

Abstract
Toll-like receptors (TLRs) are increasingly implicated in the pathogenesis of cancer. The present study describes TLR expression and function in healthy and malignant airway epithelial cells. The squamous cell carcinoma cell line Detroit-562 was compared with the healthy bronchial epithelial cell line NL-20 and primary human nasal epithelial cells (HNECs). TLR2, TLR3 and TLR5 were present in primary head and neck squamous cell carcinomas (HNSCCs). Consistent with this, Detroit-562 expressed TLR2, TLR3 and TLR5, whereas NL-20 expressed mainly TLR3 and HNECs expressed TLR2-5. In Detroit-562, Pam3CSK4, poly(I:C) and flagellin, ligands for TLR2, TLR3 and TLR5, respectively, induced an up-regulation of intercellular adhesion molecule 1 (ICAM-1), an increase in interleukin (IL)-6 and IL-8 secretion and a decrease in cell viability. Additionally, poly(I:C) affected IL-1β production and the migratory behaviour of Detroit-562. NL-20 responded with a slight increase in IL-8 secretion upon poly(I:C) stimulation. Poly(I:C) induced a small increase in IL-1β, IL-6 and IL-8 production in HNECs, while Pam3CSK4 increased viability. The TLR signalling was transcription-dependent, but the pathways involved differed among TLRs as well as cells. In Detroit-562, TLR2 and TLR5 activation was mediated via c-jun N-terminal kinase (JNK)-, p38-, phosphatidylinositol 3-kinase (PI3K)- and nuclear factor (NF)-κB-related pathways, while TLR3 was dependent on NF-κB. In NL-20, TLR3 signalled via p38, and in HNECs, NF-κB, JNK and extracellular signal-regulated kinase (ERK) appeared to be involved. We found that TLR agonists induced a robust response in HNSCCs, characterized by generation of inflammation and cell death. A similar response was not seen in normal epithelial cells. Thus, the TLR system should be considered an important target in future antitumour immunotherapy.
Keywords: apoptosis, cancer, inflammation, signal transduction, Toll-like receptors
Introduction
The airways are constantly exposed to inhaled and digested microbes. The immunological activity of their epithelial lining keeps the microflora in balance. To fulfil its function in the first line of defence, the epithelium has the ability to release antimicrobial factors and mount an inflammatory reaction.1–3 Like other cells involved in innate immunity, epithelial cells recognize conserved molecular motifs of microbial origin called pathogen-associated molecular patterns (PAMPs) by use of different pattern-recognition receptors. The Toll-like receptors (TLRs) are the most important of these,4,5 comprising 10 members (TLR1–10) in humans.6,7 Each TLR recognizes a specific microbial pattern such as lipopolysaccharide (LPS), lipopeptides, double-stranded (ds)RNA and flagellin,5,7–9 and thereby initiates an immune response.4,5 TLRs have also been implicated in the pathogenesis of several diseases, among them cancer.10,11 Further, TLR ligands have been demonstrated to have anticancer effects, and certain TLR agonists appear to promote cell death in tumours.10 Imiquimod, a TLR7/8 agonist, is used clinically for treating basal cell carcinomas, and in the past TLR3 agonists have been used as adjuvants in cancer treatment.12,13
Head and neck squamous cell carcinomas (HNSCCs) are the most frequent tumour types in the aerodigestive tract. Their detection is often late, resulting in a poor outcome for the patients.14,15 Better knowledge of the disease will be needed in order to develop new diagnostic methods and therapeutic alternatives. A recent publication demonstrated that TLR3 stimulation induces tumour proliferation in HNSCCs.16 However, the role of TLRs in HNSCC is still far from understood. The present study was designed to characterize TLR expression and function in HNSCC by comparing the tumorigenic pharyngeal epithelial cell line Detroit-562 with the healthy bronchial epithelial cell line NL-20 in addition to primary human nasal epithelial cells (HNECs).
Materials and methods
Reagents and antibodies
Tumour necrosis factor (TNF)-α was obtained from R&D Systems (Minneapolis, MN), LPS (Salmonella minnesota) from Alexis Biochemicals (Lausen, Switzerland), and Pam3CSK4, flagellin (Bacillus subtilis) and chloroquine from InvivoGen (San Diego, CA). Lyophilized poly(I:C) (InvivoGen) was reconstituted in sterile physiological salt solution, heated to 50° and then allowed to cool to room temperature to ensure proper annealing. The following monoclonal antibodies (mAbs) were used for flow cytometry analysis: intracellular adhesion molecule 1 (ICAM-1)-phycoerythrin (PE) (clone HA58) and epithelial cell adhesion molecule (EpCAM)-PE (1B7) from eBioscience (San Diego, CA), unlabelled TLR1 (GD2.F4) from Acris Antibodies (Hiddenhausen, Germany), TLR2-PE (TL2.1), TLR3-PE (40C1285), TLR4-fluorescein isothiocyanate (FITC) (HTA125) and TLR5-PE (85B152.5) from AMS Biotechnology (Abingdon, UK) and ICAM-1-fluorescein (BBIG-I1) from R&D Systems. The following Abs were used for immunohistochemistry: TLR2 (polyclonal, rabbit) and TLR3 (40C1285.6) from AMS Biotechnology and TLR5 (polyclonal, rabbit) from Abcam (Cambridge, UK). To study intracellular signalling the cells were incubated with the following inhibitors: PD98059, SB203580 (Tocris-Cookson, Bristol, UK), SP600125, LY294002 (Calbiochem, Bad Soden, Germany), MG-132 (Alexis Biochemicals) and actinomycin D (Sigma-Aldrich, St Louis, MO). All inhibitors, except actinomycin D, were reconstituted in dimethyl sulphoxide (DMSO).
HNSCC tumours
Biopsies were obtained from three patients with HNSCC during standard surgical procedure, and the use of tumour biopsies for research purposes was approved by the local Ethics Committee. All tumours were of squamous cell carcinoma origin and located in the larynx. They were embedded in paraffin, sliced into 3-μm sections, mounted on glass slides and stored at −80° until use.
Cell lines
The human bronchial epithelial cell line NL-20 (CRL-2503), obtained from the American Type Culture Collection (ATCC; Manassa, VA), is an immortalized and non-tumorigenic cell line derived from normal bronchus. This cell line was established through transfection with the origin of replication-defective SV40 large T plasmid p129. The cells were cultured in Ham’s F12 medium (Gibco, Grand Island, NY) supplemented with 2·7 g/l glucose, 5 μg/ml insulin, 10 ng/ml epidermal growth factor, 1 μg/ml transferrin and 500 ng/ml hydrocortisone (Sigma-Aldrich), 2 mm l-glutamine, 0·1 mm non-essential amino acids (NEAA) and 50 μg/ml gentamicin (Gibco), and 4% fetal bovine serum (FBS; PAN Biotech, Aidenbach, Germany).
