

Abstract
HIV-1 protease (PR) and its mutants are important antiviral drug targets. The PR flap region is critical for binding substrates or inhibitors and catalytic activity. Hence, mutations of flap residues frequently contribute to reduced susceptibility to PR inhibitors in drug resistant HIV. Structural and kinetic analyses were used to investigate the role of flap residues Gly48, Ile50 and Ile54 in the development of drug resistance. The crystal structures of flap mutants PRI50V, PRI54V, and PRI54M complexed with saquinavir (SQV) and PRG48V, PRI54V, and PRI54M complexed with darunavir (DRV) were determined at the resolutions of 1.05–1.40 Å. The PR mutants showed changes in flap conformation, interactions with adjacent residues, inhibitor binding and the conformation of the 80’s loop relative to the wild type PR. The PR contacts with darunavir were closer in PRG48V-DRV than in the wild type PR-DRV, while they were longer in PRI54M-DRV. The relative inhibition of PRs with mutations I54V and I54M was similar for saquinavir and darunavir. PRG48V was about 2-fold less susceptible to saquinavir than to darunavir, while the opposite was observed for PRI50V. The observed inhibition was in agreement with the association of G48V and I50V with clinical resistance to saquinavir and darunavir, respectively. This analysis of structural and kinetic effects of the mutants will assist in development of more effective inhibitors for drug resistant HIV.
Keywords: drug resistance, aspartic protease, saquinavir, darunavir (TMC114)
INTRODUCTION
HIV-1 (Human immunodeficiency virus type 1) protease (PR) is an effective drug target for antiviral inhibitors. Each subunit of the active PR homodimer has a glycine-rich region termed the flap comprising residues Lys45-Met-Ile-Gly-Gly-Ile-Gly-Gly-Phe-Ile-Lys55, which fold into two anti-parallel β strands. The flexible flaps participate in the binding of a substrate or inhibitor in the active site cavity of PR 1; 2; 3. The importance of residues in the flap for PR activity has been characterized through large scale mutagenesis 4. The residues Met46, Phe53 and Lys55 are the most tolerant of substitutions; Ile47, Ile50, Ile54 and Val56 only tolerate a few conservative substitutions; and Gly48, Gly49, Gly51, Gly52 are the most sensitive to mutation. Therefore, mutations in the flap residues may alter the enzyme activity, conformation and flexibility of the flap. Mutations in flap residues 46, 47, 48, 50, 53, and 54 are frequently observed in drug resistant mutants of HIV and show various levels of reduced drug susceptibility to different PR inhibitors (PIs) 5; 6. Altered flap conformations have been reported for protease and its mutants in the unliganded form 7; 8; 9 or with bound peptide 10. Previously, we have analyzed the structures, activities, inhibition and stability of PR variants with the single substitutions of flap residues M46L, G48V, I50V, and F53L 8; 11; 12; 13; 14; 15. These studies use PR from HIV-1 subtype B, which forms about 10% of global infections with HIV-1 group M compared to ~50% for subtype C 16. Recently, the PRs from subtype C and F were analyzed 17; 18. These subtypes contain 8–10 polymorphic substitutions in the sequence outside of the flaps and inhibitor binding site, which can influence catalytic activity and inhibition. The PR structures and interactions with inhibitors were very similar to those of the B subtype PR, with larger structural variations in surface residues 34–42 for the F subtype PR and 65–69 for the C subtype PR.
Saquinavir (SQV) was the first PR inhibitor to be approved by the FDA in 1995, and is still widely used in AIDS therapy, along with nine other inhibitors. The latest, darunavir (DRV, previously known as TMC114), a chemical derivative of the PR inhibitor amprenavir, was approved in 2006 for salvage therapy and is extremely potent against the wild type PR and most drug resistant mutants in vitro and in vivo 11; 19; 20; 21. Darunavir, boosted with ritonavir, is recommended for treatment-experienced patients who respond poorly to other PIs. Saquinavir was designed to target the wild type PR and its chemical structure contains a number of peptidic main chain groups mimicking a natural substrate of PR as shown in Figure 1a 22. In contrast, darunavir was designed to be less peptidic while introducing more hydrogen bond interactions with the main chain atoms of PR in order to maintain its effectiveness on PR variants 20; 23.
