI]=1). LM infection was terminated 4 hrs later by adding chloramphenicol (10 µg/ml). To examine LM infection-induced maturation of DCs, cell surface expression of CD80, CD86, and MHC II was compared between un-infected and LM-infected DCs at 4 days after infection by flow cytometry.
In vitro T cell priming assay
Naive TCR transgenic CD8 T cells were activated in vitro with LM-infected bone marrow-derived DCs as described elsewhere (32). Bone marrow-derived DCs were infected with rLM/GP33 as described above. As controls, uninfected DCs were coated with the MHC I-restricted epitope GP33 peptide (1.0 ng/ml or 0.1 ng/ml). Following infection with rLM/GP33 or coating with peptide, DCs were incubated for an additional 18 hrs before naive P14 CD8 T cells were added to the culture.
Naive P14 CD8 T cells were purified from the spleens of P14 TCR transgenic mice using T cell enrichment columns (R&D Systems MN) and labeled with carboxyfluorescein succinimidyl ester (CFSE; Molecular Probe, OR). CFSE-labeled purified P14 CD8 T cells were mixed with LM-infected or peptide-coated DCs at a ratio of 10 T cells per DC and cultured for 72 hours. After 72 hours of stimulation, CFSE fluorescence of P14 T cells was analyzed by flow cytometry.
LM-induced DC maturation in vivo
+/+ or C3−/− mice were infected with rLM/GP33. At days 3 and 4 after infection, single cell suspensions of splenocytes were prepared after collagenase digestion, as described elsewhere (33). Cells were stained with anti-CD11c, anti-CD11b, anti-CD80, anti-CD86, anti-CD40, anti-Db (MHC I), anti-I-Ab (MHC II), and anti-B220. The alteration in expression of CD80, CD86, CD40, MHC I, and MHC II molecules on subsets of DCs was assessed by flow cytometry.
Adoptive transfer of TCR transgenic P14 CD8 T cells
Ten thousand naïve GP33-specific CD8 T cells purified from the spleens of P14/Ly5.1 TCR transgenic mice were adoptively transferred into congenic +/+/Ly5.2 or C3−/−/Ly5.2 mice. Twenty-four hours after cell transfer, mice were infected with rLM/GP33.
Treatment of mice with C5aR antagonist
As a specific C5aR antagonist (C5aRa), cyclic hexapeptide AcF[OPdChaWR] was prepared and used as described before (34). Mice were injected intraperitoneally with C5aRa at a dose of 1µg of peptide/gram body weight on days 1, 3, and 5 after LM infection.
Quantitation of LCMV-specific CD8 T cells by MHC class I tetramers
Db/GP33 and Kb/GP34 MHC I tetramers were prepared as described before (35). Single cell suspensions of splenocytes were stained with anti-CD8, anti-CD44, and MHC I tetramers for 1 hr at 4C. After staining, cells were fixed in 2% paraformaldehyde and acquired on a FACSCalibur flow cytometer (Becton Dickinson, San Francisco, CA). Flow cytometry data were analyzed using the FlowJo software (TreeStar Inc, OR). FoxP3+ve regulatory CD4 T cells were visualized by staining splenocytes for cell surface CD4 and intracellular FoxP3 using a commercially available kit (eBiosciences, San Diego, CA).
Quantitation of antigen-specific CD4 and CD8 T cells by intracellular staining
Production of IFNγ by epitope-specific CD8 and CD4 T cells was assessed by intracellular cytokine staining as described before (35). Briefly, freshly explanted splenocytes were cultured ex vivo for 5 hrs with the epitope peptide, IL-2, and brefeldin A. After culture, cells were stained for cell surface CD8 or CD4 and intracellular IFNγ using the Cytofix/Cytoperm kit (BD-Pharmingen, La Jolla, CA). Cells were acquired on a FACSCalibur flow cytometer, and data were analyzed using FlowJo software.
Adoptive transfer of CD8 T cells from C3−/− mice into congenic Ly5.1 B6 mice
CD8 T cells were purified from spleens of naïve +/+ or C3−/− mice (Ly5.2) by negative selection using magnetic beads (Milltenyi Biotec). 4.8 × 106 of purified CD8 T cells were adoptively transferred into congenic C57BL/6/Ly5.1 mice by intravenous injection. Twenty-four hours after cell transfer, mice were infected with rLM/GP33 as above.
T cell proliferation assay
CD8 T cells were purified from spleens of +/+ or C3−/− mice by negative selection using magnetic bead separation technique (Milltenyi). 1 × 105 CD8 T cells were stimulated with plate-bound anti-CD3 (10µg/ml) for 48 hours in 96-well round bottom plates (36). Between 24–48 hours, cells were pulsed with 3[H] Thymidine, and T cell proliferation was assessed by measuring thymidine incorporation at 48 hours.
Statistical Analysis
The commercially available software (SYSTAT [Chicago, IL], version 10.2) was used to analyze data.
Results
C3 is required for optimal expansion of CD8 T cells in a LM infection
Here, we examined the requirement for C3 in the activation and expansion of CD8 T cells following infection of mice with an intracellular bacterial pathogen, Listeria monocytogenes (LM). Groups of wild type +/+ and C3−/− mice were infected with rLM/GP33 that expresses the CTL epitope GP33–41 of LCMV. On day 7 post-infection (PI), activation and expansion of CD8 T cells in spleens of +/+ and C3−/− mice was examined by flow cytometry. First, we compared overall CD8 T cell activation between +/+ and C3−/− mice by quantitating the number of activated (CD44hi) CD8 T cells in the spleen. As shown in Fig. 1A, the relative proportion of activated (CD44hi) CD8 T cells in spleen of C3−/− mice was reduced by ~50%, as compared to +/+ mice. The differences were more striking when total number of activated (CD44hi) CD8 T was compared between +/+ and C3−/− mice. Fig. 1B shows that the number of activated but not naive (CD44lo) CD8 T cells was substantially lower (3-fold; P<0.0001) in spleens of C3−/− mice, compared to +/+ mice. These data suggested that optimal activation of CD8 T cells induced by LM infection is dependent upon C3.
Figure 1. Activation of CD8 T cells is compromised in C3-deficient mice.
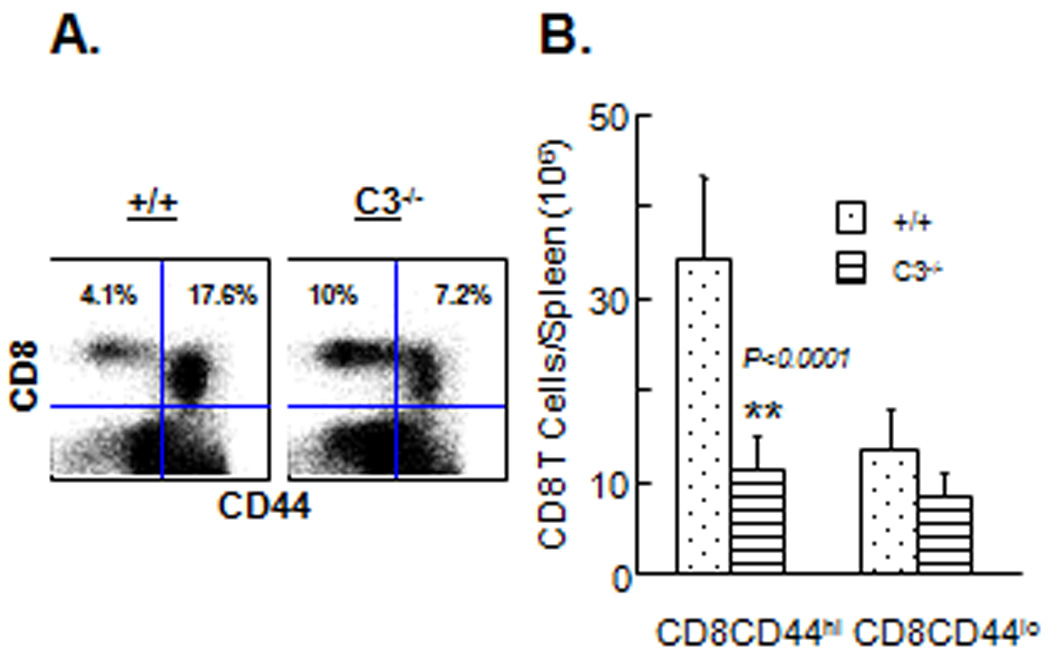
+/+ and C3−/− were infected with rLM/GP33. On the seventh day after infection, splenocytes were stained with anti-CD8 and anti-CD44 antibodies. A. Dot plots are gated on total viable splenocytes and the numbers are the percentages of naive or activated CD8 T cells amongst splenocytes. B. Total numbers of activated (CD44hi) and naïve (CD44lo) CD8 T cells in spleen on day 7 PI. Data are the averages of three to five mice per group ± SD.
Next, we examined the effect of C3 deficiency on activation of antigen-specific CD8 T cells following rLM/GP33 infection. Note that the LCMV CTL epitope GP33–41 present in rLM/GP33 is presented by both Db and Kb MHC I molecules. On day 7 PI, using MHC I tetramers, we enumerated the number of Db/GP33- and Kb/GP34-specific CD8 T cells in spleen of +/+ and C3−/− mice. As illustrated in Fig. 2A, the frequencies of both Db/GP33- and Kb/GP34-specific CD8 T cells were lower in C3−/− mice compared to +/+ mice. Fig. 2B shows that the total numbers of CD8 T cells that are specific to these two CTL epitopes were significantly lower (P=0.001) in spleens of C3−/− mice than in +/+ mice.
Figure 2. Reduced expansion of antigen-specific CD8 T cells in C3-deficient mice.