Detroit-562 (CCL-138), a human pharyngeal squamous cell carcinoma cell line, was obtained from the ATCC. This cell line was cultured in minimum essential medium (MEM) with Earl’s salts and 2 mm l-glutamine (Gibco) and supplemented with 10% FBS, 1 mm sodium pyruvate (Sigma-Aldrich), 0·1 mm NEAA, 50 μg/ml gentamicin and 0·25 μg/ml fungizone (Gibco).
Isolation of primary human nasal epithelial cells (HNECs)
Isolation of primary HNECs was accomplished as described by O’Brien et al.17 Briefly, nasal brushings were performed on six healthy non-smoking subjects. Cells were cultured in airway epithelial cell growth medium supplemented with 0·4% bovine pituitary extract, 10 ng/ml epidermal growth factor, 5 μg/ml insulin, 0·5 μg/ml hydrocortisone, 0·5 μg/ml epinephrine, 6·7 ng/ml triiodothyronine, 10 μg/ml transferrin, 0·1 ng/ml retinoic acid (PromoCell, Heidelberg, Germany) and 100 U/ml penicillin–streptomycin (Gibco). In the experiments, cells from passages 1–3 were used and they were all positive for EpCAM (> 90%), an adhesion molecule specific for epithelial cells.18 All cells were cultured in tissue culture flasks at 37° in a humidified 5% CO2 air atmosphere. The study was approved by the local Ethics Committee.
Culture conditions
Cells were plated in 24-well culture plates (2·5 × 105 cells/well) in 1 ml of complete MEM, Ham’s F12 medium or airway epithelial cell growth medium and incubated overnight. The cells were stimulated for 6 or 24 hr with various concentrations of Pam3CSK4, poly(I:C), flagellin, LPS or TNF-α. The cells and the culture supernatants were subsequently analysed using fluorescence-activated cell sorting (FACS) and enzyme-linked immunosorbent assay (ELISA). To study intracellular signalling pathways, cells were pre-incubated for 1 hr with 10 μM for either of the inhibitors PD98059, SB203580, SP600125, LY294002, MG-132, or 5 μg/ml actinomycin D. Thereafter, TLR ligands were added and the cells were incubated for an additional 24-hr period. To block uptake by TLR3, cells were pretreated with chloroquine (10 and 100 μm) for 30 min before the addition of poly(I:C).
Immunohistochemistry
Prior to staining, the tumour sections were deparaffinized in xylen, rehydrated in decreasing concentrations of ethanol and incubated with a target retrieval solution for 20 min in a microwave oven to facilitate the binding of Abs. To increase the membrane permeability, the sections were treated with 1% Triton-X-100 followed by hydrogen peroxidase to reduce the endogenous peroxidase activity. Primary mouse Abs against TLR3 and rabbit Abs against TLR2 and TLR5 (all diluted 1 : 20) were applied to the sections for 2 hr. As a negative control, Ab diluent or N-series Universal Negative Control Reagent (DakoCytomation, Copenhagen, Denmark) was used. The sections were incubated with either horseradish peroxidase (HRP)-labelled goat anti-mouse or goat anti-rabbit polymer for 30 min and subsequently 3,3′-diaminobenzidine (DAB) substrate-chromogen for 10 min for Ab detection. Sections were dehydrated with increasing concentrations of ethanol, washed with xylen and thereafter mounted in Pertex® (Histolab products AB, Gothenburg, Sweden). The staining was analysed by microscopy.
RNA extraction and real-time reverse transcription–polymerase chain reaction (RT-PCR)
Epithelial cells were lysed in RLT buffer (Qiagen, Hilden, Germany) supplemented with 1% 2-mercaptoethanol and stored at −80° until use. The RNeasy Mini Kit (Qiagen) was utilized for RNA extraction. For cDNA synthesis, the Omniscript Reverse Transcriptase Kit (Qiagen) and Oligo(dT) primer (Novagen, Nottingham, UK) were used. For each PCR reaction, 20 ng of cDNA was utilized. The real-time PCR was carried out on a Smart Cycler (Cepheid, Sunnyvale, CA) using TaqMan Universal PCR Master Mix, No AmpErase UNG and Assay-on-Demand Gene Expression products containing unlabelled primers and MGB probes for TLR1-TLR10 and β-actin (FAM™dye-labelled; Applied Biosystem, Foster City, CA). The thermal cycler was programmed to perform an initial set-up step (95° for 10 min) and 45 cycles of denaturation (95° for 15 seconds), followed by annealing/extension (60° for 1 min). To assess gene expression, the comparative threshold cycle (Ct) method was used.19 To determine the relative amount of mRNA for the TLRs, the Ct values for these genes were subtracted by the Ct values of the housekeeping gene β-actin (ΔCt). The TLR mRNA concentration is expressed in relation to 100 000 mRNA molecules of β-actin (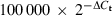 ) and presented as the mean value ± standard error of the mean (SEM).
) and presented as the mean value ± standard error of the mean (SEM).
FACS analysis
The epithelial cells were analysed on a Coulter Epics XL flow cytometer (Beckman Coulter, Marseille, France) and gated based on forward and side scatter properties. Events in the range 10 000–20 000 were collected and the data were analysed with the expo32 adc analysis software (Beckman Coulter). Cells were incubated with Abs for 20 min at room temperature, and thereafter washed and resuspended in phosphate-buffered saline (PBS). Unlabelled TLR1 mAbs was detected using the Alexa Fluor 488 mouse IgG1 labelling kit (Molecular Probes, Eugene, OR). To detect intracellular TLRs, the IntraPrep™ permeabilization reagent kit was used according to the specifications of the manufacturer (Beckman Coulter). Briefly, the cells were fixed and permeabilized using formaldehyde and saponin, respectively, prior to incubation with TLR Abs or appropriate isotype controls.
Cell survival
The Vybrant Apoptosis Assay Kit #3 from Molecular Probes was used to assess the percentages of viable, apoptotic and necrotic cells. Briefly, the cells were stained with Annexin-V-FITC (ANXV) and propidium iodide (PI) and then analysed by FACS. Viable cells were defined as ANXV− PI−; apoptotic cells as ANXV+ PI−; and necrotic cells as ANXV+ PI+.
