

Abstract
Mammalian thioredoxin reductase (TR) contains a rare selenocysteine (Sec) residue in a conserved redox active tetrapeptide of sequence Gly-Cys1-Sec2-Gly. The high chemical reactivity of the Sec residue is thought to confer broad substrate specificity to the enzyme. In addition to utilizing thioredoxin (Trx) as a substrate, other substrates are: protein disulfide isomerase, glutaredoxin, glutathione peroxidase, NK-lysin/granulsin, HIV Tat protein, H2O2, lipid hydroperoxides, vitamin K, ubiquinone, juglone, ninhydrin, alloxan, dehydroascorbate, DTNB, lipoic acid/lipoamide, S-nitrosoglutathione, selenodiglutathione, selenite, methylseleninate, and selenocystine. Here we show that the Cys2-mutant enzyme or the N- terminal reaction center alone can reduce Se-containing substrates selenocystine and selenite with only slightly less activity than the wild type enzyme, in stark contrast to when Trx is used as the substrate when the enzyme suffers a 175- to 550-fold reduction in kcat. Our data supports the use of alternative mechanistic pathways for the Se-containing substrates that bypass a critical ring-forming step when Trx is the substrate. We also show that lipoic acid can be reduced through a Sec-independent mechanism that involves the N-terminal reaction center. These results show that the broad substrate specificity of the mammalian enzyme is not due to the presence of the rare Sec residue, but is due to the catalytic power of the N-terminal reaction center. We hypothesize that the N-terminal reaction center can reduce substrates: (i) with good leaving groups such as DTNB, (ii) that are highly electrophilic such as selenite, (iii) or are activated by strain such as lipoic acid/lipoamide. We also show that the absence of Sec only changed the IC50 for aurothioglucose by a factor of 1.7 in the full-length mammalian enzyme (83 nM to 142 nM), but surprisingly the truncated enzyme showed much stronger inhibition (25 nM). This contrasts with auranofin, where the absence of Sec more strongly perturbed inhibition.
High Mr1 thioredoxin reductase (TR) is a member of the pyridine nucleotide disulfide oxidoreductase family that includes glutathione reductase (GR) and lipoamide dehydrogenase (LipDH). All three enzymes have a similar structure and mechanism (1). TR and GR use NADPH as a cofactor to transfer a pair of electrons, via FAD, to a conserved disulfide redox center, while LipDH uses NADH instead of NADPH (2). This N-terminal disulfide redox center transfers a pair of electrons to: oxidized glutathione (GSSG) in the case of GR, lipoamide/lipoic acid in the case of LipDH2, and a C-terminal, vicinal disulfide (Cys1-Cys2) in the case of TR. This vicinal disulfide bond, although part of the polypeptide chain, is functionally equivalent to GSSG and lipoamide, and acts as a shuttle to transfer electrons to what is thought to be the primary substrate – thioredoxin (Trx) (3, 4). Mammalian TR uses the rare amino acid selenocysteine (Sec, U), the so-called 21st amino acid in the genetic code (5, 6), in place of Cys2.
It is thought that substitution of Cys with Sec has imbued the mammalian enzyme with broad substrate specificity in comparison to its prokaryotic counterpart (3, 7, 8). Other macromolecules besides Trx that have been reported as substrates include: protein disulfide isomerase (PDI), glutaredoxin (Grx), glutathione peroxidase (Gpx), NK-lysin/granulsin, and HIV Tat protein (9–13). Mammalian TR also reduces many small molecule compounds such as: H2O2, lipid hydroperoxides, vitamin K, ubiquinone, juglone, ninhydrin, alloxan, dehydroascorbate, DTNB, lipoic acid/lipoamide, S-nitrosoglutathione (GSNO), selenodiglutathione, selenite, methylseleninate, and selenocystine (14–24). Numerous places in the literature cite the presence of the rare Sec residue in mammalian TR as the reason for this broad substrate specificity3. However, it is known that some of these small molecules (DTNB and ubiquinone), are at least in part turned over by a Sec-independent mechanism involving the N-terminal reaction center (16, 24). Here we demonstrate that several Se-containing substrates can be turned over by the Cys2-mutant or by the N-terminal reaction center alone with only a small loss in activity. This is in stark contrast to the steep loss in activity with macromolecular Trx as the substrate when Sec2 is mutated to Cys2 (175- to 550-fold in kcat) (24, 25), and the complete loss of Trx-reductase activity when the C-terminal reaction center is missing.
In our previous work, we investigated the differences in the thiol/disulfide exchange step between N- and C-terminal reaction centers of the mammalian Sec2-TR and the Cys2-TR from Drosophila melanogaster (DmTR) (26). The results of that study showed that a peptide containing a cyclic 8-membered disulfide was an extremely poor substrate for the N-terminal reaction center of the mammalian enzyme, but was a good substrate for the N-terminal reaction center for the Drosophila enzyme. The N-terminal reaction center of the mammalian enzyme would however reduce peptides containing either an 8-membered ring selenosulfide bond or an acyclic selenosulfide bond, demonstrating the importance of Se to the thiol/disulfide exchange step between N- and C-terminal reaction centers. We provided a geometric rationale to explain the difference between the two types of enzymes in this thiol/disulfide exchange step. In broad terms, we stated that a Cys2-TR (such as DmTR) could compensate for the lack of Sec by using ring geometry to correctly position the thiolate of Cys2 relative to the active site general acid (HisH+). This geometry would stabilize the thiolate, enabling the thiol/disulfide exchange reaction to occur between the two reaction centers in the absence of Sec. Our specific proposal was that the intervening amide between neighboring half-cystinyl residues adopted a cis conformation, and this special ring geometry allowed proton transfer from HisH+ to the thiolate of Cys2 to occur. This explanation was based in part by the observation that the S atoms of a vicinal disulfide bond were superposable with the S atoms of GSSG bound in the active site of GR when the geometry of the intervening amide bond is cis as shown in Figure 1 (27). We would like to point out that the thiolate of Cys2 could be stabilized by an ion pair mechanism, similar to a previous proposal by Wessjohann and Brandt (28, 29), and that the amide geometry of the intervening peptide bond need not be cis for this to occur.
Figure 1.
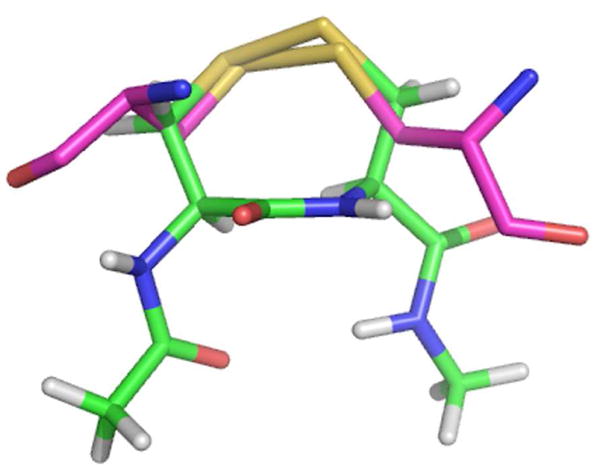
Overlay of the disulfide bond of GSSG (purple) bound in the active site of GR with a vicinal disulfide bond (cis) in a type VIa β-turn conformation (green). S atoms are shown in yellow.