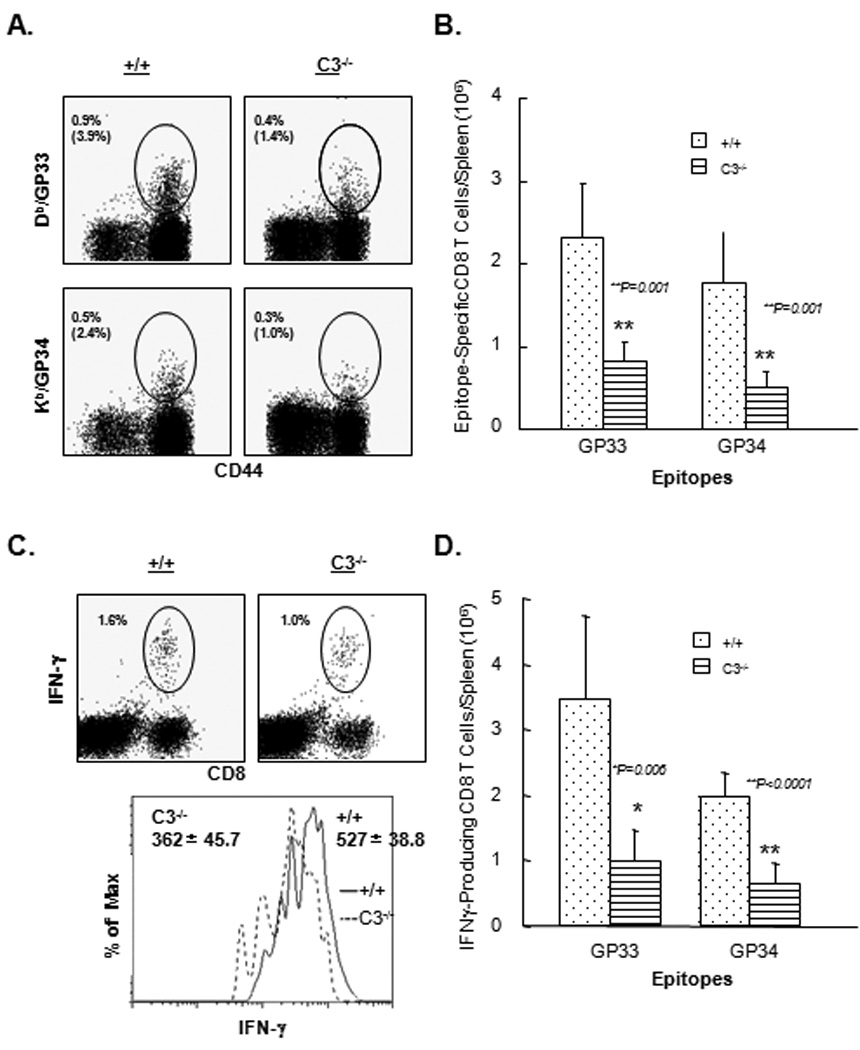
+/+ and C3−/− mice were infected with rLM/GP33, and on day 7 PI, splenocytes were stained with anti-CD8, anti-CD44, and the indicated MHC I tetramers. Dot plots in A are gated on total CD8 T cells, and the numbers represent the percentages of tetramer-binding CD8 T cells of splenocytes; numbers in parentheses are the percentages amongst total CD8 T cells. B. Total number of epitope-specific CD8 T cells in spleen. Data are from 3–5 mice/group ± SD. C. Epitope-specific IFNγ-producing CD8 T cells in spleen of +/+ and C3−/− mice. Splenocytes were stimulated with the indicated LCMV epitope peptides, and IFNγ-producing CD8 T cells were detected by intracellular staining and flow cytometry. The dot plots are gated on total splenocytes and numbers in the dot plots are the percentages of antigen-specific IFNγ producing cells amongst total splenocytes. The FACS histograms are gated on IFNγ-producing CD8 T cells, and numbers are the mean fluorescence intensities (MFI) of staining for IFNγ. D. Total number of epitope-specific IFNγ−producing CD8 T cells in spleen of +/+ and C3−/− mice. Data are the averages of three to five mice/group ± SD.
We also evaluated the effect of C3 deficiency on the cytokine-producing ability of antigen-specific CD8 T cells ex vivo, by intracellular staining for IFNγ. Dot plots of representative mice from each group (Fig. 2C) show antigen-triggered IFNγ production by GP33- and GP34-specific CD8 T cells. Note that stimulation with the GP33 peptide induces IFNγ production by both Db/GP33- and Kb/GP34-specific CD8 T cells. In contrast, stimulation with GP34 peptide only stimulates cytokine production in Kb/GP34-specific CD8 T cells. FACS dot plots in Fig. 2C show that Db/GP33- and Kb/GP34-specific CD8 T cells from both +/+ and C3−/− mice produced readily detectable levels of intracellular IFNγ. Fig. 2C also show that the mean fluorescence intensities (MFI) for IFNγ staining in C3−/− CD8 T cells was lower than +/+ CD8 T cells, which suggested that C3 deficiency also affects the quantity of IFNγ produced by antigen-specific CD8 T cells on a per cell basis. Moreover, consistent with analyses using MHC I tetramers (Fig. 2A and 2B), the frequencies and total number of IFNγ-producing GP33/GP34-specific CD8 T cells were significantly (P<0.006) reduced in spleens of C3−/− mice compared to +/+ mice (Fig. 2C and 2D).
Data in Fig. 2 show the effect of C3 deficiency on primary CD8 T cell responses to recombinant GP33 epitope peptide (in processed form) expressed by LM. It was interesting to examine whether C3 would regulate the processing and presentation of an epitope present in a recombinant protein expressed by LM. Additionally, previous work has shown that C3 dependency of CD8 T cell expansion during an acute viral infection might be epitope specific (11). To address these issues, we compared primary CD8 T cell responses to the other LCMV epitope between +/+ and C3−/− mice in response to rLM/NP that expresses the full-length nucleoprotein (NP) of LCMV. On day 7 PI, we enumerated the number of CD8 T cells that are specific to the CTL epitope NP396–404 using MHC I tetramers or intracellular staining for IFNγ (Fig. 3). Similar to GP33-specific CD8 T cell responses (Fig. 2), optimal activation and expansion of NP396-specific CD8 T cells also requires C3 activity (Fig. 3). These data suggest that C3 promotes CD8 T cell responses to MHC I-restricted epitopes regardless of whether they are expressed in processed or unprocessed form in APCs. Even though CD8 T cell activation was significantly reduced in C3−/− mice, both rLM/GP33 and rLM/NP were cleared from livers in both groups of mice (data not shown) within 7 days PI, which indicated that LM control can occur independently of C3 activity.
Figure 3. Effect of C3 deficiency on generation of CD8 T cell responses to an unprocessed epitope expressed by Listeria monocytogenes.
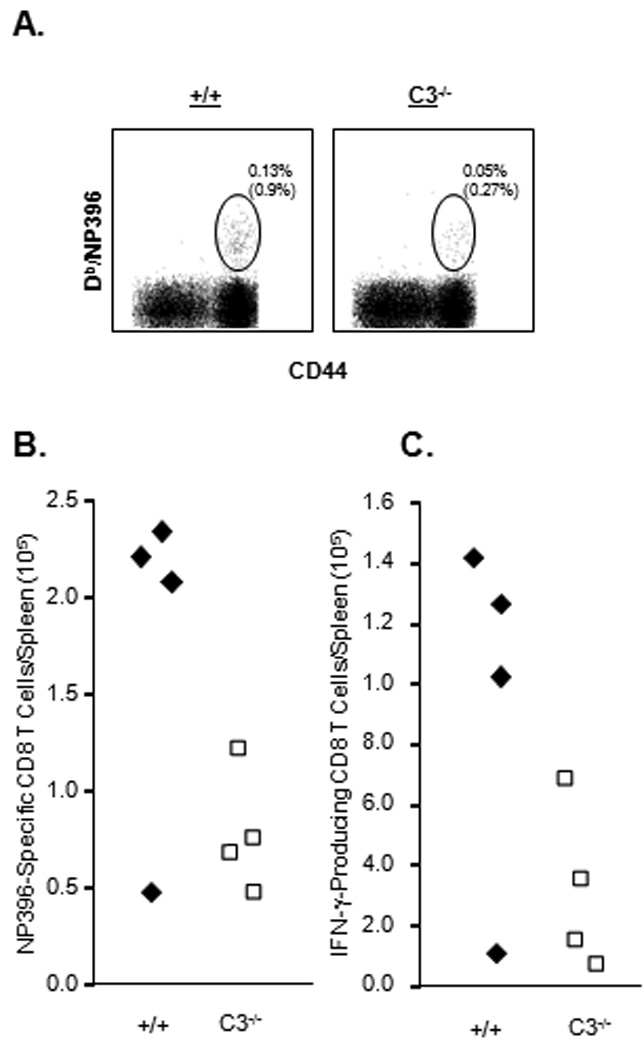
+/+ and C3−/− mice were infected with rLM/NP. On day 7 after infection, the number of CD8 T cells that are specific to the NP396 epitope was determined by staining with MHC I tetramers (A, B) or intracellular staining for IFNγ (C). A. Splenocytes were stained with anti-CD8, anti-CD44, and Db/NP396 tetramers. Dot plots in A are gated on total CD8 T cells, and numbers are the percentages of tetramer-binding cells of splenocytes; numbers in parentheses are percentages amongst total CD8 T cells. B. Absolute numbers of NP396 tetramer positive cells. C. Splenocytes were stimulated with NP396 peptide and number of IFNγ-producing CD8 T cells was determined by intracellular staining. The total number of NP396-specific CD8 T cells in the spleens is shown. Data is from 3–5 mice/group ± SD.
Activation and expansion of CD4 T cells during an LM infection is compromised in C3-deficient mice
LM infection of C57BL/6 mice is known to elicit strong CD8 and CD4 T cell responses (37). Here, we examined the requirement for C3 in CD4 T cell expansion during infection of mice with rLM/GP33 or rLM/NP. First, on day 7 after infection with LM/GP33, we assessed CD4 T cell activation by quantitating the number of activated (CD44hi) CD4 T cells in spleens of +/+ and C3−/− mice. The percentages of activated CD4 T cells in spleens of C3−/− mice were lower than in +/+ mice (Fig. 4A). Remarkably, the total number of activated CD4 T cells in C3−/− mice was ~5-fold lower than in +/+ mice (Fig. 4B); C3 deficiency did not affect the number of naive (CD44lo) CD4 T cells. Similar differences in CD4 T cell activation were observed when +/+ and C3−/− mice were infected with rLM/NP (data not shown). We also quantitated CD4 T cells that are specific to the MHC class II-restricted epitope LLO190–201 present in listeriolysin O (LLO) of LM. Fig. 4C show the total number of LLO190–201-specific CD4 T cells on day 7 after infection with rLM/GP33 or rLM/NP. These data show that expansion of LLO190–201-specific CD4 T cells was significantly lower (P<0.001) in C3−/− mice than in +/+ mice. Collectively, data presented in Fig 1–Fig 4 strongly suggested that full activation and expansion of CD8 and CD4 T cells during an LM infection requires C3 activity.
Figure 4. Activation of CD4 T cells is compromised in C3-deficient mice.