Nuclear and cytosolic protein extraction
Cells (3 × 106 cells/flask) were cultured overnight, stimulated with or without poly(I:C) for 1 hr and thereafter washed twice in cold PBS. The cells were resuspended in hypotonic buffer and incubated for 15 min on ice, followed by addition of Triton-X-100 (10%) and centrifugation for 10 min. The supernatant contained the cytosolic fraction whereas the pellet contained the nuclear fraction. The pellet was then resuspended in cell extraction buffer for 30 min on ice with vortexing every 10 min and centrifuged for 30 min. The nuclear-containing supernatant was then collected.
ELISA
ELISA plates from R&D Systems were used to determine the IL-1β (sensitivity level 1·0 pg/ml), IL-6 (0·7 pg/ml) and IL-8 (3·5 pg/ml) concentrations in the cell culture supernatants. To detect nuclear and cytosolic NF-κB expression, an ELISA from Biosource (Invitrogen, Carlsbad, CA) was utilized (< 50 pg/ml).
Migration assay
To examine cell migration, the Oris Cell Migration Assay Kit from AMS Biotechnology was used. Briefly, Detroit-562 cells were plated (1 × 105 cells/well) on a 96-well migration plate in 100 μl of complete medium with or without poly(I:C) for 24 hr to allow cell attachment. The inserts were removed (except for the reference wells), the cells were washed, and fresh culture medium with or without poly(I:C) was applied. After another 24 hr of incubation to permit cell migration, the cells were fixed, treated with Giemsa staining and analysed by microscopy. The percentage of migrated cells was determined using ImageJ (NIH, Washington, DC) and calculated as migrated area = total area − non-migrated area.
Statistics
Statistical analysis was performed using graphpad prism 5 (GraphPad Software, San Diego, CA). All data are expressed as mean ± SEM and n is equal to the number of experiments performed. For unpaired data, the statistical difference was determined using the unpaired Student’s t-test. Paired data were analysed using the paired t-test (when two sets of data were compared) and one-way repeated measures analysis of variance (anova) with Dunnett’s post-test (for comparisons of more than two data sets). A P-value ≤ 0·05 was considered statistically significant.
Results
Expression of TLR2, TLR3 and TLR5 in primary HNSCC
Immunohistochemical staining of primary HNSCC sections with Abs directed against TLR2, TLR3 and TLR5 was carried out. All three TLRs were detected within the squamous cell carcinoma with the weakest expression of TLR3 (Fig. 1a–c). The intensity of the TLR staining varied among the different tumour cells within the HNSCC, which indicates that squamous cell carcinomas are composed of a heterogenic cell population.
Figure 1.

Head and neck squamous cell carcinomas (HNSCC) express Toll-like receptor 2 (TLR2), TLR3 and TLR5. The sections show keratinized squamous cell carcinomas from the larynx. The tumour sections were stained with antibodies against (a) TLR2, (b) TLR3 and (c) TLR5. For localization, the sections were stained with 3,3′-diaminobenzidine (DAB; brown staining) and analysed by microscopy (magnification ×4, ×10 and ×40). The arrows indicate TLR-positive cells.
TLR expression profiles in Detroit-562 and NL-20
To further analyse the TLR expression and function in HNSCC, the squamous cell carcinoma cell line Detroit-562 was chosen as a model, together with the non-tumorigenic epithelial cell line NL-20. Expression of TLR1–TLR10 mRNA was investigated in both cell lines using real-time RT-PCR. Detroit-562 exhibited strong expression of TLR2, TLR3 and TLR5, whereas TLR1 and TLR4 were barely detected (Fig. 2a). TLR6 to TLR10 were not found at all. NL-20 expressed mRNAs for TLR3 and TLR4, while TLR1, TLR5 and TLR6 mRNAs were weakly expressed (Fig. 2b). The remaining TLR mRNAs were below the detection limit. To determine whether the expression of mRNA actually reflected the expression of proteins, cells were stained with mAbs against TLR1, TLR2, TLR3, TLR4 and TLR5 or appropriate isotype controls, followed by flow cytometry analysis. Detroit-562 expressed significant levels of TLR2, TLR3 and TLR5 (Fig. 2c), whereas NL-20 expressed TLR2 and TLR3 (Fig. 2d). In contrast to the mRNA analysis, TLR4 was absent in NL-20.
Figure 2.

The Toll-like receptor (TLR) expression profiles in Detroit-562 and NL-20. The epithelial cell lines Detroit-562 and NL-20 were analysed for expression of TLR mRNA and proteins using real-time reverse transcription–polymerase chain reaction (RT-PCR) and fluorescence-activated cell sorting (FACS) analysis, respectively. (a, b) TLR mRNA expression in (a) Detroit-562 and (b) NL-20. The values are expressed in relation to the housekeeping gene β-actin as 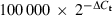 (n = 6). (c, d) Intracellular staining with monoclonal antibodies against TLRs (open histograms) or appropriate isotype controls (shaded histograms) in (c) Detroit-562 and (d) NL-20. Data show one representative out of three independent experiments.
(n = 6). (c, d) Intracellular staining with monoclonal antibodies against TLRs (open histograms) or appropriate isotype controls (shaded histograms) in (c) Detroit-562 and (d) NL-20. Data show one representative out of three independent experiments.
TLR agonists induce pro-inflammatory mediators in Detroit-562
To investigate whether the TLRs are functionally active, the epithelial cells were stimulated with various TLR agonists followed by analyses of ICAM-1 expression along with IL-1β, IL-6 and IL-8 secretion. Detroit-562 and NL-20 were stimulated with or without the ligands for TLR2, TLR3, TLR4 and TLR5, namely Pam3CSK4, poly(I:C), LPS and flagellin, respectively, for 6 or 24 hr. TNF-α was used as a positive control. By gating ICAM-1-positive cells, the mean fluorescence intensity (MFI) was determined. Pam3CSK4, poly(:IC) and flagellin concentration-dependently up-regulated ICAM-1 after 24 hr (Fig. 3a). Poly(I:C) also gave rise to a time-dependent up-regulation of ICAM-1 (Fig. 3b), whereas a 6-hr culture period was sufficient for Pam3CSK4 and flagellin to induce maximum ICAM-1 expression. LPS did not have any effect on ICAM-1 levels, which is an expected finding given the absence of TLR4 expression (data not shown). In contrast to Detroit-562, the non-tumorigenic cell line NL-20 was unable to respond with ICAM-1 expression following TLR ligand stimulation for 6 and 24 hr (data not shown).
Figure 3.