The relationship between GSSG and a vicinal disulfide bond was recognized earlier by the research group of Gino Lucente, who reported the synthesis of γ-(H-Glu-OH)-Cys-Cys-OH(ox) as a glutathione analogue (Figure 2 – top ) (30). Lucente may have developed this analogue with the idea that the geometry of the disulfide bond in this compound was similar to that of the disulfide bond of GSSG as we show in Figure 1. This same group also synthesized derivatives of 4-amino-1,2-dithiolane-4-carboxylic acid (Adt) as analogues of GSSG (Figure 2 – middle) (31). The disulfide bond of Adt is a 1,2-dithiolane, the same disulfide bond contained in lipoic acid/lipoamide. Given the strong evolutionary relationship between TR, GR, and LipDH mentioned above, Lucente may have reasoned that a 1,2-dithiolane could be functionally equivalent to GSSG, even though we realized that the 1,2-dithiolane ring of lipoic acid would be significantly more strained than that of a vicinal disulfide bond. Lucente’s GSSG analogues spurred the hypothesis that the lipoic acid reductase activity of mammalian TR could be due largely from the activity of the N-terminal reaction center. We decided to test this hypothesis and the results presented herein are very instructive about the mechanism of the thiol/disulfide exchange step between the N- and C-terminal reaction centers.
Figure 2.

Similarity of glutathione, lipoic acid, and a peptide vicinal disulfide bond. (top) Glutathione analogue γ-(H-Glu-OH)-Cys-Cys-OH(ox). (middle) Glutathione analogue 4-amino-1,2-dithiolane-4-carboxylic acid. (bottom) Overlay of the conformation of lipoic acid (white) with the same vicinal disulfide bond shown in Figure 1. Note how the S atoms of lipoic acid fit inside the ring and approximately match the positions of the S atoms of the ring. For the sake of clarity, the bond between the two S atoms of lipoic acid is omitted so that the positions of the atoms can be more clearly seen.
MATERIALS AND METHODS
Materials
NADPH, racemic (R, S) lipoic acid, lipoamide, and aurothiogluose were purchased from Sigma-Aldrich (St. Louis, MO). Auranofin was a generous gift from Dr. Pamela Cassidy (University of Utah). Selenocystine was synthesized by Dr. Alayne Schroll (St. Michael’s College, VT). Cystine was from Acros (Geel, Belgium). The production of the recombinant enzymes used in the study, except for the truncated mutant mTRΔ24, has been previously reported (24, 27, 32). All other chemicals and reagents were purchased from either Fisher Scientific (Fair Lawn, NJ) or Sigma-Aldrich (St. Louis, MO) and were of reagent grade or better.
Purification of Enzymes and Synthesis of mTRΔ2
We have previously reported the construction and purification of truncated forms of mTR3 and DmTR (27, 32). These truncated forms are either missing the final eight amino acids containing the C-terminal redox center (denoted as mTR3Δ8 and DmTRΔ8), or in the case of the mouse enzyme missing only the final 3 amino acids (denoted as mTR3Δ3). Here we also report the construction of a truncated mutant of mTR3 missing its final two amino acids (Sec-Gly). This was accomplished by expressing mTRΔ3 as a TR-intein-chitin binding domain fusion protein in Escherichia coli cells. Cell lysate was applied to a chitin-agarose column, and TR was cleaved from the intein with L-cysteine present in the buffer. Since L-cysteine contains both a thiol group and an amino group, the use of this thiol as an intein cleavage reagent results in the incorporation of one additional Cys residue into the target protein (TR), as the presence of the amino group will cause rearrangement of the nascent thioester to produce a stable amide linkage. The use of the TR-intein fusion protein for protein engineering studies in our lab has been previously detailed (32).
Lipoic Acid/Lipoamide Reductase Assay
Racemic (R, S) lipoic acid and lipoamide were both assayed with various TRs to determine activity, which was monitored spectrophotometrically by the decrease in absorbance at 340 nm due to consumption of NADPH by the enzyme. The assay consisted of assay buffer (100 mM potassium phosphate buffer, pH 7.0, 1 mM EDTA, 150 μM NADPH), and varying concentrations of lipoic acid or lipoamide in a 500 μL final assay volume. The concentrations of enzymes in these assays are given in Table S1 of the Supporting Information.
Reduction of oxidized glutathione and trans-4,5-Dihydroxy-1,2-dithiane
Both mTRΔ8 and DmTRΔ8 were assayed to determine their activity towards GSSG and the oxidized form of dithiothreitol (trans-4,5-dihydroxy-1,2-dithiane – DTT(ox)). Assays consisted of 5 mM GSSG or 5 mM DTT(ox) in assay buffer, with either 963 nM mTRΔ8 or 835 nM DmTRΔ8 in a final volume of 500 μL. Activity was monitored by the decrease in absorbance at 340 nm for both small molecule disulfides.
pH Rate Profiles For mTRΔ8 and DmTRΔ8
In order to determine the optimum pH of the truncated TRs for enzymatic reduction of lipoic acid, two truncated mutants missing their final eight amino acids (mTRΔ8 and DmTRΔ8) were assayed in 100 mM citrate, sodium phosphate, or Tris buffers with a range of pH values from 4.0 to 10.0 with a constant concentration of 7.5 mM racemic lipoic acid (from ethanol stock solution). The 500 μL assays also contained 1 mM EDTA, 150 μM NADPH, and either 48 nM mTRΔ8 or 56 nM DmTRΔ8.
Reduction of Selenocystine and Cystine
Each solid was dissolved in a small volume of 1 M sodium hydroxide, which was then nearly neutralized by the addition of almost the same volume of 1 M hydrochloric acid (resulting in a slightly basic solution). The total volume was then increased to 1 mL by addition of deionized water. Activity towards substrate was measured using assays containing 500 mM potassium phosphate, pH 7.0, 10 mM EDTA, 200 μM NADPH, and 91 μM selenocystine or cystine in a final volume of 1 mL. In the selenocystine assay, the following enzyme concentrations were utilized: mTR-GCUG (27.2 nM), mTR-GCCG (22.8 nM), mTRΔ8 (530 nM), and DmTR-SCCS (16.5 nM). In the cystine assay, the enzyme concentrations utilized were: mTR-GCUG (135 nM), mTR-GCCG (227.5 nM), mTRΔ8 (530 nM), and DmTR-SCCS (330 nM).
Reduction of selenite
A stock solution of sodium selenite was prepared by dissolving the solid in 100 mM phosphate buffer, and then assayed with various TRs to determine activity. Assays contained assay buffer and various concentrations of selenite ranging from 0 – 1 mM in a final volume of 500 μL. Enzyme concentrations utilized were: mTR-GCUG (23.8 nM), mTR-GCCG (29.2 nM), mTRΔ2 (40 nM), mTRΔ8 (24.2 nM), DmTR-SCCS (33 nM), and DmTRΔ8 (68 nM).
Inhibition of TR By Gold Compounds
Inhibition of TR activity was assessed by 5,5′dithio-bis(2-nitrobenzoic acid) (DTNB) reduction. Each 500 μL assay contained 100 mM sodium phosphate, pH 7.4, 2 mM EDTA, 200 μM NADPH, 3 mM DTNB and varying concentrations of either auranofin or aurothioglucose. IC50 values were determined by plotting percent activity versus uninhibited enzyme. Enzyme concentrations for these inhibition assays were: mTR-GCUG (4 nM), mTR-GCCG (4 nM), and mTRΔ8 (2 nM).