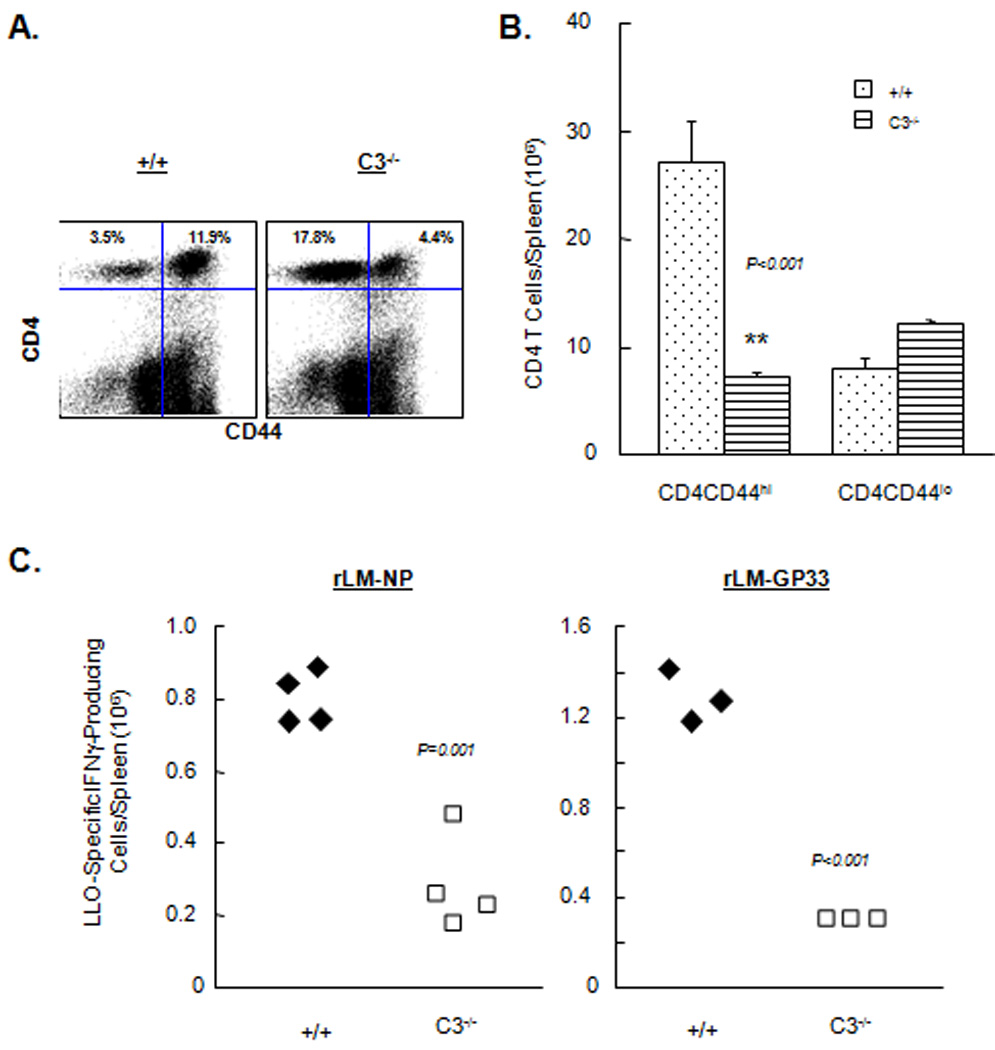
+/+ and C3 −/− mice were infected rLM/GP33 or rLM/NP, and on day 7 PI, activation of CD4 T cells was assessed in the spleen. A and B. Activation of CD4 T cells. Splenocytes were stained with anti-CD4 and anti-CD44 and the number of activated (CD44hi) and naive (CD44lo) CD4 T cells was determined by flow cytometry. Dot plots in A are gated on total viable splenocytes and the numbers are percentages of cells in the respective quadrant of total splenocytes. Data in B are from 3–5 mice/group ± SD. C. Activation of antigen-specific CD4 T cells. Splenocytes from rLM/GP33- or- rLM/NP-infected mice were stimulated ex vivo with the MHC II-restricted peptide LLO190-201, and the number of IFNγ-producing CD4 T cells was determined by intracellular staining. Each symbol in C and D represents data from individual mice.
Requirement of C5aR signaling for CD8 T cell activation in LM infection
In the complement activation cascade, C3 cleavage leads to the generation of C5 convertase, which in turn cleaves C5 into C5a and C5b (38, 39). Among the two cleavage products of C5, C5a is an anaphylatoxin known to exert potent pro-inflammatory effects by binding to its receptor on leukocytes, and also act as a chemoattractant for DCs (38, 40). Receptors for C5a are known to be expressed on T cells (41, 42), and it has been reported that C5aR signaling might be required for optimal CD8 T cell expansion in influenza virus-infected mice (12). However, it is yet to be determined whether C3-dependent augmentation of T cell expansion in a systemic infection like LM also requires C5aR signaling. Therefore, we next examined whether C5a/C5aR interactions are important for activation of CD8 T cell responses in LM infection. The experimental approach was to block C5a/C5aR interactions in vivo by treating LM-infected mice with a C5aR antagonist (C5aRa), which binds specifically to cell surface C5aR. Treatment of mice with C5aRa has been shown to effectively inhibit C5a–dependent activities in vivo (12, 34). Cohorts of rLM/GP33-infected C57BL/6 mice were treated with C5aRa or vehicle PBS during expansion phase of the T cell response to LM. On day 7 PI, the activation and expansion of CD8 T cells was analyzed by flow cytometry as described above. As shown in Fig. 6, the number of activated CD44hi CD8 T cells and GP33-specific CD8 T cells in the spleen of C5aRa-treated mice was comparable to those in control PBS-treated mice. Similar to CD8 T cell responses, C5aRa treatment did not significantly affect the activation of CD4 T cells during an LM infection (Fig. 7). Taken together, data in Fig. 6–Fig. 7 indicated that C5aRa treatment did not significantly affect either the CD8 or CD4 responses to LM infection in mice.
Figure 6. Effect of treatment with C5aR antagonist on CD4 T cell responses to LM in mice.
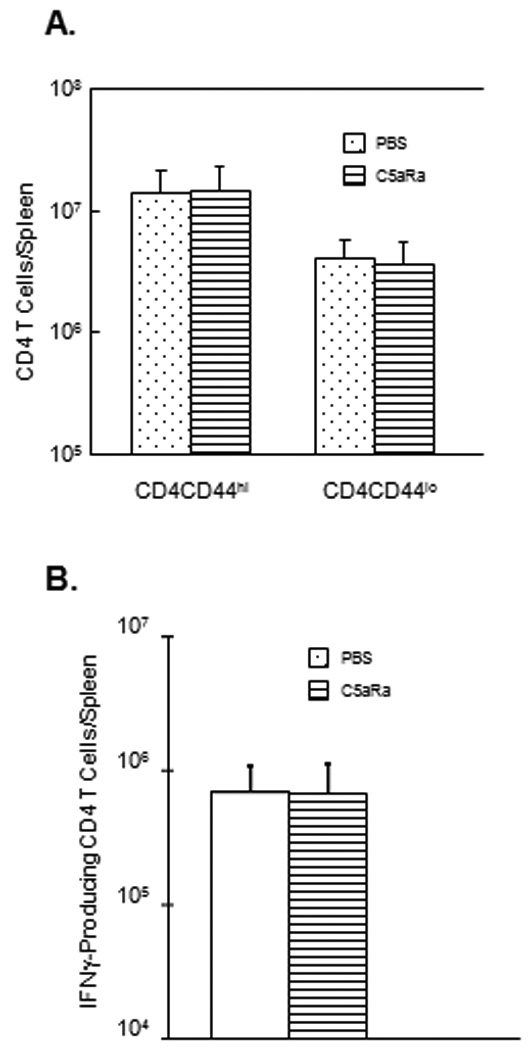
Cohorts of rLM/GP33-infected C57BL/6 mice were treated with vehicle control PBS or C5aRa on days 1, 3, and 5 PI. A. On day 7 PI, the total number of activated (CD44hi) and naïve (CD44lo) CD4 T cells in spleen was determined by flow cytometry. B. Total number of LLO190-specific CD4 T cells in spleen was quantitated by intracellular cytokine staining. Data are derived from 5 mice/group ± SD.
Figure 7. CD8 and CD4 T cell responses to LM in C5aR-deficient mice.
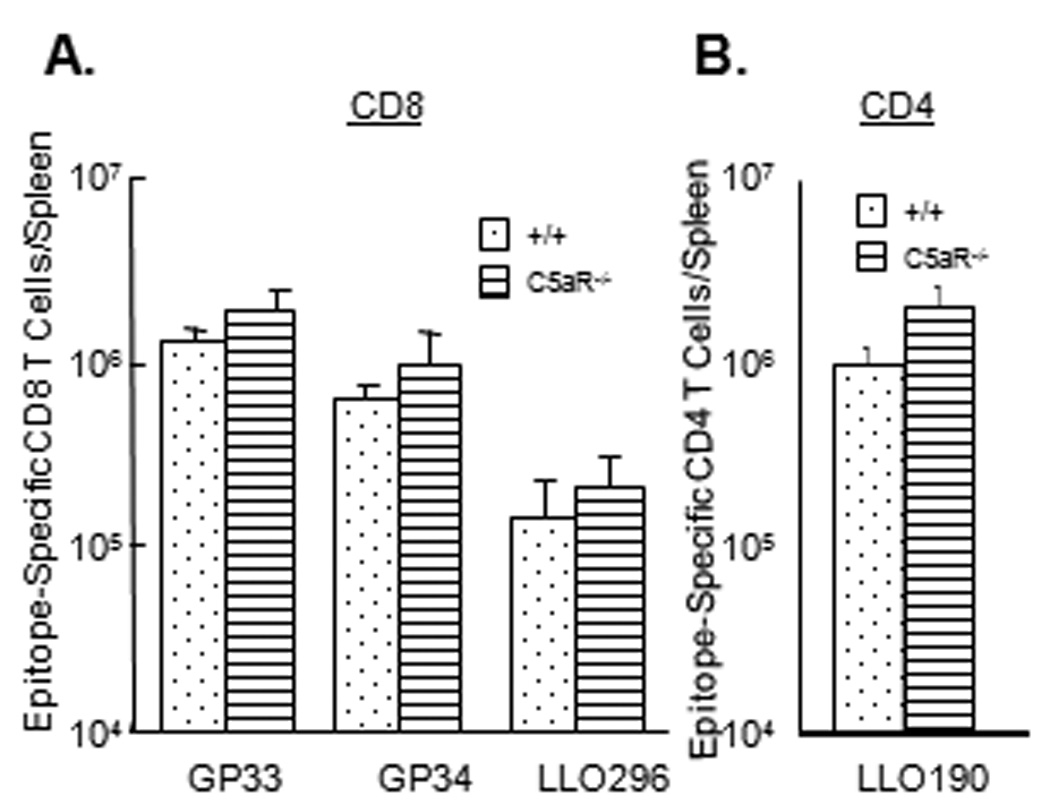
Groups of wild type C57BL/6 (+/+) and C5aR−/− mice were infected with rLM/GP33. On day 7 PI, activation of CD8 T cells (A) and CD4 T cells (B) was assessed in the spleen. A. Splenocytes were stimulated ex vivo with the indicated epitope peptides, and the number of epitope-specific IFNγ-producing CD8 T cells was determined by intracellular staining. B. The number of LLO190-specific CD4 T cells was quantitated by intracellular staining for IFNγ. Data are the mean of 4–5 mice/group ± SD.