Toll-like receptor (TLR) agonists induce up-regulation of intercellular adhesion molecule 1 (ICAM-1) in Detroit-562. The cells were stimulated with or without Pam3CSK4, poly(I:C), flagellin or tumour necrosis factor (TNF)-α (10 ng/ml) for 6 or 24 hr. Thereafter, the cells were stained with monoclonal antibodies against ICAM-1 and analysed by fluorescence-activated cell sorting (FACS). The mean fluorescence intensity (MFI) of the ICAM-1-positive cells was calculated. (a) ICAM-1 expression after stimulation for 24 hr. (b) Time-dependent up-regulation of ICAM-1 by poly(I:C) (C = control; n = 4; *P < 0·05; **P < 0·01; ***P < 0·001).
Stimulation with Pam3CSK4, flagellin and particularly poly(I:C) gave rise to an increase in IL-1β, IL-6 and IL-8 levels after a 24-hr culture period (Fig. 4a–c). A similar increase was also seen after 6 hr, although it was less pronounced (data not shown). Parallel to the inability of LPS to induce ICAM-1 expression, cytokine secretion was not affected (data not shown). Consequently, stimulation of Detroit-562 with LPS was not further explored. In cultures with NL-20, neither Pam3CSK4, LPS nor flagellin enhanced IL-1β, IL-6 or IL-8 release (data not shown). Poly(I:C), in contrast, induced increased IL-8 secretion and a barely detectable increase in IL-6 production (control, 3·46 ± 0·27; 1 μg/ml, 8·73 ± 1·41; 10 μg/ml, 7·83 ± 1·35) after 24 hr. No effect was seen on cytokine levels after 6 hr (data not shown).
Figure 4.

Toll-like receptor (TLR) ligands induce cytokine secretion. Cells were stimulated with or without Pam3CSK4, poly(I:C), flagellin or tumour necrosis factor (TNF)-α (10 ng/ml) for 24 hr. Thereafter, interleukin (IL)-1β, IL-6 and IL-8 production was measured in the cell culture supernatants by enzyme-linked immunosorbent assay (ELISA). (a) IL-1β, (b) IL-6 and (c) IL-8 secretion by Detroit-562 (n = 6). (d) IL-8 secretion by NL-20 after poly(I:C) stimulation (C = control; n = 6; *P < 0·05; **P < 0·01; ***P < 0·001).
Decreased viability in Detroit-562 after TLR agonist stimulation
Previous studies have shown that TLR3 and TLR7/8 agonists induce apoptosis in human cancer cells.13,20 We therefore endeavoured to assess whether the TLR ligands could affect epithelial cell survival. After stimulation for 24 hr, cells were stained with ANXV and PI, and subsequently analysed by flow cytometry to detect viable, apoptotic and necrotic cells. A significantly lower viability, and conversely higher apoptosis and necrosis, were seen in Detroit-562 stimulated with Pam3CSK4 and poly(I:C) compared with untreated controls. Flagellin gave rise to a slight increase in apoptosis (Table 1). As NL-20 cells were unresponsive to Pam3CSK4, LPS and flagellin, viability experiments were only performed with poly(I:C). However, in contrast to Detroit-562, no difference in viability, apoptosis or necrosis was recorded after stimulation.
Table 1.
Decreased viability in Detroit-562 after Toll-like receptor (TLR) ligand stimulation
| Detroit-562 |
NL-20 |
||||||
|---|---|---|---|---|---|---|---|
| Ligand | Concentration (μg/ml) | Viable | Apoptotic | Necrotic | Viable | Apoptotic | Necrotic |
| Pam3CSK4 | 0·1 | 45·01 ± 3·29* | 32·32 ± 2·10* | 20·25 ± 2·43 | ND | ND | ND |
| 1 | 47·22 ± 3·91 | 30·00 ± 1·52 | 20·19 ± 2·08 | ND | ND | ND | |
| Poly(I:C) | 1 | 45·96 ± 2·75* | 29·16 ± 1·12 | 22·05 ± 2·61 | 88·39 ± 2·07 | 3·23 ± 0·54 | 7·65 ± 1·42 |
| 10 | 39·20 ± 4·51** | 29·27 ± 3·16 | 27·27 ± 2·14** | 89·06 ± 0·69 | 2·76 ± 0·64 | 7·16 ± 0·33 | |
| Flagellin | 0·1 | 54·03 ± 4·89 | 26·29 ± 2·33* | 17·35 ± 2·26 | ND | ND | ND |
| 1 | 47·93 ± 3·84 | 30·19 ± 2·76 | 20·01 ± 3·28 | ND | ND | ND | |
| Control | 0 | 55·96 ± 4·84 | 25·34 ± 2·88 | 16·79 ± 2·42 | 89·40 ± 1·04 | 3·67 ± 0·73 | 6·24 ± 0·40 |
ND, not done.
n = 4; *P < 0·05; **P < 0·01.
TLR3-induced activation is completely abolished by chloroquine
To ensure that the effects of poly(I:C) are mediated via TLR3 and not through the other dsRNA receptors, protein kinase receptor (PKR) and retinoic acid-inducible gene-I (RIG-1),21 cells were incubated with chloroquine for 30 min and thereafter stimulated with poly(I:C) for another 24 hr. Chloroquine inhibits endosomal acidification and therefore blocks TLR3 without affecting PKR and RIG-1.22 The effect of chloroquine on IL-8 production was investigated, as poly(I:C) induced the strongest effect on this cytokine. Both cell lines cultured with chloroquine displayed an IL-8 concentration identical to that of the control (data not shown). We could therefore conclude that the response poly(I:C) induces in both Detroit-562 and NL-20 is generated exclusively via TLR3.
Poly(I:C) has a dual and time-dependent effect on migration of Detroit-562
As the motility of cancer cells can be regarded as a measure of their ability to cause tumour metastases, the capacity of poly(I:C) to induce cell migration was investigated in Detroit-562. Cells were plated on a 96-well Oris migration plate and cultured with or without poly(I:C). Cells were supplemented with poly(I:C) during either the attachment phase or the migration phase, termed early and late addition, respectively. Upon early addition of poly(I:C), a concentration-dependent decrease in migration was seen. When added at a late phase, a clear increase in migration was produced (Fig. 5a–d).
Figure 5.