RESULTS AND DISCUSSION
Activity of TR Towards Se-Containing Substrates
The importance of the Sec residue to catalysis has been shown in several studies and mutation of Sec to Cys causes a large drop in the rate of Trx reduction, 175- to 550-fold in kcat (24, 25). In contrast, when we tested the Sec ⇒ Cys mutant of TR using selenocystine as the substrate, the activity was only 3.7-fold lower than that of the wild type (WT) enzyme as shown by the data in Table 1. Similarly, there is only a ~3-fold difference in activities between the mammalian enzyme and DmTR, a TR that has a Cys residue in the 2nd position of the dyad instead of Sec. The data in Table 1 also shows that the truncated enzyme missing the C-terminal reaction center reduces the diselenide bond of selenocystine at a dramatically lower rate, while the mutant in which only Cys1 is present in the C-terminal reaction center reduces selenocystine at an intermediate rate. This demonstrates that the enzyme uses a pathway for reduction of this substrate that depends on the use of the C-terminal reaction center, but this mechanism must be distinct from the pathway that the enzyme uses for the reduction of the disulfide bond of Trx. This distinctiveness is demonstrated by: (i) the fact that substitution of Sec2 with Cys2 results in a large decrease in the rate of reduction of Trx, (ii) the truncated enzyme in which only Cys1 is present will not reduce Trx at all (data not shown). These two points are in marked contrast when selenocystine is the substrate as shown in Table 1.
Table 1.
Activities of Various TRs Towards Selenocystine and Cystinea

|
Performed at pH 7.0 and 91 μM substrate concentration.
The reduction of selenocystine by TR was first described in (29).
We also tested sodium selenite as a substrate for the WT enzyme and the same mutants discussed above and the results are summarized in Table 2. A similar, but not identical pattern is exhibited. Similar to when selenocystine is used as the substrate, the Cys2-mutant enzyme has only ~ 2-fold less activity than the WT enzyme when selenite is the substrate. However the truncated enzyme missing the C-terminal reaction center (mTRΔ8) still had very significant activity with selenite – only 6-fold lower activity than the WT enzyme, and the mutant missing Sec2, but still containing Cys1 has the same activity as the truncation mutant where both Cys1 and Sec2 are absent. These last two results are different than when selenocystine is the substrate.
Table 2.
Activities of Various TRs Towards Selenitea

|
Assays were done at pH 7.0.
The data in Tables 1 and 2 also show that DmTR reduces selenocystine and selenite at considerable rates. This observation together with the fact that the Cys2-mutant of the mammalian enzyme reduces these Se-containing substrates at rates comparable to the WT enzyme demonstrates that the nucleophilicity of Sec, relative to Cys, is not an important factor for some reactions catalyzed by TR, as we have previously contended (26). Further, the presence of Sec in TR is not entirely responsible for the broad substrate specificity of the enzyme.
The data presented above leads to the interesting observation that Se can be removed from the enzyme, resulting in only a small decline in substrate turnover rate, if a Se atom is present in the substrate. This model can be rationalized by understanding the types of bonds that the N-terminal reaction center can reduce. As shown in Figure 3A, the C-terminal reaction center reduces macromolecular Trx and becomes oxidized, forming a cyclic S1-Se2 bond as an 8- membered ring. This selenosulfide bond is essentially an internal substrate for the N-terminal redox center and can be reduced by the N-terminal reaction center because it is polarized and has a low pKa leaving group (Se) (36). As has been previously demonstrated, the disulfide bond of DTNB can be reduced directly by the N-terminal reaction center. This is because the S-S bond of DTNB is highly polarized due to the presence of symmetrical 2-nitrobenzoate groups bonded to each S atom, and as a result of this polarization this S-S bond is highly electrophilic. In addition, this strong polarization results in a leaving group pKa of the thionitrobenzoate anion of 4.75 (37). These two observations lead us to hypothesize that the N-terminal reaction center prefers to reduce substrates of the form S-Y, where Y is a good leaving group. We explain this hypothesis in mechanistic terms in Figure 3B showing our proposed mechanism for the reduction of selenocystine by TR. This mechanism explains why Se-containing substrates are turned over by the Cys2-mutant at only slightly lower rates than the Sec2-WT enzyme, in direct contrast when Trx is reduced by the Cys2-mutant. In the case in which Sec2 is mutated to Cys2 with Trx as the substrate, the mutant enzyme still utilizes a ring formation pathway but now an 8-membered S1- S2 ring is formed as an intermediate. This type of disulfide is unreactive towards thiol-disulfide exchange with the N-terminal reaction center, as we have already previously demonstrated (26). Whereas when selenocystine is the substrate (Figure 3B), the enzyme could utilize a pathway that bypasses ring formation, but still allows for formation of a S1-Sesub bond (of the type S-Y, though in this case the selenosulfide bond that forms is between enzyme and substrate instead of an internal selenosulfide bond), and this S1-Sesub bond is reactive towards thiol/disulfide exchange with the N-terminal reaction center.
Figure 3.

Comparison of the current, accepted mechanism of the reduction of Trx (A) with our proposed alternate mechanistic pathways with Se-containing substrates (B and C). In (A) The Se atom of Sec2 attacks the disulfide bond of Trx to form a mixed selenosulfide bond (Se2-Ssub) between TR and Trx (middle panel). The S atom of Cys1 then attacks the Se atom of the mixed selenosulfide bond to form a cyclic S1-Se2 bond as an 8-membered ring. The thiolate of CysIC must then attack either S1 (path 1) or Se2 (path 2). Several groups have proposed path 2, while we previously have argued for path 1 (26, 27, 33–35). The first step of the reduction of selenocystine (B) initially begins just like the reduction of Trx (compare first panels of A and B), but after the mixed diselenide bond forms between TR and substrate (Se2-Sesub – middle panel), the S atom of Cys1 attacks the Se atom of the substrate to form an acyclic S1-Sesub bond. This is the preferred substrate for the N-terminal reaction center (compare the last panels of A and B and note the similarity). We previously demonstrated the mammalian TR reduces acyclic S-Se bonds with only a moderate reduction in activity compared to the cyclic, 8-membered selenosulfide bond (26). The data indicates that the mechanism for the reduction of selenite (C) must be somewhat different in comparison to selenocystine. Once a mixed diselenide bond (Se2-Sesub) forms between TR and substrate, CysIC may attack Sesub directly. In this case Sesub is a good electron sink due in part from the electron withdrawing effect of the bonded oxygen atoms and also to the electrophilic nature of Se itself. This logic may bolster the case for path 2 – attack at Se in (A). If this is true, we still argue for a leaving group role for Se in the subsequent reduction of the SIC-Se2 bond. Note that CysIC is the interchange cysteine and CysCT is the charge-transfer cysteine.
The mechanistic situation with selenite as substrate is somewhat different. Selenite, like the S-S bond of DTNB, is highly electrophilic and the data in Table 2 shows that it can be reduced by the N-terminal reaction center directly, with only a small loss in activity compared to the full-length enzyme when Sec2 is present. The electrophilicity of selenite is a direct consequence of the Se atom, as we were unable to detect activity using sulfite as the substrate (data not shown). Because the truncated enzyme where only Cys1 is present had the same activity as the Δ8 enzyme, the mechanism shown in Figure 3B is unlikely to be used for selenite. Instead, the thiolate of CysIC could attack the mixed diselenide bond between enzyme and substrate directly. The proposed mechanism also bypasses ring formation (Figure 3C), but in a different way than for selenocystine. The reason for higher activity for the enzymes with an intact C-terminal reaction center is that this allows for further polarization of the bond between enzyme and substrate, whether the 2nd residue of the dyad is Cys or Sec. We explain the difference in enzyme mechanisms for selenocystine and selenite as being due to the difference in polarization in the bond formed between enzyme and substrate (compare the middle panels of Figures 3B and 3C). The three oxygen atoms on selenite confer significant polarization to the mixed diselenide bond, allowing for direct attack by CysIC, whereas in the case of selenocystine, a relatively unpolarized diselenide would form between enzyme and substrate and this diselenide bond is unreactive towards exchange with the N-terminal reaction center. Thus the S atom of Cys1 is needed to form the required S-Y bond. This idea is supported by the fact that the Δ8 enzyme cannot directly reduce selenocystine.