CD8 and CD4 T cell responses to LM in C5aR-deficient mice
Although less likely, the inability of C5aRa treatment to inhibit T cell responses (Fig. 6–Fig. 7) might be attributed to the ineffectiveness of blocking the binding of C5a to C5aR. To address this issue, we investigated the role of C5a in promoting T cell expansion by infecting +/+ and C5aR-deficient mice (C5aR−/−) with rLM/GP33. On day 7 PI, we assessed activation of CD8 T cells that are specific to the LCMV epitope GP33 or LM epitope LLO296 in the spleen of +/+ and C5aR−/− mice using intracellular cytokine staining. The expansion of GP33- and LLO296-specific CD8 T cells in the spleen of C5aR−/− mice was comparable to those in +/+ mice (Fig. 8A). Analyses using MHC I tetramers provided similar results for the Db/GP33 epitope of LCMV (data not shown). Data in Fig. 8B show that C5aR deficiency had a minimal impact on the activation and expansion of LLO190-specific CD4 T cells. Thus, studies in C5aR−/− mice provided convincing evidence that C5a/C5aR interactions are dispensable for primary activation and expansion of CD8 and CD4 T cells following a systemic infection with LM.
Figure 8.
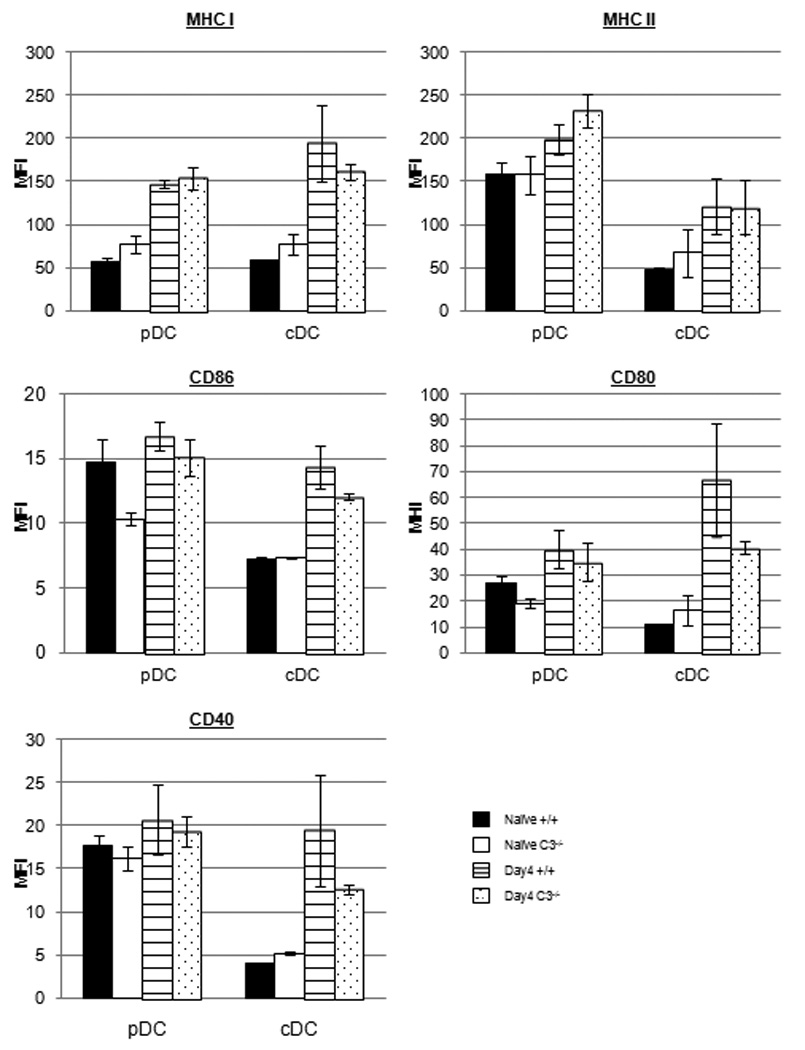
LM-induced maturation of DCs in C3-deficient mice. Groups of wild type (+/+) and C3−/− mice were left uninfected or infected with rLM/GP33. At day 4 after infection, DCs were isolated from spleens and stained with anti-CD11c, anti-B220, anti-Db (MHC I), anti-IAb (MHC II), anti-CD80, anti-CD86, and anti-CD40. Following staining, the expression of MHC I, MHC II, CD80, CD86, and CD40 on plasmacytoid (pDCs; CD11c+veB220+ve) and conventional (cDCs; CD11c+veB220−ve) DCs was assessed by flow cytometry. The data are the mean fluorescent intensities (MFIs) of staining for the indicated molecule from analysis of 3 mice/group.
LM-induced maturation of C3-deficient DCs in vitro and in vivo
Following antigen uptake, under the influence of innate inflammatory signals, immature DCs mature into potent antigen presenting cells and present antigen to naïve T cells in the secondary lymphoid organs. There is evidence that C3 promotes T cell responses by enhancing the antigen presenting function of professional APCs like DCs and macrophages (40, 43–48). Therefore, we hypothesized that reduced T cell responses to LM in C3−/− mice could be due to impairment in maturation of DCs and/or effective antigen presentation to naïve T cells. In order to examine the role of C3 in LM infection-induced maturation of DCs, bone marrow-derived DCs from +/+ or C3−/− mice were infected with rLM/GP33, and upregulation of CD80, CD86, and MHC class II molecules was assessed by flow cytometry. These studies showed that,infected DCs from +/+ mice expressed high levels of all three surface molecules, as compared to uninfected controls (data not shown). Importantly, LM-induced upregulation of CD80, CD86, and MHC II in C3-deficient DCs was comparable to +/+ DCs (not shown), which indicated that C3 is not required for LM infection driven maturation of DCs, in vitro.
Next, we investigated the role of C3 in priming of naïve CD8 T cells by DCs in vitro. CFSE-labeled naïve TCR transgenic CD8 P14 T cells were cultured with uninfected, rLM/GP33-infected, or GP33 peptide-coated DCs, and antigen-driven proliferation of P14 cells was assessed by flow cytometry. These analyses showed that 40 to 46 % of P14 CD8 T cells divided upon exposure to rLM/GP33-infected DCs from +/+ or C3−/− mice. Similarly, 50–53% of P14 CD8 T cells divided when stimulated with +/+ or C3-deficient GP33-coated DCs (not shown). Additionally, only proliferating P14 CD8 T cells present in cultures of LM-infected +/+ or C3−/− DCs upregulated CD44 expression; as expected all P14 CD8 T cells cultured with uninfected DCs maintained their CD44lo (naïve) phenotype (data not shown). These results suggested that the C3 deficiency did not affect the ability of DCs to process and/or present antigens to naïve CD8 T cells in vitro.
Infection with LM has been shown to induce maturation of DCs (49). Therefore, we assessed whether C3 deficiency affected DC maturation induced by LM infection in +/+ and C3−/− mice in vivo. Plasmacytoid and conventional DCs isolated from spleen of LM-infected mice at days 3 and 4 PI were assessed for maturation as above directly ex vivo. The expression levels for MHC I, MHC II, CD40, CD80, and CD86 on plasmacytoid or conventional DCs from naïve/uninfected C3−/− mice were largely similar to those in spleen of +/+ naïve/uninfected mice (Fig. 8). LM infection triggered upregulation in the expression of these molecules, especially on conventional DCs, from both +/+ and C3−/− mice at days 3 (not shown) and 4 PI (Fig. 8). Notably, the levels of MHC I, CD40, CD80, and CD86 expressed on the surface of C3−/− DCs were lower but not significantly (P<0.05) different, as compared to DCs isolated from +/+ mice. These data suggested that C3 deficiency did not significantly affect the maturation of DCs in vivo, following an acute LM infection.
Activation and expansion of wild type monoclonal TCR transgenic CD8 T cells in C3-deficient mice
Next, we tested whether C3-deficient APCs are able to activate and expand wild type antigen-specific CD8 T cells in vivo. We adoptively transferred 104 purified wild type naïve/Ly5.1 GP33-specific TCR transgenic P14 CD8 T cells into congenic +/+/Ly5.2 or C3−/−/Ly5.2 mice, which were subsequently infected with rLM/GP33. Seven days after rLM/GP33 infection, the activation and expansion of donor P14/Ly5.1 CD8 T cells in spleens of +/+/Ly5.2 and C3−/−/Ly5.2 recipient mice was assessed by flow cytometry. As shown in Fig. 9A and 9B, the frequencies and total number of P14/Ly5.1 CD8 T cells in spleens of C3−/− mice tended to be lower, but not significantly different compared to those in +/+ mice. Thus, deficiency of C3 in non-T cells did not significantly compromise the activation and expansion of wild type CD8 T cells following infection with rLM/GP33. These data also suggested that C3−/− APCs are capable of activating wild type CD8 T cells in vivo.
Figure 9.
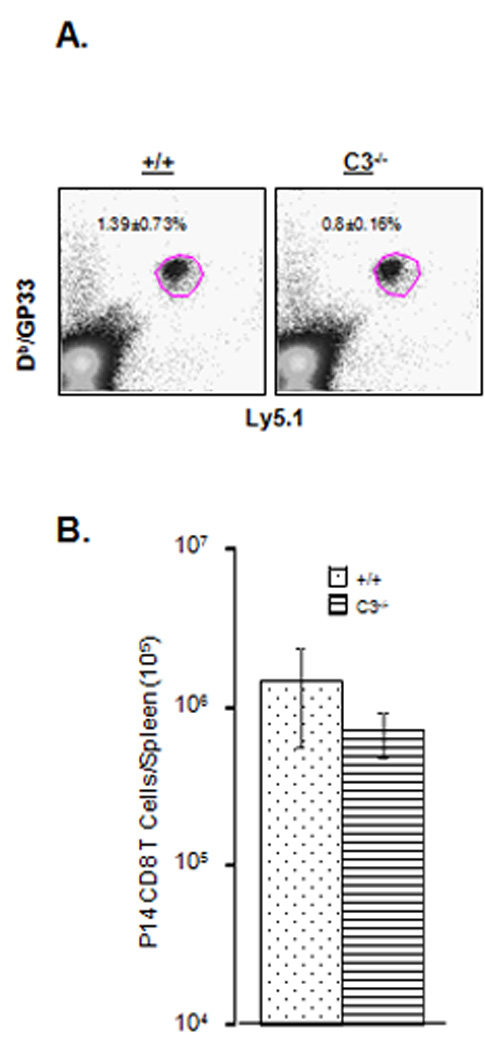
Activation and expansion of adoptively transferred TCR transgenic CD8 T cells in LM-infected C3-deficient mice. Purified naïve GP33-specific P14/Ly5.1 CD8 T cells were adoptively transferred into congenic +/+/Ly5.2 or C3−/−/Ly5.2 mice, which were subsequently infected with rLM/GP33. On the seventh day after infection, splenocytes were stained with anti-CD8, anti-Ly5.1, and Db/GP33 tetramers. The number of Ly5.1+ve tetramer-binding P14 CD8 T cells was quantitated by flow cytometry. Dot plots in A are gated on total splenocytes, and the numbers are the percentages of P14 CD8 T cells among splenocytes. B. Total number of P14 CD8 T cells in +/+ and C3−/− mice. Data in B are from 4 mice/group and representative of two independent experiments.