Poly(I:C) affects migration of Detroit-562. Cells were plated on a cell migration plate (100 000 cells/well) and incubated in the absence or presence of poly(I:C) (early addition). After 24 hr, the inserts were removed, and the cells were washed and cultured in medium with or without poly(I:C) (late addition) for another 24 hr. Thereafter, cells were stained with Giemsa staining and analysed by microscopy. Results show migration for (a) untreated cells, (b) early addition of poly(I:C), and (c) late addition of poly(I:C). (d) Percentage of migrated area (n = 6; *P < 0·05; **P < 0·01).
TLR-mediated effects in primary human nasal epithelial cells (HNECs)
NL-20 is a transfected cell line that originates from the bronchi. Primary human epithelial cells were obtained from the nose in order to provide an additional match to the pharyngeal cell line Detroit-562. These primary cells were also used to verify the differences we have found in TLR responsiveness between Detroit-562 and NL-20. HNECs were found to display a TLR profile restricted to TLR1–TLR5 (Fig. 6a). In this respect, the data for HNECs resembled the data obtained from Detroit-562 and NL-20, but with expression levels more similar to the former. HNECs were then stimulated with Pam3CSK4, poly(I:C) and flagellin for 24 hr with a subsequent analysis of ICAM-1 expression, IL-1β, IL-6 and IL-8 release and cell viability. As found for NL-20, none of the ligands seemed to affect ICAM-1 expression. However, poly(I:C) induced an increase in IL-1β, IL-6 and IL-8 secretion, and flagellin gave rise to a barely noticeable, yet significant, increase in IL-6 levels (Fig. 6b–e). Neither poly(I:C) nor flagellin affected the survival of HNECs, whereas Pam3CSK4 gave rise to a small but significant increase in the percentage of viable cells (Table 2).
Figure 6.

Responses induced by Toll-like receptor (TLR) ligands in human nasal epithelial cells (HNECs). (a) HNECs were analysed for expression of TLR mRNA using real-time reverse transcription–polymerase chain reaction (RT-PCR). The values are expressed in relation to the housekeeping gene β-actin (n = 5). (b) The cells were cultured with or without Pam3CSK4, poly(I:C), flagellin or tumour necrosis factor (TNF)-α (10 ng/ml) for 24 hr. Thereafter, cells were stained with monoclonal antibodies against intercellular adhesion molecule 1 (ICAM-1) and analysed by fluorescence-activated cell sorting (FACS). The mean fluorescence intensity (MFI) of the ICAM-1-positive cells was calculated (n = 4). (c, d, e) After stimulation, the cell culture supernatants were analysed for (c) interleukin (IL)-1β, (d) IL-6 and (e) IL-8 production by enzyme-linked immunosorbent assay (ELISA) (C = control; n = 6; *P < 0·05; **P < 0·01).
Table 2.
Pam3CSK4 increased the viability of human nasal epithelial cells (HNECs)
| HNECs | ||||
|---|---|---|---|---|
| Ligand | Concentration (μg/ml) | Viable | Apoptotic | Necrotic |
| Pam3CSK4 | 0·1 | 72·20 ± 2·77*** | 13·55 ± 3·29 | 5·76 ± 1·18 |
| 1 | 72·35 ± 2·35*** | 14·59 ± 3·00 | 5·39 ± 1·02 | |
| Poly(I:C) | 1 | 69·44 ± 1·25 | 17·36 ± 2·31 | 5·25 ± 0·55 |
| 10 | 68·14 ± 2·34 | 17·04 ± 2·74 | 6·06 ± 0·52 | |
| Flagellin | 0·1 | 68·48 ± 2·41 | 17·16 ± 1·84 | 6·73 ± 0·66 |
| 1 | 68·26 ± 3·12 | 18·79 ± 3·68 | 5·64 ± 0·52 | |
| Control | 0 | 68·19 ± 2·73 | 18·00 ± 2·54 | 6·19 ± 1·09 |
n = 4; ***P < 0·001.
TLR signalling in Detroit-562, NL-20 and HNECs is dependent on different mitogen-activated protein kinase (MAPK) pathways
To study the signalling pathways activated by the TLR ligands, cells were pretreated with various MAPK inhibitors or actinomycin D for 1 hr. Thereafter, TLR ligands were added and the cells were cultured for another 24 hr. The effect of the different inhibitors on IL-8 secretion was analysed, as the TLR agonists in general exerted the strongest effect on IL-8 release. Incubation with the transcriptional inhibitor actinomycin D showed that the TLR responses in both cell lines were transcription-dependent (data not shown). More specifically, it was found that TLR2 and TLR5 activation of Detroit-562 was affected by the inhibitors SB203580, SP600125, LY294002 and MG-132; SB203580, SP600125 and LY294002 inhibit p38 MAPK, c-jun N-terminal kinase 1/2 (JNK1/2) and phosphatidylinositol 3-kinase (PI3K), respectively, and MG-132 suppresses IκBα degradation, which prevents nuclear factor kappa B (NF-κB) nuclear translocation (Fig. 7a,b). No effect was seen with PD98059, which inhibits extracellular signal-regulated kinase (ERK) activation. In contrast, TLR3 signalling seemed to be solely dependent on NF-κB (Fig. 7c). To ensure that the blocking effects of the inhibitors were not attributable to unspecific mechanisms, cells were incubated with the various inhibitors in the absence of TLR ligands. None of the inhibitors was found to exhibit any unspecific effects on Detroit-562. In NL-20, however, the poly(I:C)-induced effects were abolished by SB203580, suggesting a p38 MAPK-mediated activation (Fig. 7d). However, no firm conclusions regarding the involvement of NF-κB and JNK in NL-20 signalling could be drawn from these experiments as they were found to have unspecific effects on IL-8 production. Because of the large variations between the different donors and the relatively small increase in IL-8 seen upon poly(I:C) stimulation, it was difficult to dissect which signalling pathways were activated in HNECs. However, it appeared as if NF-κB, JNK and ERK were involved (Fig. 7e).
Figure 7.

Mitogen-activated protein kinase (MAPK)-dependent Toll-like receptor (TLR) signalling in Detroit-562, NL-20 and human nasal epithelial cells (HNECs). Cells were pretreated with vehicle [dimethyl sulphoxide (DMSO)] or MAPK inhibitors for 1 hr, and further incubated for 24 hr with TLR ligands. The supernatants were then collected and analysed for interleukin (IL)-8 by enzyme-linked immunosorbent assay (ELISA). (a, b, c) Detroit-562 stimulated with (a) 1 μg/ml Pam3CSK4, (b) 1 μg/ml flagellin or (c) 10 μg/ml poly(I:C). (d) NL-20 and (e) HNECs stimulated with 10 μg/ml poly(I:C) (n = 4; *P < 0·05; **P < 0·01; ***P < 0·001). ERK, extracellular signal-regulated kinase; JNK, c-jun N-terminal kinase; PI3K, phosphatidylinositol 3-kinase; NF-κB, nuclear factor kappa B.