Further evidence that the mechanisms of these two substrates are different is evidenced by the pH vs. activity profiles. The profile for selenite is broad with a pH optimum between pH 6 and 7, while the profile for selenocystine is also broad, but the optimum is between 7.5 and 8. These profiles are shown in the Supporting Information as Figures S1A and S1B, respectively. It is also interesting to view the profiles of these two substrates for the mammalian Δ8 enzyme so that the effect of pH on activity can be determined for the N-terminal reaction center only. For selenite, the pH optimum is near pH 6.0 and the profile is significantly sharper than the full-length enzyme. For selenocystine, the pH optimum is near 8.0 for the truncated enzyme (shown in the Supporting Information as Figures S2A and S2B, respectively). As reported previously, the pH optimum of the Δ8 enzyme with DTNB as substrate is also shifted significantly towards acidic pH (26). Both selenite and DTNB are highly electrophilic compounds. It is tempting to infer that the reason for the occurrence of Se in the mammalian enzyme is that there is a certain electrophilic threshold required by the N-terminal reaction center for substrate reduction (note sulfite is a poor substrate), whether it is Se in an external substrate like selenite, or Se in the S1- Se2 bond of the C-terminal selenosulfide motif (internal substrate). The electrophilicity of Se is a well established principle in organic chemistry (38). This idea is complementary to the leaving group concept for Se in TR we introduced previously.
Our proposed leaving group concept is further supported by the result in Table 1 showing that selenocystine is a much better substrate in comparison to cystine. These two substrates are highly similar in terms of dihedral angles, overall charge, and size (39). A major difference between these two small molecule substrates is leaving group pKa upon Se–Se or S–S bond scission, as the pKa of a selenolate is ~5.2 versus ~8.3 for that of a thiolate (36, 40). Thus cystine unlike DTNB, could not be reduced by the truncated Δ8 enzyme (direct reduction by the N-terminal reaction center), because its S-S bond is not polarized and lacks a good leaving group. Our proposed mechanisms also explain why cystine cannot be reduced by the WT-enzyme. If the pathway outlined in Figure 3B is used to reduce cystine, a mixed S1-Ssub bond would form between enzyme and substrate and this is not of the type S-Y. This S1-Ssub bond would then be unreactive towards exchange with the N-terminus, just as is the case when an 8-membered S1-S2 ring forms in the Cys2 mutant. If we imagine that cystine could be reduced using the pathway shown in Figure 3C, a mixed Se2-S1 bond would form between enzyme and substrate and this has the reverse context compared to a S1-Se2 bond, making exchange very slow. Moreover, if this were the case, the thiolate of CysIC would have to attack a S atom of cysteine, which compared to the Se atom of selenite, is not very electrophilic. Our proposed mechanisms explain why these Se-containing substrates are largely independent of the presence of the C-terminal Sec residue.
Leaving group pKa as a determining factor in substrate utilization by mammalian TR is also seen with other known, small molecule substrates such as we have pointed out with DTNB above. This concept explains why GSSG is not a substrate (its leaving group thiol has a relatively high pKa), while analogues of glutathione such as selenodiglutatione (GS-Se-SG), and S-nitrosoglutathione are both utilized as substrates by mammalian TR (17, 20). This is most likely due to the low pKa of the selenolate in GS-Se-SG and HNO in GSNO (pKa = 4.7) (41).
Disulfide Reductase Activity of TR
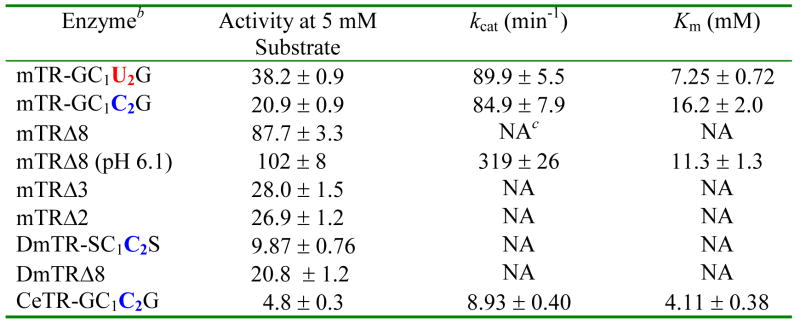
A summary of the kinetic data using lipoic acid as a substrate for full-length and truncated TRs is given in Table 3 (the mitochondrial TR from C. elegans (CeTR2) is included as part of our analysis). As the data in Table 3 demonstrates, the assumption that Sec is needed to catalyze the reduction of lipoic acid is found to be untrue upon comparison of the full-length WT Sec2-containing enzyme to the full-length Cys2-mutant enzyme as the kcat values are nearly identical. The Km value does increase 2.3-fold in the Cys2 mutant, however. The truncated mammalian enzymes in this study also turned over lipoic acid, consistent with our hypothesis that the reduction of the 1,2-dithiolane ring of lipoic acid is largely due to the reactivity of the N-terminal reaction center. Like our results with small molecule Se-containing substrates above, this result demonstrates again that Sec is not needed to reduce some substrates and the broad substrate specificity is not due to the presence of the Sec residue. However, while the truncated mammalian enzymes would turn over lipoic acid, they did not display saturation kinetics, so we are unable to report a Km value for these mutants. Comparing the activity of mTRΔ8 to the full-length enzyme, we see that this truncated enzyme has higher activity than the WT enzyme, demonstrating that the substrate has greater access to the N-terminal reaction center in the mutant than in the WT enzyme.
Table 3.
Lipoic Acid Reductase Activity of Various full-length and truncated TRsa.
|
For assay conditions, please see the text under Lipoic Acid/Lipoamide Reduction Assay.
See footnote 4.
Not applicable for these assays because saturation kinetics were not observed.
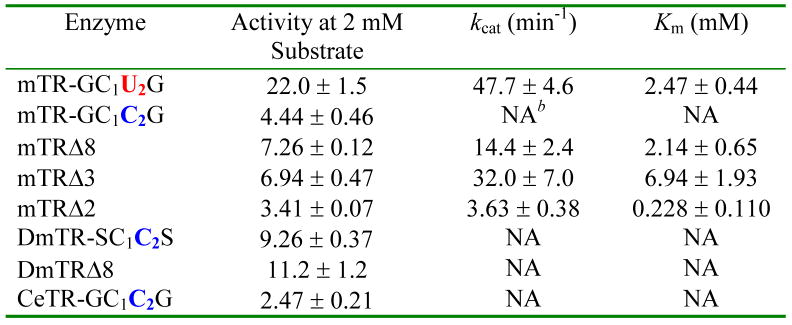
Interestingly, we found that mTRΔ8 had significantly higher activity at pH 6.1. At this pH, mTRΔ8 did show saturation kinetics, with kcat increasing nearly 3.6-fold compared to the WT enzyme at pH 7.0. The pH rate profiles for mTRΔ8 and the full-length enzyme with lipoic acid as a substrate are shown in Figure 4A. As can be seen in the profile, there is a very sharp drop in activity below pH 6.1 for both enzymes and an explanation for this behavior is currently unknown. A possible explanation is that as the pH becomes lower than 6.0, the thiolate of CysIC becomes protonated, making thiol/disulfide exchange slow between enzyme and the 1,2 dithiolane of lipoic acid. We previously reported the pKa of CysIC as 5.8 (26). In contrast to the truncated mammalian enzyme, DmTRΔ8 did not show a sharp increase in activity near pH 6 and had a very similar profile to that of its full-length counterpart as shown in Figure 4B. However, if we use lipoamide as a substrate, we see lower activity overall (lower kcat), but also tighter binding as reflected by a nearly 3-fold drop in Km (Table 4). The pH optimum using lipoamide as a substrate is ~ 8.5 for this truncated enzyme (see Figure S3 in the Supporting Information). The difference in activities between lipoic acid and lipoamide seems to be the affinity the enzyme has for the negatively charged lipoic acid versus the neutral lipoamide. The data indicates that this affinity is not preferential binding because the neutral substrate has a lower Km than the charged substrate. We posited that the carboxylate group of lipoic acid was acting as a general acid/general base catalyst. This hypothesis was tested by adding 50 mM acetate to the lipoamide assay buffer solution. The presence of acetate in the reaction buffer resulted in 23.6% higher activity using lipoamide as a substrate at pH 6.1 and 17.5% higher activity at pH 7.0, respectively. This data is consistent with an acid/base catalytic role for the carboxylate group of lipoic acid, but the higher activity could also be the result of a specific binding interaction between substrate and enzyme.