Characterization of T cells, B cells, and DCs in spleen of naïve uninfected C3−/− mice
It is possible that a defect in the peripheral T cell compartment of C3−/− mice might underlie ineffective activation and expansion of T cells during an immune response. To address this issue, we compared the number of T cells, B cells, and DCs in spleens of uninfected +/+ and C3−/− mice. As shown in Fig. 10A, the numbers of naïve (CD44lo) or activated (CD44hi) phenotype CD8 and CD4 T cells in spleen of C3−/− mice were similar to those in wild type mice. Additionally, the numbers of B cells and FOXp3+ve regulatory CD4 T cells were unaffected by C3 deficiency. Moreover, C3 deficiency did not cause detectable alterations of DC subsets in spleen (Fig. 10B). Taken together, these data suggested that C3 deficiency did not affect the number of T cells, B cells, or DCs in the periphery. Therefore, lower CD8 and CD4 T cell responses to LM cannot be linked to a deficiency in the number of T cells or DCs in C3−/− mice, prior to infection.
Figure 10.
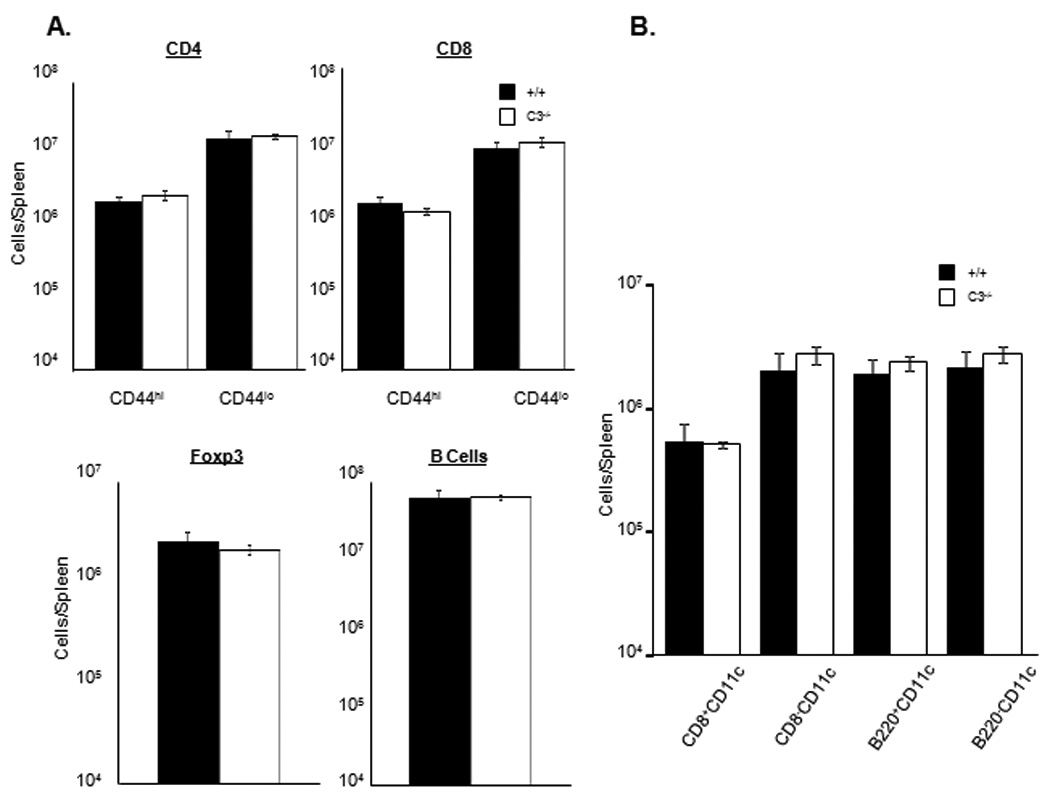
Characterization of T cells, B cells, and DCs in spleen of uninfected C3-deficient mice. A. Single cell suspensions of splenocytes from uninfected +/+ or C3−/− mice were stained with anti-CD8, anti-CD4, anti-CD44, anti-CD19, or anti-B220 antibodies. The numbers of naïve (CD44lo) or activated/memory (CD44hi) phenotype CD4 or CD8 T cells, and CD19+veB220+ve B cells were enumerated by flow cytometry. To stain for regulatory T cells, splenocytes were stained for cell surface CD4 and intracellular Foxp3 using a commercially available kit (e-bioscience, San Diego, Ca). B. Splenocytes were stained with anti-CD11c, anti-CD8, and anti-B220 antibodies. Following staining, the percentages of DC subsets were quantitated by flow cytometry. Data are from 3 mice/group.
Activation and expansion of C3−/− CD8 T cells in a C3-sufficient lymphoid environment of wild type mice
Although the numbers of naïve and activated phenotype T cells in C3−/− mice were similar to those in +/+ mice, it is possible that T cells that have matured in a C3-deficient environment might have intrinsic defects in activation and expansion following antigenic stimulation. Here we tested whether CD8 T cells from C3−/− mice respond to antigenic stimulation in a C3-sufficient environment. We adoptively transferred purified CD8 T cells from C3−/− (Ly5.2) or +/+ (Ly5.2) mice into congenic +/+ Ly5.1 mice, which were subsequently infected with rLM/GP33. At day 7 after infection, we quantitated the activation and expansion of donor Ly5.2+ve and recipient (Ly5.1+ve) GP33-specific CD8 T cells in spleen of LM-infected Ly5.1 mice. LM infection triggered strong activation of GP33-specific CD8 T cells in Ly5.1 recipient mice. The numbers of endogenous Ly5.1+ve GP33-specific CD8 T cells in spleen of mice that were recipients of C3−/− CD8 T cells were similar to those in spleen of mice that were recipients of +/+ CD8 T cells (Fig. 11). Importantly, the activation and expansion of donor Ly5.2+ve C3−/− GP33-specific CD8 T cells was comparable to those of Ly5.2+ve +/+ GP33-specific +/+ CD8 T cells in adoptively transferred recipients. These data illustrated that CD8 T cells from C3−/− mice are not intrinsically defective but are fully capable of antigen-driven activation and expansion in a C3-sufficient environment.
Figure 11.
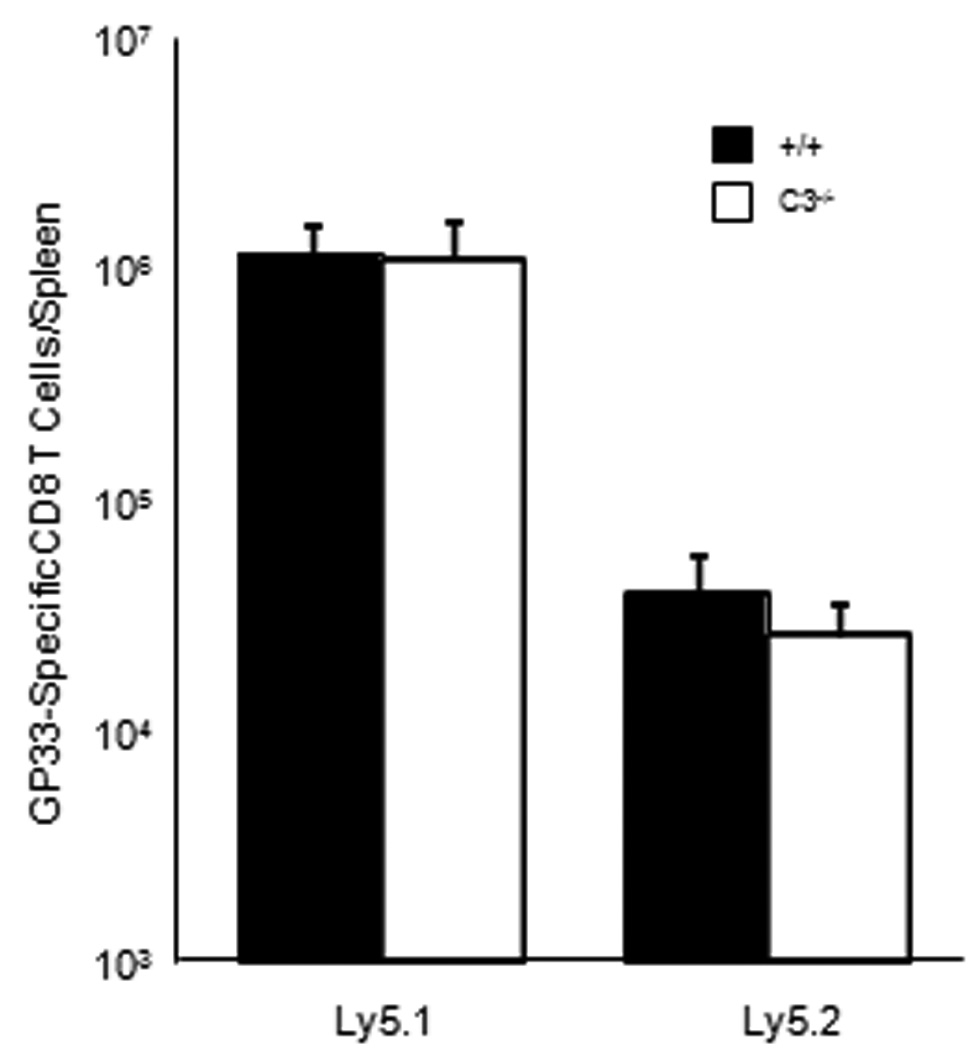
Activation and expansion of adoptively transferred C3-deficient CD8 T cells. CD8 T cells were purified from spleen of +/+ or C3−/− mice by magnetic bead separation (Milltenyi). 4.8 × 106 Ly5.2+ve CD8 T cells from +/+ or C3−/− mice were adoptively transferred into congenic Ly5.1/C57BL/6 mice, which were subsequently infected with rLM/GP33; Ly5.1 mice receiving CD8 T cells from +/+ and C3−/− mice are labeled as +/+ and C3−/− respectively. At day 7 after infection, the numbers of Ly5.1+ve (CD8 T cells of endogenous or recipient origin) and Ly5.2+ve (donor-derived CD8 T cells) GP33-specific CD8 T cells were quantitated in spleen by intracellular cytokine staining for IFN-γ. Data are from 4 mice/group and representative of two independent experiments.