Figure 1.

(a) The chemical structures of saquinavir and darunavir. (b) Structure of HIV-1 PR dimer with the locations of mutated residues Gly48 (cyan), Ile50 (red), Ile54 (green) indicated by spheres for main chain atoms in both subunits. Darunavir is shown in sticks colored by atom type. The flap residues (45–55) and the 80’s loop (78–82) are colored in blue and purple, respectively.
In this study, PR variants with the individual flap mutations G48V, I50V, I54V and I54M were analyzed to gain insight into their role in the development of drug resistance. G48V is one of the primary drug resistant mutations selected during treatment with saquinavir 24; 25. I50V arises in treatment with amprenavir, and also confers resistance to darunavir 5. Mutations of I54M and I54V are commonly observed during therapy with multiple PR inhibitors 5; 26; 27; 28. Several mutations of Ile54 are present in isolates with reduced susceptibility to saquinavir. Mutations I54M and I54L are frequent in clinical isolates resistant to darunavir 29. Moreover, Met was the most frequently identified substitution of residue 54 after treatment with amprenavir, which is chemically related to darunavir 28. Residue 50 lies at the tip of the PR flap, while residues 48 and 54 are located on opposite β strands of the flap (Figure 1b). Previously, the crystal structure of the double mutant G48V/L90M with saquinavir was analyzed 30, and we reported the structure of the PRI50V mutant with darunavir 11. Here, the crystal structures of flap mutants PRG48V, PRI50V, PRI54V, and PRI54M were solved in complexes with saquinavir and darunavir. Comparison of the mutant and wild type structures revealed changes in the flap conformation, interactions between flap residues from the two PR subunits, inhibitor binding and conformation of residues 78–82 (the 80’s loop). The kinetic data are discussed in relation to the structural changes. This analysis confirmed the important roles of residues in the flaps and enhanced our understanding of the drug resistant mechanisms used by the flap mutants.
RESULTS AND DISCUSSION
Kinetics
The wild type HIV-1 PR in these studies contains mutations Q7K, L33I, and L63I to diminish autoproteolysis and C67A and C95A to prevent cysteine-thiol oxidation, and showed almost identical kinetic parameters, stability and dimer dissociation as the unmutated wild type PR 31. Kinetic parameters were measured for the resistant mutants and the wild type PR using the fluorescence substrate based on the p2-NC cleavage site of HIV-1 (Table 1). The mutants PRG48V, PRI50V and PRI54V had reduced catalytic efficiency (kcat/Km) of about 10–40% of wild type PR value, while the catalytic efficiency of PRI54M was similar to that of PR. The mutants showing reduced activity are likely to be less effective during viral replication.
Table 1.
Kinetic parameters for substrate hydrolysis and inhibition of darunavir and saquinavir.