Figure 4.

Reduction of lipoic acid as a function of pH. (A) mTRΔ8 (closed squares) and full-length mTR (open squares). The truncated enzyme has an optimum at pH 6.1, while the full-length enzyme has an optimum near 7.5. (B) DmTRΔ8 (closed circles) and full-length DmTR (open circles). Both enzymes have similar profiles with pH optima near 7.5.
Table 4.
Lipoamide Reductase Activity of Various Full-length and Truncated TRsa.
|
For assay conditions, please see the text under Lipoic Acid/Lipoamide Reduction Assay.
Not applicable for these assays because saturation kinetics were not observed.
The overall results clearly demonstrate that the disulfide bond of lipoic acid/lipoamide is capable of being reduced by the N-terminal redox center and this suggests to us that in the holoenzyme reduction of lipoic acid (and other substrates) can occur via the N-terminal redox center. We speculate that some substrates are in competitive equilibrium with the C-terminal selenosulfide ring for interaction with the N-terminal redox center. This model allows for reduction of small molecule substrates to take place at either site (Figure 5). This model is similar to the one put forth by Fujiwara and coworkers for the reduction of DTNB by TR (42). Previously it has been assumed that the reduction of lipoic acid is dependent upon the presence of Sec in the C-terminal redox center (18).
Figure 5.

Proposed model for the interaction of small molecule substrates with TR. Both the oxidized C-terminal tetrapeptide (Gly-Cys-Sec-Gly) and small molecule disulfide (such as lipoic acid) can bind in the tetrapeptide binding pocket and are thus in competitive equilibrium for interacting with the N-terminal C1VNVGC2 redox center. These equilibrium constants are represented by KSM1, for small molecules, and KTP, for the C-terminal tetrapeptide. Both of these equilibrium constants are composed of individual rate constants in the forward and reverse directions that describe the rate of reduction of either the small molecule disulfide or the C-terminal vicinal selenosulfide bond. Please note that here the 8-membered ring of the C-terminus is shown with trans amide geometry in the chair-chair conformation. Depending on the redox state of the holoenzyme, the reduction of lipoic acid can take place via the reduced C-terminal tetrapeptide (described by equilibrium constant KSM2). Thus lipoic acid can be reduced via two modes of interaction. While not proven here, the overall rate of reduction is most likely a combination of the two different pathways (and thus combination of rate constants). A similar model has been previously proposed for the reduction of DTNB (20).
Since lipoic acid/lipoamide must also bind in the same place occupied by the C-terminal tail containing either a vicinal disulfide bond in the case of DmTR or a vicinal selenosulfide bond in the case of the mammalian enzyme, the results also show a clear difference in the preference of ring size of various disulfide substrates with each type of enzyme as summarized in Tables 5 and 6. As shown by the data in Table 5, the mammalian enzyme clearly prefers a small-ringed, disulfide substrate. This can be seen in comparing the activities of lipoic acid/lipoamide (5-membered ring) to DTT(ox) (6-membered ring) and a peptide containing a vicinal disulfide bond (8-membered ring). The mammalian enzyme can utilize an 8-membered ring substrate, but only if a Se atom is present in the substrate. The preference of the mammalian enzyme for the 5-membered 1,2-dithiolane ring of lipoic acid is in spite of the much higher pKa value for both sulfhydryl groups (10.7) (43) compared to the 8-membered ring of the peptide containing a vicinal disulfide bond (8.3) (40). We put forth two reasons for the higher activity of the 1,2-dithiolane ring as a substrate in comparison to the other ring sizes with the truncated mammalian enzyme. First, seminal work by Whitesides and coworkers demonstrated that due to the large ring strain imparted on the 1,2-dithiolane ring system by the highly compressed C-S-S- C dihedral angle (35°), that a 1,2-dithiolane is 650-fold more reactive towards thiol/disulfide exchange reactions compared to a 1,2-dithiane ring (such as DTT(ox)) (46, 47). Second, the leaving group sulfur atom in this exchange reaction must be positioned to be protonated by the enzymic general acid (His463′). The fact that the 8-membered ring of the peptide containing a Cys1-Cys2 vicinal disulfide is a very poor substrate could be interpreted as meaning the ring is relatively unstrained in this peptide, or the thiolate of Cys2 is not in the correct position to be stabilized by His463′, either by proton transfer (our previous argument) (26, 27) or by electrostatic stabilization.
Table 5.
Activity of mTRΔ8 Toward Cyclic and Acyclic Substrates.
| Substrate | Type of Bond Broken | Ring Size | Leaving Group pKa | Activity at 5 mM Substrateb |
|---|---|---|---|---|
| Lipoic Acida | S–S | 5 | 10.7d | 87.7 |
| Lipoic Acid (pH 6.1) | S–S | 5 | 10.7d | 102 |
| DTT(ox) | S–S | 6 | 9.2e | 0.234 |
| PTVTGCCG(ox)c | S–S | 8 | 8.3f | 0.153 |
| PTVTGCUG(ox)c | S–Se | 8 | 5.2g | 793 |
| Glutathione | S–S | Acyclic | 9.42h | 0.0723 |
| DTNB | S–S | Acyclic | 4.75i | 2430 |
Table 6.
Activity of DmTRΔ8 Toward Cyclic and Acyclic Substratesa.
| Substrate | Type of Bond Broken | Ring Size | Leaving Group pKa | Activity at 5 mM Substrateb |
|---|---|---|---|---|
| Lipoic Acid | S–S | 5 | 10.7 | 20.8 |
| DTT(ox) | S–S | 6 | 9.2 | 0.109 |
| PTPASCCS(ox) | S–S | 8 | 8.5 | 233 |
| PTPASCUS (ox) | S–Se | 8 | 5.2 | 514d |
| Glutathione | S–S | Acyclic | 9.42 | NDe |
| DTNB | S–S | Acyclic | 4.75 | 519 |
These assays were done at pH 7.0.
The units are mol NADPH/min/mol TR.
This data was taken from (26).
The activity reported here is with 1 mM oxidized peptide.
No detectable activity.