Proliferation of C3-deficient CD8 T cells in vitro
It was of interest to examine whether C3 regulates T cell proliferation directly, independent of its effects on antigen presenting cells. To explore this possibility, we asked whether C3 is required for CD8 T cells to undergo proliferative expansion in vitro upon stimulation via the TCR, independent of the contribution of C3 derived from APCs. As illustrated in Fig. 12, the proliferation of purified C3-deficient CD8 T cells induced by plastic-immobilized anti-CD3 was significantly lower, as compared to +/+ CD8 T cells. These findings show that C3 promotes proliferation of CD8 T cells induced by signaling via the TCR. Taken together, these data suggested that sub-optimal CD8 T cell responses to LM in C3−/− mice might be linked at least in part linked to lack of C3 effects on T cells.
Figure 12.
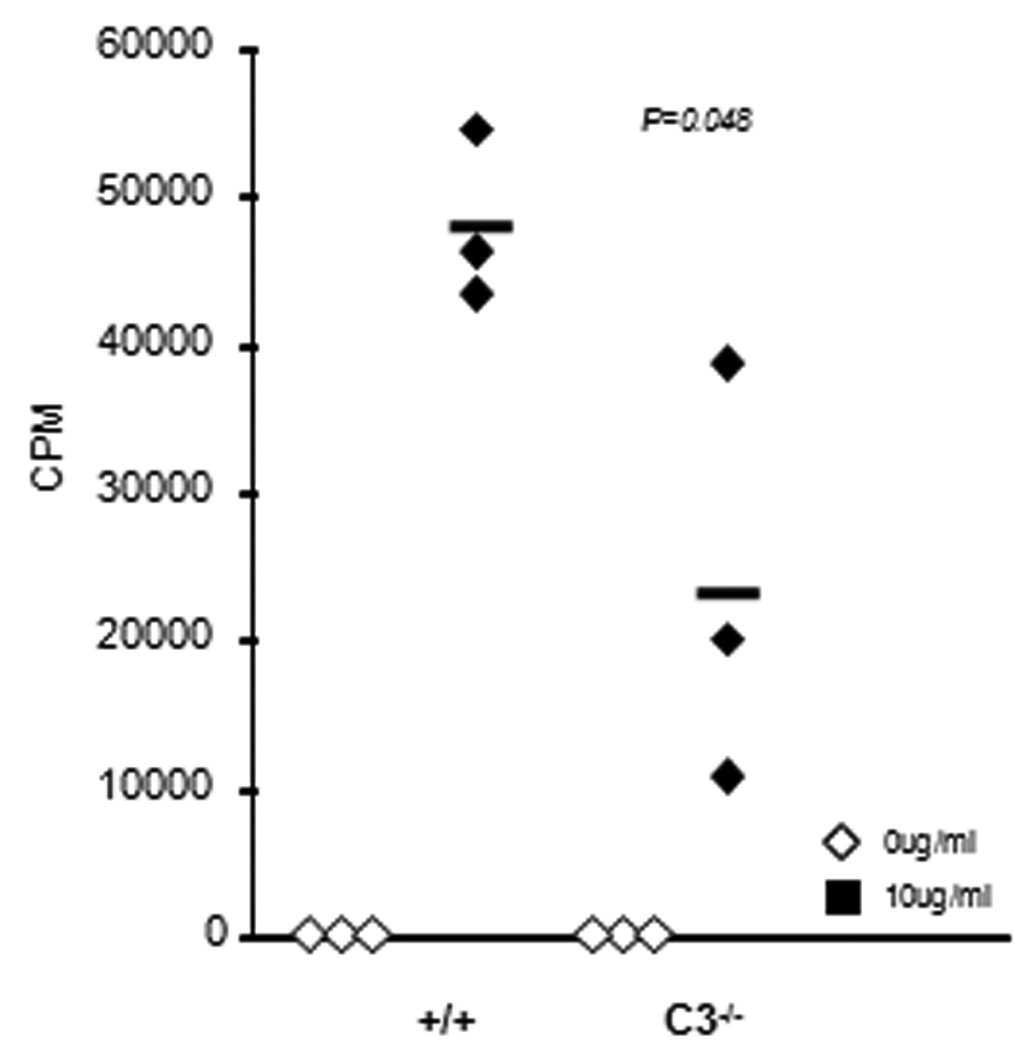
Proliferative responses of C3-deficient CD8 T cells to anti-CD3 stimulation in vitro. CD8 T cells were purified from spleen of +/+ or C3−/− mice by negative selection using magnetic bead separation kit (Milltenyi). Purified CD8 T cells were unstimulated or stimulated with plastic-immobilized anti-CD3 antibodies in a 96-well round-bottomed plate. Cultures were pulsed with 3[H] Thymidine between 24–48 hours after stimulation. 3[H] incorporation by proliferating CD8 T cells was assessed at 48 hours after stimulation. Each symbol represents data from individual mice, and the data is representative of two independent experiments.
Discussion
In response to infections, complement system can be activated by three different pathways namely classical, alternative, and lectin. Regardless of the pathway involved, activation of C3 is a necessary event in the step-wise progression of the cascade of enzymatic reactions that results in the generation of biologically active molecules, which exert distinct effects during the immune response (8, 39, 50). In addition to the well studied role in B cell activation, several studies have shown that complement do regulate T cell responses (10, 11, 51). Although complement activation has been shown to occur in mice infected with LM (26), the role of complement components in the elicitation of T cell responses is unknown. Here, we show that complement component C3 is essential for optimal activation and expansion of antigen-specific CD8 and CD4 T cells during infection of mice with LM. Further studies to understand the mechanisms demonstrate that reduced T cell responses to LM in C3−/− mice is not linked to defective maturation of DCs or a deficiency for C5a in vivo. By performing adoptive transfer of wild type CD8 T cells into C3−/− mice or C3−/− CD8 T cells into wild type mice, we show that autocrine or paracrine sources of C3 might be sufficient to drive clonal expansion of CD8 T cells in vivo. Finally, we report that C3 augments antigen receptor-triggered proliferation of purified CD8 T cells in vitro, which would suggest that lack of direct effects of C3 on T cells might blunt T cell responses of C3−/− mice to LM infection.
In the present study, complete absence of C3 resulted in a marked reduction in the expansion of antigen-specific CD4 and CD8 T cell responses to LM in mice. Similarly, optimal expansion of virus-specific CD8 T cells during infections with influenza virus and LCMV requires C3 activity (10, 11). Additionally, T cell-dependent acute rejection of renal grafts is promoted by C3 activity (14). The mechanism(s) that are involved in promoting T cell expansion by C3 is not completely understood. It has been reported that CD8 T cell responses to influenza virus in mice is significantly reduced by deficiency of either C3 or C5a, which suggested that C3 might promote T cell responses via C5a (12). In contrast to studies with influenza infection, our studies clearly showed that both CD4 and CD8 T cell responses to LM were normal in the apparent absence of C5aR signaling. These findings indicated that requirement for C5a/C5aR interactions might be dictated by the nature of the infecting organism and the associated pathogenesis. Since C5aR has been found to be expressed locally in tissues such as lung and liver, C5a/C5aR signaling might be more critical for tissue-specific host defense in the peripheral tissues like lung during an influenza virus infection, but not in a systemic LM infection (29, 52, 53).
It has been reported that complement might augment vaccine-induced CD8 T cell immunity to leishmaniasis via natural antibodies and IL-4 (13), and complement activation products iC3b/C3dg can bind to antigen/natural antibody complexes and promote antigen uptake by APCs (54, 55). Since absence of B cells did not appear to affect CD8 T cell responses to LM in mice, it is unlikely that C3 promotes T cell responses to LM by antibody-dependent mechanisms (56).
It is known that innate immune mechanisms, especially those mediated by neutrophiland macrophage-mediated phagocytosis are important in control of LM infection (19, 57). Complement receptor 3 has been implicated in opsonization, phagocytosis, and killing of C3b–bound LM by listericidal macrophages. Therefore, in C3−/− mice, lack of C3b–dependent uptake of LM by phagocytes might impede efficient antigen processing and presentation to T cells, resulting in suboptimal expansion of T cells. This hypothesis is supported by reports which show that: 1) efficiency of antigen processing and presentation by professional APCs to T cells is greatly enhanced by tagging antigens to C3 fragments (58, 59); 2) complement factors can directly interact with T cells and modulate the function of antigen presenting T cells, which are crucial for T cell expansion (10, 11, 46, 60, 61); 3) C3 deposition on APCs augments T cell proliferation (62); 4) C3-deficient macrophages and DCs do not effectively stimulate alloreactive T cells in vitro. Our studies clearly show that LM infection-induced maturation of C3-deficient DCs was comparable to +/+ DCs. Notably, the ability of LM-infected DCs from C3−/− mice to activate naïve CD8 T cells was similar to those of +/+ DCs. Therefore, C3 deficiency does not appear to impair antigen processing and/or presentation of listerial antigens to naïve CD8 T cells at least in vitro. Similarly, C3-deficient DCs infected with Mycobacterium bovis strain bacille Calmette-Guérin (BCG) did not exhibit detectable defects in activation of naïve T cells (data not shown). Moreover, the maturation of DCs induced by LM infection appears to be largely normal in C3−/− mice. Importantly, normal activation and expansion of adoptively transferred TCR transgenic CD8 T cells in LM-infected C3−/− mice supports our interpretation that APC-derived C3 may not be required for optimal antigen processing and/or presentation in vivo. It is unknown why DCs require C3 to optimally stimulate alloreactive T cells, but not after LM infection. One possibility is that LM could trigger in infected DCs a broad spectrum of pattern recognition receptors whose downstream effects are redundant with complement-mediated effects. Hence, in LM-infected DCs, C3 function could become redundant and therefore dispensable during T cell activation.
Apart from their ability to enhance antigen presentation by professional APCs, anaphylatoxins C3a and C5a have potent pro-inflammatory and chemotactic effects. Activated T lymphocytes have been shown to express functional receptors for C3a (C3aR) that are known to influence immune cell trafficking in inflammation (50, 61, 63). Hence, it is possible that deficiency of C3a in C3−/− mice disrupts proper trafficking of DCs and T cells in vivo during the T cell response to LM. This in turn could adversely influence the recruitment and activation of naïve T cells in the secondary lymphoid organs.