To further explore the importance of NF-κB in TLR3 signalling in the tumorigenic cell line, cells were stimulated with or without poly(I:C) for 1 hr. Thereafter, nuclear proteins were extracted in conjunction with cytosolic proteins followed by analyses of NF-κB translocation by use of ELISA. Detroit-562 showed a much higher ratio of NF-κB in the nucleus when stimulated with poly(I:C) (P = 0·02), compared with the unstimulated control (Fig. 8a). TLR3 activation in NL-20 and HNECs, in contrast, did not induce any significant NF-κB translocation (NL-20, P = 0·19; HNECs, P = 0·21) (Fig. 8b,c). Taken together, these data show differences in signalling pathways not only between the different ligands, but also between the three cell types stimulated via identical TLRs.
Figure 8.

Nuclear factor (NF)-κB-dependent Toll-like receptor 3 (TLR3) activation in Detroit-562. Cells were stimulated with or without poly(I:C) (10 μg/ml) for 1 hr. Proteins were extracted from the nucleus as well as the cytosol and further analysed for NF-κB by enzyme-linked immunosorbent assay (ELISA). NF-κB was determined in (a) Detroit-562, (b) NL-20 and (c) human nasal epithelial cells (HNECs) (nuclear/total) (n = 3–4; *P < 0·05).
Discussion
There are several reports indicating that TLRs might have a role in the pathogenesis of cancer.13,20 The present study investigated the differences in TLR expression and function in the tumorigenic pharyngeal epithelial cell line Detroit-562 as compared with the healthy bronchial epithelial cell line NL-20 along with primary human epithelial cells isolated from the nose. Expression of TLR2, TLR3 and TLR5 in squamous cell carcinomas was identified using immunohistochemical staining of HNSCC sections. Detroit-562 was found to express high levels of TLR2, TLR3 and TLR5. Stimulation with Pam3CSK4, poly(I:C) and flagellin gave rise to very strong activation of Detroit-562, manifested as an up-regulation of ICAM-1 expression, increased IL-1β, IL-6 and IL-8 secretion and decreased viability. Further, the migratory behaviour of these cells appeared to be affected by poly(I:C). NL-20, however, only expressed TLR3 and responded weakly to TLR stimulation, demonstrated by the release of a small amount of IL-8 in response to poly(I:C). Results from studies using HNECs showed weak activation upon poly(I:C) stimulation, which further confirmed the impression that tumorigenic airway epithelial cells are more TLR responsive than healthy cells.
Ex vivo staining of HNSCCs showed the presence of TLR2, TLR3 and TLR5. Consistent with these findings, the squamous cell carcinoma cell line Detroit-562 displayed high levels of expression of TLR2, TLR3 and TLR5 at both the mRNA and protein levels. The non-tumorigenic cell line NL-20 showed weak mRNA expression of TLR3 and TLR4, whereas FACS analysis demonstrated the presence of TLR2 and TLR3. However, the functional analyses revealed that TLR3 was the only active receptor in NL-20. There were differences not only in expression patterns, but also in expression levels between the two cell lines. Previous studies have shown that epithelial cells from nasal polyps express all TLRs to different extents,3 that TLR2, TLR3, TLR5 and TLR6 have the highest levels of expression in airway epithelium,2 and that HNSCC cell lines express only TLR3.16 The discrepancy between these and the present data might be explained by the use of different primers for real-time RT-PCR, as well as by the way in which the data were analysed and presented. Some authors presented a picture of a gel, whereas others simply depicted the inverted Ct values without comparing the results with those for a housekeeping gene. Moreover, the epithelial cell activation in response to Pam3CSK4, poly(I:C) and flagellin demonstrated in the present study has been confirmed previously by several groups.2,23,24
Changes in ICAM-1 expression together with IL-1β, IL-6 and IL-8 secretion were chosen as markers of epithelial cell activation. These markers are important mediators in inflammation where they are essential for the recruitment and transmigration of leucocytes. They have also been shown to be expressed by tumour cells.25–27 Upon stimulation with Pam3CSK4, poly(I:C) and flagellin, Detroit-562 responded with very strong up-regulation of ICAM-1 and increased production of IL-1β, IL-6 and IL-8. The largest effect was seen with poly(I:C). In contrast, NL-20 responded with only a small increase in IL-8 secretion after stimulation with poly(I:C). This indicates that predominantly TLR3, but also TLR2 and TLR5, are highly active receptors in the malignant cell line. Similar results were reported recently, demonstrating that the human pulmonary carcinoma cell line NCI-H292 has higher TLR expression than normal human bronchial epithelial cells, and that the TLR ligand-induced IL-8 and vascular endothelial growth factor (VEGF) production is higher in the tumorigenic cell line.24 These differences support the present data, indicating that normal airway epithelium initiates a less robust innate immune response to PAMPs than carcinogenic epithelial cells. Furthermore, experiments using chloroquine, which blocks uptake of poly(I:C) by TLR3, show that these effects exclusively are mediated through TLR3 and not PKR or RIG-1.21 Taken together, our data suggest that carcinoma cells upon TLR activation promote a strong epithelial immune response by inducing recruitment of leucocytes and at the same time facilitating trans-epithelial leucocyte migration.
Stimulation with the TLR agonists, preferably poly(I:C), significantly decreased the viability of Detroit-562, while this was not seen with NL-20 or HNECs. This suggests a role of TLR ligands in inducing apoptosis in malignant epithelial cells without affecting the normal epithelium. This is corroborated by previous reports demonstrating that TLR7/8 and TLR3 agonists have the ability to induce apoptosis in human cancer cells.13,20 However, there are contrasting data showing that activation of TLR2, TLR4 and TLR9 might enhance tumour proliferation.28,29 Furthermore, a recent study by Pries et al.16 demonstrated that TLR3 participates in HNSCC proliferation by activation of NF-κB and oncogenes such as c-Myc. These discrepancies suggest that the role of TLRs in squamous cell carcinomas might be very complex, an impression supported by recent genetic data on TLR polymorphism in tumour cells.30 It may be that external stimuli can induce either tumour progression or tumour regression depending on which TLR is activated and which cell type carries the receptor.