The situation with DmTRΔ8 (a Cys-TR) contrasts with that of the mammalian enzyme as it shows a preference for 8-membered ring substrates if we compare the same series of disulfides (Table 6). However, lipoic acid is still turned over 190-fold faster in comparison to DTT(ox), but the overall rate is lower in comparison to mTRΔ8. This lower activity with lipoic acid is probably a reflection of the higher pKa of the attacking thiolate in DmTRΔ8 in comparison to mTRΔ8 (6.5 for Cys57 – CysIC in DmTR vs. 5.8 for Cys52 – CysIC in mTR) (26). If we use the same line of reasoning as above to explain the higher activity of the 8-membered ring of the Cys1-Cys2 vicinal disulfide peptide substrate in comparison to the other disulfide substrates in Table 6, it would mean that there is either a large degree of strain energy in the ring of the enzyme/peptide complex – rendering this peptide substrate highly reactive, or the conformation of the ring allows for correct positioning for the stabilization of the thiolate of Cys2. We also find that the turnover rate of a homologous peptide in which a Se atom substitutes for a sulfur atom (while still maintaining the size of the ring at 8 atoms) is significantly higher –greater than 2-fold. This fact implies that the presence of a Se atom imparts higher activity to the peptide substrate because of the lower pKa of a selenolate compared to a thiolate, since Se for S substitution would most likely decrease ring strain as a result of a larger ring size. Both types of truncated enzymes can utilize acyclic substrates if the substrate contains a low pKa leaving group. This is seen upon comparing the linear, acyclic substrates GSSG and DTNB. The low pKa leaving group of DTNB compensates for a lack of ring strain as well as the “incorrect” position of the leaving group thiolate relative to reactive groups in the tetrapeptide binding pocket.
Inhibition of TR By Gold Compounds
Conventional wisdom also holds that the reason for the very strong inhibition of TR by various organogold compounds is that the Se atom of the mammalian enzyme makes a very strong coordinate covalent bond with Au (48, 49). However, it is well known that thiols also have a strong interaction with Au (50). Given that we have tested several strongly held beliefs about the mammalian enzyme as discussed above, we decided to examine the ability of the mammalian enzyme and the truncated mammalian enzyme to be inhibited by auranofin and aurothioglucose. The results are summarized in Table 7 and the activity/inhibition curves are given in Figure 6. The results show that mutation of Sec to Cys causes the IC50 to increase 4-fold in the case of auranofin and 1.7-fold in the case of aurothioglucose, indicating that the Se atom interacts with the Au atom in both of these inhibitors. To further assess the role of the Se atom in binding these inhibitors, we tested the truncated enzymes ability to be inhibited by both compounds. The results show that the binding of auranofin is strongly perturbed by the elimination of the C-terminal tail containing the Gly- Cys1-Sec2-Gly tetrapeptide, as the IC50 increases from 75 nM to 850 nM. In the case of aurothioglucose, the truncation mutant shows tighter binding than the WT enzyme. This demonstrates that a significant portion of the binding interaction of the Au atom of aurothioglucose is with the two thiol groups of the CICVNVGCCT (N-terminal) active site. We recently demonstrated that the pKa values of these two thiol groups are: pKaIC = 5.8 and pKaCT = 5.02 (26). Thus, at pH 7, these thiol groups are strongly ionized and can attack the more positively charged Au atom of aurothioglucose (in comparison to auranofin) and form a strong complex. In addition, aurothioglucose is more compact than auranofin and can more easily fit into the tetrapeptide binding site of the enzyme (see Figure 7 for the structures of these compounds).
Table 7.
Inhibition of Mouse Mitochondrial TR and TR Mutants By Gold Compounds.

|
Taken from (35)
Figure 6.

Inhibition plots of TR using auranofin as an inhibitor (A) or aurothioglucose as an inhibitor (B) for mTRΔ8 (open triangles), mTR-GCCG (open squares), and mTR-GCUG (closed circles). IC50 values are calculated from this plot and reported in Table 6.
Figure 7.
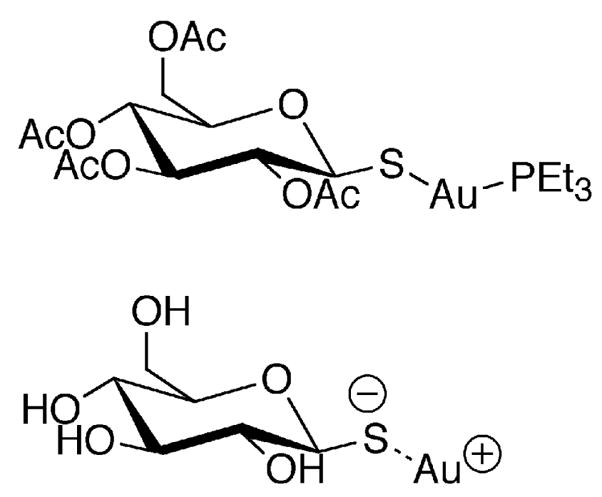
Structures of gold containing inhibitors of TR: auranofin (top), aurothioglucose (bottom).
The data in Table 7 potentially explains some of the differential effects that have been observed with these inhibitors. In a recent study it was shown that when cells were treated with aurothioglucose that there was no increase in the amount of oxidized Trx and there was also no increase in the production of reactive oxygen species (51). In contrast, cells treated with auranofin underwent apoptosis in a mechanism that depended on pro-apoptotic members of the Bcl-2 family (Bax/Bak). The auranofin treated cells exhibited signs of mitochondrial oxidative stress such as increased levels of peroxiredoxin 3 (52). Arnér and coworkers have recently characterized what they have termed as “SecTRAPs” (selenium compromised thioredoxin reductase derived apoptotic proteins). A SecTRAP is a form of TR in which the Sec residue has become modified with a cellular electrophile. The modification of the Sec residue apparently unmasks a new function of the enzyme, and it is this new function that causes apoptosis. This group also demonstrated that this new function of TR induced oxidative stress inside the cell and that this activity was dependent upon the CVNVGC active site (53). Our potential explanation is that auranofin complexes with the Se atom of TR forming a SecTRAP and enhances the function of the N-terminal reaction center and this new function leads to cellular apoptosis, while aurothioglucose completely inhibits the activity of the CVNVGC active site so that the toxic effects are not observed.
CONCLUSION
The presence of a rare Sec residue in mammalian TR is the frequently cited reason for the broad substrate specificity of the enzyme. This investigation has shown that this precept is not true for two reasons. First, Se-containing substrates can be reduced by the Cys2-mutant or by the N-terminal reaction center alone with only a slight decrease in activity. Second, some substrates such as lipoic acid/lipoamide can be reduced directly by the N-terminal reaction center as has been shown for ubiquinone and DTNB. Therefore, not all of the reported activities of the enzyme are due to the presence of this exotic amino acid. These two observations show that the enzyme is more defined by the types of bonds that the N-terminal reaction center will reduce than the by the presence of Sec. Our hypothesis is that the N-terminal reaction center will reduce bonds of the type S-Y, where Y is a good leaving group. This definition can be expanded to include highly electrophilic compounds such as selenite and disulfide bonds that are highly strained such as is the case with lipoic acid. We also suggest that substrates reduced by the N-terminal reaction center require a certain electrophilic threshold in order to be efficient substrates of the enzyme. This view highlights the electrophilic character of Se in the conserved redox active tetrapeptide, Gly-Cys1-Sec2-Gly, in the C-terminal reaction center. Given the findings presented here, a detailed investigation should be undertaken to determine which activities of the enzyme are due to the presence of a Se atom and which are due solely to the activity of the N-terminal reaction center. The data we presented with inhibitors aurothioglucose and auranofin also shows the need to re-examine the mechanism of inhibition of known inhibitors as they may either inhibit the enzyme via the Sec residue or inhibit the chemistry of the N-terminal reaction center.