Binding of complement activation products to complement receptors CR1 and CR2 is a strategy by which complement modulates immune responses. CR1/CR2 are predominantly expressed on antigen presenting cells including B cells, and CR1/CR2 signaling in B cells has been shown to regulate activation threshold, antigen uptake, processing and presentation, isotype switching, and generation of memory B cells (4, 58, 64, 65). In addition to B cells, CR1/CR2 expression on T cells has been reported, and believed to mediate stable binding between the APC and T cells via C3 (62, 66). Hence, C3 could regulate T cell responses by interacting directly with T cells or indirectly via antigen presenting cells. In other models of infection, C3-promoted CD8 T cell responses are minimally affected by CR1/CR2 deficiency (10, 11). It remains to be determined whether C3 promotes T cell responses to LM via CR1/CR2.
CD46, membrane-cofactor protein (MCP) is a cell-surface receptor that is expressed on all nucleated cells, and ligands for CD46 include complement products C3b and C4b. The effects on T cell expansion induced by binding of C3b or C4b depends upon the isoform of CD46. Only when both isoforms of CD46 are coexpressed on a T cell, stimulatory effects appear to dominate over the suppressive effects (67). Therefore, it is possible that in LM-infected mice, C3 deficiency led to abrogation of CD46/C3b interactions, and reduced expansion of antigen-specific CD8 T cells. However, emerging role of CD46-induced regulatory T cells makes it more difficult to separate the effect on T cell activation and regulation (45, 68).
To reiterate, reduced expansion of CD8 and CD4 T cells in LM-infected C3−/− mice could be due to defects in T cells and/or non-T cells. Data presented in this manuscript show that activation and expansion of adoptively transferred wild type TCR transgenic CD8 T cells was largely intact in LM-infected C3−/− mice, which indicated that C3 deficiency in non-T cells did not significantly affect antigen processing/presentation to wild type CD8 T cells in vivo. We explored whether impaired T cell responses in C3−/− mice (11, 12) could be a sequel to developmental defects in the peripheral T cell repertoire. However, the peripheral T and B cell compartment, including Foxp3+ve regulatory T cells and DCs are unaffected by C3 deficiency. Additionally, C3-deficient CD8 T cells do not appear to be intrinsically defective because they exhibit normal activation and expansion upon transfer into wild type mice. These two lines of evidence strongly suggest that lower T cell responses to LM in C3−/− mice is not likely linked to a defective T cell compartment.
How do C3 deficiency blunt T cell responses to LM? First, C3 produced by APCs themselves or other cells could augment their antigen presenting abilities. Second, C3 derived form APCs could act on T cells directly to augment their proliferation. Third, T cell-derived C3 (41) could activate themselves and/or APCs in an autocrine or paracrine fashion respectively. Our studies indicated that: 1) CD8 T cell activation and expansion can occur when responding T cells can produce C3 but not the APCs; 2) responding CD8 T cells are not dependent upon autocrine C3 for activation and proliferation. It is likely that regardless of the cellular source, autocrine or paracrine C3 will support full expansion of CD8 T cells in vivo. However, it is unclear whether direct effects of C3 on T cells promote T cell responses in vivo. Our in vitro studies showed that C3 might promote the proliferative responses of purified CD8 T cells to TCR stimulation, by a mechanism that is distinct from its effect on professional APCs. This finding suggest that lower CD8 T cell responses to LM in C3−/− mice might be linked at least in part to the lack of C3-induced effects on antigen-stimulated T cells. Future studies using C3aR-deficient CD8 T cells might be able to resolve this question in vivo.
In summary, in this manuscript we provide strong evidence that C3 plays a critical role in enhancing T cell responses to an intracellular bacterial infection by promoting proliferative expansion of antigen-triggered CD8 T cells. These findings have implications in rational design of effective vaccines and treatment of T cell-dependent autoimmune disorders.
Figure 5. Effect of treatment with C5aR antagonist on CD8 T cell responses to LM in mice.
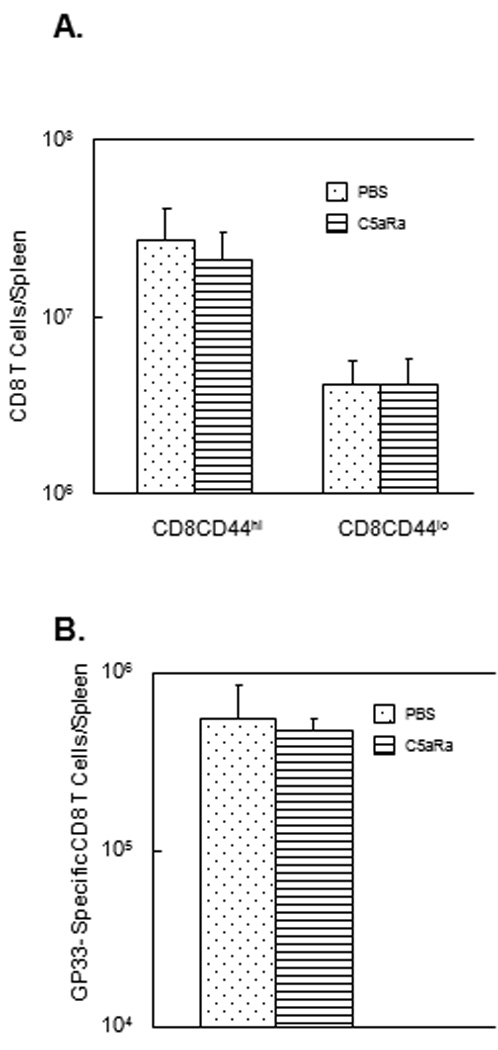
C57BL/6 mice were infected with rLM/GP33 and treated with the C5aR antagonist (C5aRa) or vehicle PBS on days 1, 3, and 5 post-infection. A. On day 7 PI, splenocytes were stained with anti-CD8 and anti-CD44 antibodies and the number of activated (CD44hi) and naive (CD44lo) CD8 T cells were determined by flow cytometry. Data in A are from 5 mice/group ± SD. B. Antigen-specific CD8 T cells in spleen. On day 7 PI, splenocytes were stained with anti-CD8 and Db/GP33 tetramers, and the number of tetramer-binding CD8 T cells was enumerated by flow cytometry. Data in B are from 5 mice/group ± SD.
Acknowledgements
We thank Erin Hemmila-Plisch and Marlese Koenlin for excellent technical assistance.
This work was supported by Public Health Service Grants from the NIH to M. Suresh (AI48785, AI59804, and AI068841) and John D. Lambris (GM062134 and AI68730).
References
- 1.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171:715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll MC, Fischer MB. Complement and the immune response. Curr Opin Immunol. 1997;9:64–69. doi: 10.1016/s0952-7915(97)80160-4. [DOI] [PubMed] [Google Scholar]
- 4.Carroll MC, Prodeus AP. Linkages of innate and adaptive immunity. Curr Opin Immunol. 1998;10:36–40. doi: 10.1016/s0952-7915(98)80028-9. [DOI] [PubMed] [Google Scholar]
- 5.Fearon DT, Carter RH. The CD19/CR2/TAPA-1 complex of B lymphocytes: linking natural to acquired immunity. Annu Rev Immunol. 1995;13:127–149. doi: 10.1146/annurev.iy.13.040195.001015. [DOI] [PubMed] [Google Scholar]
- 6.Davies KA, Schifferli JA, Walport MJ. Complement deficiency and immune complex disease. Springer Semin Immunopathol. 1994;15:397–416. doi: 10.1007/BF01837367. [DOI] [PubMed] [Google Scholar]
- 7.Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. 2000;76:227–324. doi: 10.1016/s0065-2776(01)76021-x. [DOI] [PubMed] [Google Scholar]
- 8.Walport MJ. Complement Second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 9.Rus H, Cudrici C, Niculescu F. The role of the complement system in innate immunity. Immunol Res. 2005;33:103–112. doi: 10.1385/IR:33:2:103. [DOI] [PubMed] [Google Scholar]
- 10.Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat Med. 2002;8:373–378. doi: 10.1038/nm0402-373. [DOI] [PubMed] [Google Scholar]
- 11.Suresh M, Molina H, Salvato MS, Mastellos D, Lambris JD, Sandor M. Complement component 3 is required for optimal expansion of CD8 T cells during a systemic viral infection. J Immunol. 2003;170:788–794. doi: 10.4049/jimmunol.170.2.788. [DOI] [PubMed] [Google Scholar]
- 12.Kim AH, Dimitriou ID, Holland MC, Mastellos D, Mueller YM, Altman JD, Lambris JD, Katsikis PD. Complement C5a receptor is essential for the optimal generation of antiviral CD8+ T cell responses. J Immunol. 2004;173:2524–2529. doi: 10.4049/jimmunol.173.4.2524. [DOI] [PubMed] [Google Scholar]
- 13.Stager S, Alexander J, Kirby AC, Botto M, Rooijen NV, Smith DF, Brombacher F, Kaye PM. Natural antibodies and complement are endogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat Med. 2003;9:1287–1292. doi: 10.1038/nm933. [DOI] [PubMed] [Google Scholar]
- 14.Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8:582–587. doi: 10.1038/nm0602-582. [DOI] [PubMed] [Google Scholar]
- 15.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 16.Mocci S, Dalrymple SA, Nishinakamura R, Murray R. The cytokine stew and innate resistance to L monocytogenes. Immunol Rev. 1997;158:107–114. doi: 10.1111/j.1600-065x.1997.tb00996.x. [DOI] [PubMed] [Google Scholar]
- 17.Ladel CH, Flesch IE, Arnoldi J, Kaufmann SH. Studies with MHC-deficient knock-out mice reveal impact of both MHC I- and MHC II-dependent T cell responses on Listeria monocytogenes infection. J Immunol. 1994;153:3116–3122. [PubMed] [Google Scholar]
- 18.Unanue ER. Studies in listeriosis show the strong symbiosis between the innate cellular system and the T-cell response. Immunol Rev. 1997;158:11–25. doi: 10.1111/j.1600-065x.1997.tb00988.x. [DOI] [PubMed] [Google Scholar]
- 19.Unanue ER. Inter-relationship among macrophages, natural killer cells and neutrophils in early stages of Listeria resistance. Curr Opin Immunol. 1997;9:35–43. doi: 10.1016/s0952-7915(97)80156-2. [DOI] [PubMed] [Google Scholar]
- 20.Harty JT, Bevan MJ. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 1995;3:109–117. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 21.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 22.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 23.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 24.Drevets DA, Leenen PJ, Campbell PA. Complement receptor type 3 mediates phagocytosis and killing of Listeria monocytogenes by a TNF-alpha- and IFN-gamma-stimulated macrophage precursor hybrid. Cell Immunol. 1996;169:1–6. doi: 10.1006/cimm.1996.0083. [DOI] [PubMed] [Google Scholar]