To assess whether poly(I:C) might have an impact on the formation of metastases, the motility of Detroit-562 was assessed in a migration assay. The effects exerted by poly(I:C) were found to be highly dependent on when it was applied, thus reflecting the stage of the tumour cells. If poly(I:C) was added early when the cells were allowed to attach and proliferate, cell migration was decreased. In contrast, if the cells were stimulated during the migratory phase, poly(I:C) gave rise to enhanced migration. The effects seen on migratory behaviour might also be an indirect artefact of the poly(I:C)-induced cell activation demonstrated by the increases in ICAM-1, IL-1β, IL-6 and IL-8, possibly also in concert with increases in other mediators. It has been reported that ICAM-1 plays a role in the migration of tumour cells.31 Previous studies indicate that high expression of ICAM-1 is correlated with metastases and a poor prognosis.32 There are also contrasting data indicating that up-regulation of ICAM-1 on tumour cells induced by IFN-γ would lead to an enhanced immune response against the tumour itself.33 Our data suggest that the migratory behaviour of cancer cells is very complex, and even though cells are more motile this does not signify that they are more inclined to form metastases. Also, formation of metastases is an extremely intricate phenomenon, involving interactions with many different tissues, such as blood vessels, connective tissues and components within various organs.34
The possibility cannot be excluded that the differential responses observed between Detroit-562 and NL-20 could be attributable to tissue specificity, given that the cell lines are derived from the pharynx and bronchus, respectively. Also, the non-tumorigenic cell line NL-20 is immortalized with the SV40-large T antigen, and might not therefore be an appropriate control for studying signalling pathways and apoptosis. Therefore, primary HNECs were obtained in order to confirm that malignant cells are more responsive to TLR agonists and use different signalling pathways, as well as to obtain a better match with Detroit-562. Although HNECs exhibited a similar expression pattern to Detroit-562, their responsiveness to the different TLR ligands differed significantly. HNECs were, similarly to NL-20, almost unresponsive to the different TLR ligands and responded only with induction of IL-1β, IL-6 and IL-8 upon poly(I:C) stimulation. In addition, a small increase in cell survival was seen with Pam3CSK4.
Incubation with actinomycin D showed that TLR signalling in Detroit-562 and NL-20 was transcription-dependent. By studying the signal transduction pathways induced by the various TLR agonists, we found differences among the different TLRs, but also between the various cells stimulated through the same TLR. Signalling through TLR2 and TLR5 in Detroit-562 was mediated by a combinatorial activation of the JNK, p38 MAPK, PI3K and NF-κB pathways, whereas only NF-κB was involved upon TLR3 activation. The intracellular pathways of TLRs are dependent on which adaptor molecule the TLR associates with;9 TLR3 utilizes the MyD88-independent pathway, whereas TLR2 and TLR5 signalling is MyD88-dependent.1,5,35 Therefore, it was not surprising that different signalling pathways were used. Interestingly, activation of TLR3 in NL-20 was mediated via p38 MAPK and possibly also NF-κB. Upon poly(I:C) stimulation in HNECs, ERK, JNK and NF-κB signalling pathways seemed to be involved, although large variation was seen between the different donors. Although we were not able to make any firm conclusions as to the involvement of NF-κB in NL-20 and HNECs, it appears that TLR3 signalling in healthy epithelial cells is dependent on several pathways. In Detroit-562, in contrast, TLR3 seems to be completely dependent on NF-κB for activation.
In TLR signalling, NF-κB is an important transcription factor regulating genes encoding proteins implicated in inflammation, immunity and cell proliferation as well as apoptosis.10,36 NF-κB and pro-inflammatory agents have previously been found to be expressed in HNSCC cell lines and tumour samples.27,37,38 Van Waes et al.39 have shown that inhibition of NF-κB induces apoptosis in patients with advanced HNSCC. Our data, however, demonstrate that stimulation of TLR3 induces NF-κB activation and a simultaneous decrease in tumour cell survival. This indicates that NF-κB activation in tumours might not only be involved in tumour progression, but also in tumour regression.
The present study demonstrates clear differences in the expression patterns, expression levels and functional abilities of TLRs in normal and malignant airway epithelial cells. In squamous cell carcinomas, activation of TLRs, predominantly TLR3, induces a strong epithelial innate immune response and provokes cell death. This suggests a dual action of the TLR agonists in HNSCCs – as immune stimulators and as apoptosis inducers. Hence, the TLR system has to be considered as an important target in anti-tumour immunotherapy.
Acknowledgments
We would like to acknowledge Ulla Pettersson Westin for help with isolating nasal epithelial cells. This study was financially supported by grants from the Swedish Medical Research Council and Karolinska Institutet.
Disclosure
There are no conflicts of interest regarding this study.
References
- 1.Greene CM, McElvaney NG. Toll-like receptor expression and function in airway epithelial cells. Arch Immunol Ther Exp (Warsz) 2005;53:418–27. [PubMed] [Google Scholar]
- 2.Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–64. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- 3.Muir A, Soong G, Sokol S, Reddy B, Gomez MI, Van Heeckeren A, Prince A. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol. 2004;30:777–83. doi: 10.1165/rcmb.2003-0329OC. [DOI] [PubMed] [Google Scholar]
- 4.Tsan MF. Toll-like receptors, inflammation and cancer. Semin Cancer Biol. 2006;16:32–7. doi: 10.1016/j.semcancer.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med. 2006;84:712–25. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- 6.Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 7.Lien E, Ingalls RR. Toll-like receptors. Crit Care Med. 2002;30:S1–11. [PubMed] [Google Scholar]