Supplementary Material
Acknowledgments
We would like to thank Dr. Brian Eckenroth for constructing Figure 1 and part of Figure 2.
Footnotes
These studies were supported by National Institutes of Health Grant GM070742 to RJH.
C. elegans, Caenorhabditis elegans; CeTR2, mitochondrial TR from Caenorhabditis elegans; Cys, cysteine; DmTR, TR from Drosophila melanogaster; DTNB, 5,5′dithio-bis(2-nitrobenzoic acid); DTT, dithiothreitol; EDTA, ethylenediaminetetraacetic acid; Gly, glycine; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; lipoamide dehydrogenase, LipDH; Mr, molecular ratio; mTR3, mitochondrial TR from mouse; NADPH, β-nicotinamide adenine dinucleotide phosphate, reduced; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; Sec, selenocysteine; Ser, serine; TB, terrific broth; TR, thioredoxin reductase; Tris, tris-(hydroxymethyl)aminomethane; Trx, thioredoxin; U, the one letter code for Sec; WT, wild type.
LipDH normally converts dihydrolipoamide to lipoamide, but can also catalyze the reverse reaction, albeit with lower efficiency.
We have catalogued many of the statements in the literature that attribute the broad substrate specificity of mammalian TR to the presence of a Sec residue in the Supporting Information.
Throughout this report we use a nomenclature of TR-AA1AA2AA3AA4 to denote the source of the enzyme and its C-terminal tetrapeptide sequence. In addition, we use the nomenclature TRΔ2, TRΔ3, etc to denote the number of amino acids missing from the truncated enzyme in question.
SUPPORTING INFORMATION AVAILABLE
There is one Table and three Figures in the Supporting Information. Table S1 tabulates the concentration of the type of substrate, type of TR, and concentration of enzyme used in the assays with lipoic acid and lipoamide. Figures S1 and S2 show the pH rate profiles for WT and truncated enzymes with substrates selenite and selenocystine, respectively. Figure S3 shows the pH rate profile for mTRΔ8 with lipoamide as a substrate. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH., Jr The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc Natl Acad Sci USA. 1997;94:3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams CH. Lipoamide Dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase - a family of flavoenzyme transhydrogenases. In: Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. CRC Press; Boca Raton: 1992. pp. 121–211. [Google Scholar]
- 3.Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000;346(Pt 1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong L, Arnér ES, Ljung J, Aslund F, Holmgren A. Rat and calf thioredoxin reductase are homologous to glutathione reductase with a carboxyl-terminal elongation containing a conserved catalytically active penultimate selenocysteine residue. J Biol Chem. 1998;273:8581–8591. doi: 10.1074/jbc.273.15.8581. [DOI] [PubMed] [Google Scholar]
- 5.Bock A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F. Selenocysteine: The 21st amino acid. Mol Microbiol. 1991;5:515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 6.Atkins JF, Gesteland RF. The twenty-first amino acid. Nature. 2000;407:463–465. doi: 10.1038/35035189. [DOI] [PubMed] [Google Scholar]
- 7.Bar-Noy S, Gorlatov SN, Stadtman TC. Overexpression of wild type and SeCys/Cys mutant of human thioredoxin reductase E. coli the role of selenocysteine in the catalytic activity. Free Radic Biol Med. 2001;30:51–61. doi: 10.1016/s0891-5849(00)00448-2. [DOI] [PubMed] [Google Scholar]
- 8.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bjornstedt M, Xue J, Huang W, Akesson B, Holmgren A. The thioredoxin and glutaredoxin systems are efficient electron donors to human plasma glutathione peroxidase. J Biol Chem. 1994;269:29382–29384. [PubMed] [Google Scholar]
- 10.Kalantari P, Narayan V, Natarajan SK, Muralidhar K, Gandhi UH, Vunta H, Henderson AJ, Prabhu KS. Thioredoxin reductase-1 negatively regulates HIV-1 transactivating protein Tat-dependent transcription in human macrophages . J Biol Chem. 2008;283:33183–33190. doi: 10.1074/jbc.M807403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersson M, Holmgren A, Spyrou G. NK-lysin, a disulfide-containing effector peptide of T-lymphocytes, is reduced and inactivated by human thioredoxin reductase. Implication for a protective mechanism against NK-lysin cytotoxicity. J Biol Chem. 1996;271:10116–10120. doi: 10.1074/jbc.271.17.10116. [DOI] [PubMed] [Google Scholar]
- 12.Bjorkhem-Bergman L, Jonsson-Videsater K, Paul C, Bjornstedt M, Andersson M. Mammalian thioredoxin reductase alters cytolytic activity of an antibacterial peptide. Peptides. 2004;25:1849–1855. doi: 10.1016/j.peptides.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Bellisola G, Fracasso G, Ippoliti R, Menestrina G, Rosen A, Solda S, Udali S, Tomazzolli R, Tridente G, Colombatti M. Reductive activation of ricin and ricin A-chain immunotoxins by protein disulfide isomerase and thioredoxin reductase. Biochem Pharmacol. 2004;67:1721–1731. doi: 10.1016/j.bcp.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 14.May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J Biol Chem. 1997;272:22607–22610. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- 15.May JM, Morrow JD, Burk RF. Thioredoxin reductase reduces lipid hydroperoxides and spares alpha-tocopherol. Biochem Biophys Res Commun. 2002;292:45–49. doi: 10.1006/bbrc.2002.6617. [DOI] [PubMed] [Google Scholar]
- 16.Cenas N, Nivinskas H, Anusevicius Z, Sarlauskas J, Lederer F, Arnér ES. Interactions of quinones with thioredoxin reductase: a challenge to the antioxidant role of the mammalian selenoprotein. J Biol Chem. 2004;279:2583–2592. doi: 10.1074/jbc.M310292200. [DOI] [PubMed] [Google Scholar]
- 17.Nikitovic D, Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem. 1996;271:19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 18.Arnér ES, Nordberg J, Holmgren A. Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem Biophys Res Commun. 1996;225:268–274. doi: 10.1006/bbrc.1996.1165. [DOI] [PubMed] [Google Scholar]
- 19.Bjornstedt M, Kumar S, Bjorkhem L, Spyrou G, Holmgren A. Selenium and the thioredoxin and glutaredoxin systems. Biomed Environ Sci. 1997;10:271–279. [PubMed] [Google Scholar]
- 20.Bjornstedt M, Kumar S, Holmgren A. Selenodiglutathione is a highly efficient oxidant of reduced thioredoxin and a substrate for mammalian thioredoxin reductase. J Biol Chem. 1992;267:8030–8034. [PubMed] [Google Scholar]
- 21.Gromer S, Gross JH. Methylseleninate is a substrate rather than an inhibitor of mammalian thioredoxin reductase. Implications for the antitumor effects of selenium. J Biol Chem. 2002;277:9701–9706. doi: 10.1074/jbc.M109234200. [DOI] [PubMed] [Google Scholar]