- 25.Nakane A, Okamoto M, Asano M, Kohanawa M, Minagawa T. An anti-CD3 monoclonal antibody protects mice against a lethal infection with Listeria monocytogenes through induction of endogenous cytokines. Infect Immun. 1993;61:2786–2792. doi: 10.1128/iai.61.7.2786-2792.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drevets DA, Campbell PA. Roles of complement and complement receptor type 3 in phagocytosis of Listeria monocytogenes by inflammatory mouse peritoneal macrophages. Infect Immun. 1991;59:2645–2652. doi: 10.1128/iai.59.8.2645-2652.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drevets DA, Leenen PJ, Campbell PA. Complement receptor type 3 (CD11b/CD18) involvement is essential for killing of Listeria monocytogenes by mouse macrophages. J Immunol. 1993;151:5431–5439. [PubMed] [Google Scholar]
- 28.Circolo A, Garnier G, Fukuda W, Wang X, Hidvegi T, Szalai AJ, Briles DE, Volanakis JE, Wetsel RA, Colten HR. Genetic disruption of the murine complement C3 promoter region generates deficient mice with extrahepatic expression of C3 mRNA. Immunopharmacology. 1999;42:135–149. doi: 10.1016/s0162-3109(99)00021-1. [DOI] [PubMed] [Google Scholar]
- 29.Hopken UE, Lu B, Gerard NP, Gerard C. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature. 1996;383:86–89. doi: 10.1038/383086a0. [DOI] [PubMed] [Google Scholar]
- 30.Shedlock DJ, Whitmire JK, Tan J, MacDonald AS, Ahmed R, Shen H. Role of CD4 T cell help and costimulation in CD8 T cell responses during Listeria monocytogenes infection. J Immunol. 2003;170:2053–2063. doi: 10.4049/jimmunol.170.4.2053. [DOI] [PubMed] [Google Scholar]
- 31.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brzoza KL, Rockel AB, Hiltbold EM. Cytoplasmic entry of Listeria monocytogenes enhances dendritic cell maturation and T cell differentiation and function. J Immunol. 2004;173:2641–2651. doi: 10.4049/jimmunol.173.4.2641. [DOI] [PubMed] [Google Scholar]
- 33.Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med. 2002;196:1627–1638. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finch AM, Wong AK, Paczkowski NJ, Wadi SK, Craik DJ, Fairlie DP, Taylor SM. Low-molecular-weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. J Med Chem. 1999;42:1965–1974. doi: 10.1021/jm9806594. [DOI] [PubMed] [Google Scholar]
- 35.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 36.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lara-Tejero M, Pamer EG. T cell responses to Listeria monocytogenes. Curr Opin Microbiol. 2004;7:45–50. doi: 10.1016/j.mib.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 39.Walport MJ. Complement First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 40.Soruri A, Riggert J, Schlott T, Kiafard Z, Dettmer C, Zwirner J. Anaphylatoxin C5a induces monocyte recruitment and differentiation into dendritic cells by TNF-alpha and prostaglandin E2-dependent mechanisms. J Immunol. 2003;171:2631–2636. doi: 10.4049/jimmunol.171.5.2631. [DOI] [PubMed] [Google Scholar]
- 41.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, Medof ME. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsoukas CD, Lambris JD. Expression of EBV/C3d receptors on T cells: biological significance. Immunol Today. 1993;14:56–59. doi: 10.1016/0167-5699(93)90059-T. [DOI] [PubMed] [Google Scholar]
- 43.Sozzani S, Sallusto F, Luini W, Zhou D, Piemonti L, Allavena P, Van Damme J, Valitutti S, Lanzavecchia A, Mantovani A. Migration of dendritic cells in response to formyl peptides, C5a, and a distinct set of chemokines. J Immunol. 1995;155:3292–3295. [PubMed] [Google Scholar]
- 44.Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421:388–392. doi: 10.1038/nature01315. [DOI] [PubMed] [Google Scholar]
- 45.Longhi MP, Harris CL, Morgan BP, Gallimore A. Holding T cells in check--a new role for complement regulators? Trends Immunol. 2006;27:102–108. doi: 10.1016/j.it.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 46.Morgan BP, Marchbank KJ, Longhi MP, Harris CL, Gallimore AM. Complement: central to innate immunity and bridging to adaptive responses. Immunol Lett. 2005;97:171–179. doi: 10.1016/j.imlet.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 47.Peng Q, Li K, Patel H, Sacks SH, Zhou W. Dendritic cell synthesis of C3 is required for full T cell activation and development of a Th1 phenotype. J Immunol. 2006;176:3330–3341. doi: 10.4049/jimmunol.176.6.3330. [DOI] [PubMed] [Google Scholar]
- 48.Zhou W, Patel H, Li K, Peng Q, Villiers MB, Sacks SH. Macrophages from C3-deficient mice have impaired potency to stimulate alloreactive T cells. Blood. 2006;107:2461–2469. doi: 10.1182/blood-2005-08-3144. [DOI] [PubMed] [Google Scholar]
- 49.Westcott MM, Henry CJ, Cook AS, Grant KW, Hiltbold EM. Differential susceptibility of bone marrow-derived dendritic cells and macrophages to productive infection with Listeria monocytogenes. Cell Microbiol. 2007;9:1397–1411. doi: 10.1111/j.1462-5822.2006.00880.x. [DOI] [PubMed] [Google Scholar]
- 50.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verschoor A, Brockman MA, Gadjeva M, Knipe DM, Carroll MC. Myeloid C3 determines induction of humoral responses to peripheral herpes simplex virus infection. J Immunol. 2003;171:5363–5371. doi: 10.4049/jimmunol.171.10.5363. [DOI] [PubMed] [Google Scholar]
- 52.Haviland DL, McCoy RL, Whitehead WT, Akama H, Molmenti EP, Brown A, Haviland JC, Parks WC, Perlmutter DH, Wetsel RA. Cellular expression of the C5a anaphylatoxin receptor (C5aR): demonstration of C5aR on nonmyeloid cells of the liver and lung. J Immunol. 1995;154:1861–1869. [PubMed] [Google Scholar]
- 53.Schieferdecker HL, Schlaf G, Jungermann K, Gotze O. Functions of anaphylatoxin C5a in rat liver: direct and indirect actions on nonparenchymal and parenchymal cells. Int Immunopharmacol. 2001;1:469–481. doi: 10.1016/s1567-5769(00)00038-2. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen CH, Leslie RG, Jepsen BS, Kazatchkine MD, Kaveri SV, Fischer E. Natural autoantibodies and complement promote the uptake of a self antigen, human thyroglobulin, by B cells and the proliferation of thyroglobulin-reactive CD4(+) T cells in healthy individuals. Eur J Immunol. 2001;31:2660–2668. doi: 10.1002/1521-4141(200109)31:9<2660::aid-immu2660>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 55.Thornton BP, Vetvicka V, Ross GD. Function of C3 in a humoral response: iC3b/C3dg bound to an immune complex generated with natural antibody and a primary antigen promotes antigen uptake and the expression of co-stimulatory molecules by all B cells, but only stimulates immunoglobulin synthesis by antigen-specific B cells. Clin Exp Immunol. 1996;104:531–537. doi: 10.1046/j.1365-2249.1996.57761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen H, Whitmire JK, Fan X, Shedlock DJ, Kaech SM, Ahmed R. A specific role for B cells in the generation of CD8 T cell memory by recombinant Listeria monocytogenes. J Immunol. 2003;170:1443–1451. doi: 10.4049/jimmunol.170.3.1443. [DOI] [PubMed] [Google Scholar]
- 57.Czuprynski CJ, Canono BP, Henson PM, Campbell PA. Genetically determined resistance to listeriosis is associated with increased accumulation of inflammatory neutrophils and macrophages which have enhanced listericidal activity. Immunology. 1985;55:511–518. [PMC free article] [PubMed] [Google Scholar]
- 58.Cherukuri A, Cheng PC, Pierce SK. The role of the CD19/CD21 complex in B cell processing and presentation of complement-tagged antigens. J Immunol. 2001;167:163–172. doi: 10.4049/jimmunol.167.1.163. [DOI] [PubMed] [Google Scholar]
- 59.Jacquier-Sarlin MR, Gabert FM, Villiers MB, Colomb MG. Modulation of antigen processing and presentation by covalently linked complement C3b fragment. Immunology. 1995;84:164–170. [PMC free article] [PubMed] [Google Scholar]
- 60.Tsuji RF, Kawikova I, Ramabhadran R, Akahira-Azuma M, Taub D, Hugli TE, Gerard C, Askenase PW. Early local generation of C5a initiates the elicitation of contact sensitivity by leading to early T cell recruitment. J Immunol. 2000;165:1588–1598. doi: 10.4049/jimmunol.165.3.1588. [DOI] [PubMed] [Google Scholar]
- 61.Nataf S, Davoust N, Ames RS, Barnum SR. Human T cells express the C5a receptor and are chemoattracted to C5a. J Immunol. 1999;162:4018–4023. [PubMed] [Google Scholar]
- 62.Kerekes K, Prechl J, Bajtay Z, Jozsi M, Erdei A. A further link between innate and adaptive immunity: C3 deposition on antigen-presenting cells enhances the proliferation of antigen-specific T cells. Int Immunol. 1998;10:1923–1930. doi: 10.1093/intimm/10.12.1923. [DOI] [PubMed] [Google Scholar]
- 63.Werfel T, Kirchhoff K, Wittmann M, Begemann G, Kapp A, Heidenreich F, Gotze O, Zwirner J. Activated human T lymphocytes express a functional C3a receptor. J Immunol. 2000;165:6599–6605. doi: 10.4049/jimmunol.165.11.6599. [DOI] [PubMed] [Google Scholar]
- 64.Prechl J, Erdei A. Immunomodulatory functions of murine CR1/2. Immunopharmacology. 2000;49:117–124. doi: 10.1016/s0162-3109(00)80297-0. [DOI] [PubMed] [Google Scholar]
- 65.Fearon DT, Carroll MC. Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annu Rev Immunol. 2000;18:393–422. doi: 10.1146/annurev.immunol.18.1.393. [DOI] [PubMed] [Google Scholar]
- 66.Delibrias CC, Mouhoub A, Fischer E, Kazatchkine MD. CR1(CD35) and CR2(CD21) complement C3 receptors are expressed on normal human thymocytes and mediate infection of thymocytes with opsonized human immunodeficiency virus. Eur J Immunol. 1994;24:2784–2788. doi: 10.1002/eji.1830241131. [DOI] [PubMed] [Google Scholar]
- 67.Marie JC, Astier AL, Rivailler P, Rabourdin-Combe C, Wild TF, Horvat B. Linking innate and acquired immunity: divergent role of CD46 cytoplasmic domains in T cell induced inflammation. Nat Immunol. 2002;3:659–666. doi: 10.1038/ni810. [DOI] [PubMed] [Google Scholar]
- 68.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]