- 8.Parker LC, Prince LR, Sabroe I. Translational mini-review series on toll-like receptors: networks regulated by toll-like receptors mediate innate and adaptive immunity. Clin Exp Immunol. 2007;147:199–207. doi: 10.1111/j.1365-2249.2006.03203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaisho T, Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol. 2006;117:979–87. doi: 10.1016/j.jaci.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 10.Killeen SD, Wang JH, Andrews EJ, Redmond HP. Exploitation of the Toll-like receptor system in cancer: a doubled-edged sword? Br J Cancer. 2006;95:247–52. doi: 10.1038/sj.bjc.6603275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–9. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 12.Urosevic M, Dummer R, Conrad C, Beyeler M, Laine E, Burg G, Gilliet M. Disease-independent skin recruitment and activation of plasmacytoid predendritic cells following imiquimod treatment. J Natl Cancer Inst. 2005;97:1143–53. doi: 10.1093/jnci/dji207. [DOI] [PubMed] [Google Scholar]
- 13.Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176:4894–901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 14.Rhee JC, Khuri FR, Shin DM. Advances in chemoprevention of head and neck cancer. Oncologist. 2004;9:302–11. doi: 10.1634/theoncologist.9-3-302. [DOI] [PubMed] [Google Scholar]
- 15.Pries R, Nitsch S, Wollenberg B. Role of cytokines in head and neck squamous cell carcinoma. Expert Rev Anticancer Ther. 2006;6:1195–203. doi: 10.1586/14737140.6.9.1195. [DOI] [PubMed] [Google Scholar]
- 16.Pries R, Hogrefe L, Xie L, Frenzel H, Brocks C, Ditz C, Wollenberg B. Induction of c-Myc-dependent cell proliferation through toll-like receptor 3 in head and neck cancer. Int J Mol Med. 2008;21:209–15. [PubMed] [Google Scholar]
- 17.O’Brien GJ, Riddell G, Elborn JS, Ennis M, Skibinski G. Staphylococcus aureus enterotoxins induce IL-8 secretion by human nasal epithelial cells. Respir Res. 2006;7:115. doi: 10.1186/1465-9921-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balzar M, Winter MJ, de Boer CJ, Litvinov SV. The biology of the 17-1A antigen (Ep-CAM) J Mol Med. 1999;77:699–712. doi: 10.1007/s001099900038. [DOI] [PubMed] [Google Scholar]
- 19.Mansson A, Adner M, Cardell LO. Toll-like receptors in cellular subsets of human tonsil T cells: altered expression during recurrent tonsillitis. Respir Res. 2006;7:36. doi: 10.1186/1465-9921-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inglefield JR, Larson CJ, Gibson SJ, Lebrec H, Miller RL. Apoptotic responses in squamous carcinoma and epithelial cells to small-molecule toll-like receptor agonists evaluated with automated cytometry. J Biomol Screen. 2006;11:575–85. doi: 10.1177/1087057106288051. [DOI] [PubMed] [Google Scholar]
- 21.Sen GC, Sarkar SN. Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev. 2005;16:1–14. doi: 10.1016/j.cytogfr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Kumar A, Zhang J, Yu FS. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology. 2006;117:11–21. doi: 10.1111/j.1365-2567.2005.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritter M, Mennerich D, Weith A, Seither P. Characterization of toll-like receptors in primary lung epithelial cells: strong impact of the TLR3 ligand poly(I:C) on the regulation of toll-like receptors, adaptor proteins and inflammatory response. J Inflamm (Lond) 2005;2:16. doi: 10.1186/1476-9255-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koff JL, Shao MX, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1068–75. doi: 10.1152/ajplung.00025.2008. [DOI] [PubMed] [Google Scholar]
- 25.Williams MR, Kataoka N, Sakurai Y, Powers CM, Eskin SG, McIntire LV. Gene expression of endothelial cells due to interleukin-1 beta stimulation and neutrophil transmigration. Endothelium. 2008;15:73–165. doi: 10.1080/10623320802092443. [DOI] [PubMed] [Google Scholar]
- 26.Lyons AJ, Jones J. Cell adhesion molecules, the extracellular matrix and oral squamous carcinoma. Int J Oral Maxillofac Surg. 2007;36:671–9. doi: 10.1016/j.ijom.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Malhotra PS, Thomas GR, et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999;5:1369–79. [PubMed] [Google Scholar]
- 28.Kundu SD, Lee C, Billips BK, et al. The toll-like receptor pathway: a novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate. 2008;68:223–9. doi: 10.1002/pros.20710. [DOI] [PubMed] [Google Scholar]
- 29.Huang B, Zhao J, Shen S, et al. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. 2007;67:4346–52. doi: 10.1158/0008-5472.CAN-06-4067. [DOI] [PubMed] [Google Scholar]
- 30.Tahara T, Arisawa T, Wang F, et al. Toll-like receptor 2 −196 to 174del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci. 2007;98:1790–4. doi: 10.1111/j.1349-7006.2007.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson JP. Cell adhesion molecules in the development and progression of malignant melanoma. Cancer Metastasis Rev. 1999;18:345–57. doi: 10.1023/a:1006304806799. [DOI] [PubMed] [Google Scholar]
- 32.Maruo Y, Gochi A, Kaihara A, Shimamura H, Yamada T, Tanaka N, Orita K. ICAM-1 expression and the soluble ICAM-1 level for evaluating the metastatic potential of gastric cancer. Int J Cancer. 2002;100:486–90. doi: 10.1002/ijc.10514. [DOI] [PubMed] [Google Scholar]
- 33.Scher RL, Koch WM, Richtsmeier WJ. Induction of the intercellular adhesion molecule (ICAM-1) on squamous cell carcinoma by interferon gamma. Arch Otolaryngol Head Neck Surg. 1993;119:432–8. doi: 10.1001/archotol.1993.01880160080012. [DOI] [PubMed] [Google Scholar]
- 34.Chin D, Boyle GM, Kane AJ, Theile DR, Hayward NK, Parson PG, Coman WB. Invasion and metastasis markers in cancers. Br J Plast Surg. 2005;58:466–74. doi: 10.1016/j.bjps.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 35.Sandor F, Buc M. Toll-like receptors. II. Distribution and pathways involved in TLR signalling. Folia Biol (Praha) 2005;51:188–97. [PubMed] [Google Scholar]
- 36.Allen CT, Ricker JL, Chen Z, Van Waes C. Role of activated nuclear factor-kappaB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck. 2007;29:959–71. doi: 10.1002/hed.20615. [DOI] [PubMed] [Google Scholar]
- 37.Ondrey FG, Dong G, Sunwoo J, et al. Constitutive activation of transcription factors NF-(kappa)B, AP-1, and NF-IL6 in human head and neck squamous cell carcinoma cell lines that express pro-inflammatory and pro-angiogenic cytokines. Mol Carcinog. 1999;26:119–29. doi: 10.1002/(sici)1098-2744(199910)26:2<119::aid-mc6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 38.Zhang PL, Pellitteri PK, Law A, et al. Overexpression of phosphorylated nuclear factor-kappa B in tonsillar squamous cell carcinoma and high-grade dysplasia is associated with poor prognosis. Mod Pathol. 2005;18:924–32. doi: 10.1038/modpathol.3800372. [DOI] [PubMed] [Google Scholar]
- 39.Van Waes C, Chang AA, Lebowitz PF, et al. Inhibition of nuclear factor-kappaB and target genes during combined therapy with proteasome inhibitor bortezomib and reirradiation in patients with recurrent head-and-neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2005;63:1400–12. doi: 10.1016/j.ijrobp.2005.05.007. [DOI] [PubMed] [Google Scholar]