- 22.Holmgren A, Lyckeborg C. Enzymatic reduction of alloxan by thioredoxin and NADPH-thioredoxin reductase. Proc Natl Acad Sci U S A. 1980;77:5149–5152. doi: 10.1073/pnas.77.9.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Bjornstedt M, Holmgren A. Selenite is a substrate for calf thymus thioredoxin reductase and thioredoxin and elicits a large non-stoichiometric oxidation of NADPH in the presence of oxygen. Eur J Biochem. 1992;207:435–439. doi: 10.1111/j.1432-1033.1992.tb17068.x. [DOI] [PubMed] [Google Scholar]
- 24.Eckenroth BE, Lacey BM, Lothrop AP, Harris KM, Hondal RJ. Investigation of the C-terminal redox center of high-Mr thioredoxin reductase by protein engineering and semisynthesis. Biochemistry. 2007;46:9472–9483. doi: 10.1021/bi7004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong L, Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J Biol Chem . 2000;275:18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- 26.Lacey BM, Eckenroth BE, Flemer S, Jr, Hondal RJ. Selenium in thioredoxin reductase: A mechanistic perspective. Biochemistry. 2008;47:12810–12821. doi: 10.1021/bi800951f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckenroth BE, Rould MA, Hondal RJ, Everse SJ. Structural and biochemical studies reveal differences in the catalytic mechanisms of mammalian and Drosophila melanogaster thioredoxin reductases. Biochemistry. 2007;46:4694–4705. doi: 10.1021/bi602394p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandt W, Wessjohann LA. The functional role of selenocysteine (Sec) in the catalysis mechanism of large thioredoxin reductases: proposition of a swapping catalytic triad including a Sec-His-Glu state. Chembiochem. 2005;6:386–394. doi: 10.1002/cbic.200400276. [DOI] [PubMed] [Google Scholar]
- 29.Gromer S, Wessjohann LA, Eubel J, Brandt W. Mutational studies confirm the catalytic triad in the human selenoenzyme thioredoxin reductase predicted by molecular modeling. Chembiochem. 2006;7:1649–1652. doi: 10.1002/cbic.200600080. [DOI] [PubMed] [Google Scholar]
- 30.Calcagni A, Lucente G, Luisi G, Pinnen F, Rossi D. Novel glutathione analogues containing the dithiol and disulfide form of the Cys-Cys dyad. Amino acids. 1999;17:257–265. doi: 10.1007/BF01366924. [DOI] [PubMed] [Google Scholar]
- 31.Morera E, Nalli M, Pinnen F, Rossi D, Lucente G. 4-Amino-1,2-dithiolane-4-carboxylic acid (Adt) as cysteine conformationally restricted analogue. Synthetic protocol for Adt containing peptides. Bioorg Med Chem Lett. 2000;10:1585–1588. doi: 10.1016/s0960-894x(00)00281-x. [DOI] [PubMed] [Google Scholar]
- 32.Eckenroth B, Harris K, Turanov AA, Gladyshev VN, Raines RT, Hondal RJ. Semisynthesis and characterization of mammalian thioredoxin reductase. Biochemistry. 2006;45:5158–5170. doi: 10.1021/bi0517887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biterova EI, Turanov AA, Gladyshev VN, Barycki JJ. Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc Natl Acad Sci U S A. 2005;102:15018–15023. doi: 10.1073/pnas.0504218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng Q, Sandalova T, Lindqvist Y, Arnér ES. Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J Biol Chem. 2009;284:3998–4008. doi: 10.1074/jbc.M807068200. [DOI] [PubMed] [Google Scholar]
- 35.Bauer H, Massey V, Arscott LD, Schirmer RH, Ballou DP, Williams CH., Jr The mechanism of high Mr thioredoxin reductase from Drosophila melanogaster. J Biol. 2003;278:33020–33028. doi: 10.1074/jbc.M303762200. [DOI] [PubMed] [Google Scholar]
- 36.Huber RE, Criddle RS. Comparison of the chemical properties of selenocysteine and selenocystine and their sulfur analogs. Arch Biochem Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- 37.Danehy JP, Elia VJ, Lavelle CJ. Alkaline decomposition of organic disulfides. IV Limitation on the use of Ellman’s reagent 2,2′Dinitro-5,5′dithiodibenzoic acid. J Org Chem. 1971;36:1003–1005. [Google Scholar]
- 38.Back TG. Electrophilic Selenium Reactions. In: Liotta D, editor. Organoselenium Chemistry. Wiley-Interscience; New York: 1987. pp. 1–125. [Google Scholar]
- 39.Muttenthaler M, Alewood PF. Selenopeptide chemistry. J Pept Sci. 2008;14:1223–1239. doi: 10.1002/psc.1075. [DOI] [PubMed] [Google Scholar]
- 40.Danehy JP, Noel CJ. The relative nucleophilic character of several mercaptans toward ethylene oxide. J Am Chem Soc. 1960;82:2511–2515. [Google Scholar]
- 41.Graetzel M, Taniguchi S, Henglein A. Pulse radiolytic study of short-lived byproducts of nitric oxide-reduction in aqueous solution. Ber Bunsenges Phys Chem. 1970;74:1003–1010. [Google Scholar]
- 42.Fujiwara N, Fujii T, Fujii J, Taniguchi N. Roles of N-terminal active cysteines and C-terminal cysteine-selenocysteine in the catalytic mechanism of mammalian thioredoxin reductase. J Biochem. 2001;129:803–812. doi: 10.1093/oxfordjournals.jbchem.a002923. [DOI] [PubMed] [Google Scholar]
- 43.Gascoigne IM, Radda GK. The reaction of dihydrolipoic acid with flavins. Biochim Biophys Acta. 1967;131:498–507. doi: 10.1016/0005-2728(67)90009-6. [DOI] [PubMed] [Google Scholar]
- 44.Singh R, Lamoureux GV, Lees WJ, Whitesides GM. Reagents for rapid reduction of disulfide bonds. Methods Enzymol. 1995;251:167–173. doi: 10.1016/0076-6879(95)51119-9. [DOI] [PubMed] [Google Scholar]
- 45.Tajc SG, Tolbert BS, Basavappa R, Miller BL. Direct determination of thiol pKa by isothermal titration microcalorimetry. J Am Chem Soc. 2004;126:10508–10509. doi: 10.1021/ja047929u. [DOI] [PubMed] [Google Scholar]
- 46.Singh R, Whitesides GM. Degenerate intermolecular thiolate-disulfide interchange involving cyclic five-membered disulfides is faster by ~103 than that involving six- or seven-membered disulfides. J Am Chem Soc. 1990;112:6304–6309. [Google Scholar]
- 47.Burns JA, Whitesides GM. Predicting the stability of cyclic disulfides by molecular modeling: Effective concentrations in thiol-disulfide interchange and the design of strongly reducing dithiols. J Am Chem Soc. 1990;112:6296–6303. [Google Scholar]
- 48.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem. 1998;273:20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 49.Hill KE, McCollum GW, Boeglin ME, Burk RF. Thioredoxin reductase activity is decreased by selenium deficiency. Biochem Biophys Res Commun. 1997;234:293–295. doi: 10.1006/bbrc.1997.6618. [DOI] [PubMed] [Google Scholar]
- 50.Kean WF, Kean IR. Clinical pharmacology of gold. Inflammopharmacology. 2008;16:112–125. doi: 10.1007/s10787-007-0021-x. [DOI] [PubMed] [Google Scholar]
- 51.Watson WH, Heilman JM, Hughes LL, Spielberger JC. Thioredoxin reductase-1 knock down does not result in thioredoxin-1 oxidation. Biochem Biophys Res Commun. 2008;368:832–836. doi: 10.1016/j.bbrc.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cox AG, Brown KK, Arnér ES, Hampton MB. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem Pharmacol. 2008;76:1097–1109. doi: 10.1016/j.bcp.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 53.Anestal K, Prast-Nielsen S, Cenas N, Arnér ES. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PLoS ONE. 2008;3:e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.