| Mutant | Km (μM) | kcat (/min) | kcat /Km (/min.μM) | Relative kcat/ Km | Ki (nM) | Relative Ki | Ki (nM) | Relative Ki |
|---|---|---|---|---|---|---|---|---|
| DRV | SQV | |||||||
| PR | 30±5 | 194±23 | 7.4±1.2 | 1.0 | 0.58±0.10 | 1 | 0.42±0.07 | 1 |
| PRG48V | 91±8 | 220±9 | 2.4±0.2 | 0.3 | 17±2 | 29 | 36±3 | 86 |
| PRI50V | 109±4 | 68±2 | 0.62±0.02 | 0.1 | 18±1 | 31 | 10±1 | 24 |
| PRI54V | 43±9 | 134±28 | 3.1±0.7 | 0.4 | 5±1 | 8 | 6±1 | 15 |
| PRI54M | 41±3 | 301±22 | 7.3±0.5 | 1.0 | 1.6±0.1 | 3 | 2.2±0.3 | 5 |
The four mutants and the wild type PR were assayed for inhibition by saquinavir and darunavir (Table 1). Compared to wild type PR, mutant PRG48V was poorly inhibited by saquinavir and darunavir with 90- and 30-fold higher inhibition constants (Ki), respectively. The Ki values of PRI50V for saquinavir and darunavir were increased 30-fold and 24-fold, and the Ki values of PRI54V were moderately increased by 8-fold and 15-fold for saquinavir and darunavir, relative to wild type PR values. In contrast, PRI54M showed similar relative Ki values of 3 and 5for the two inhibitors. Therefore, the mutant PRG48V was the most resistant to those inhibitors, while PRI54M was the most similar to the wild type enzyme. PRI50V was inhibited more effectively by saquinavir than by darunavir, in agreement with the presence of the I50V mutation in HIV isolates resistant to darunavir and its rarity in saquinavir treatment. However, PRG48V was more susceptible to darunavir than saquinavir, in agreement with the presence of G48V in clinical isolates failing saquinavir therapy. Hence, darunavir is likely to be a better drug for patients carrying HIV PR with the G48V mutation selected by saquinavir treatment, while saquinavir may be better for resistant HIV with the I50V mutation.
Dimer Stability
The dimer stability of the four flap mutants (PRG48V, PRI50V, PRI54V, and PRI54M) and the wild type PR was evaluated by assessing the PR activity with increasing concentrations of denaturing urea. When the PR activity drops to 50% of the initial value, the concentration of urea is defined as UC50. The UC50 value of PRI50V was reduced to 60% of that for the wild type PR, while the UC50 values of the other three mutants were very similar to that of the wild type PR (90–110%). Earlier, we demonstrated that another flap mutant PRF53L had reduced UC50 of 60% of the value for the wild type PR 13. Therefore, the mutations of flap residues Ile50 and Phe53 adversely affect the PR dimer stability, while mutations G48V and I54V/M had no significant effect on the dimer stability in this assay.
Crystal Structures
The crystal structures of PRI50V, PRI54V, and PRI54M complexed with saquinavir and PRG48V, PRI54V, and PRI54M complexed with darunavir were determined at resolutions of 1.05–1.40 Å. No crystals were obtained for the complex of PRG48V with saquinavir, and the structure of PRI50V with darunavir was reported previously 11. Three data sets (PRI54M-SQV, PRI54V-SQV, PRI54V-DRV) reached atomic resolution (1.05 Å). The data collection and refinement statistics are provided in Table 2. All six structures crystallized in isomorphous unit cells and the same space group P21212. The R-factors were refined to the range of 0.12 to 0.16. One asymmetric unit accommodated one PR dimer, with residues labeled 1–99 and 1′–99′ for each subunit. The electron density maps clearly showed the correct mutations in all the complexes as illustrated in Figure 2. Most of the atoms were clearly visible in the electron density maps. An example from the flap region of PRI54V-SQV is shown in Figure 2. Darunavir was bound at the active site of PR with two pseudosymmetric orientations in the complexes with PRG48V, PRI54V, and PRI54M, while saquinavir showed a single orientation in the complexes of PRI54V-SQV and two orientations in complexes of PRI54M-SQV and PRI50V-SQV. The omit maps of saquinavir (in PRI54V-SQV) and darunavir (in PRI54V-DRV) are shown in Figure 3. Alternate conformations were modeled for side chain and main chain atoms when observed in the electron density map. Met54 showed alternate conformations of the side chain only in the PRI54M-SQV complex (Figure 2) and Val54 showed alternate conformations of the side chain only in PRI54V-SQV complex. As frequently observed in near atomic resolution crystal structures, residue 50 showed alternate conformations for both main chain and side chain atoms in all complexes, except in the lower resolution structure of PRG48V-DRV, 11; 13; 22. In addition, water molecules (161–223) and other solvent molecules including glycerol, phosphate, sodium ions and chloride ions with full or partial occupancy were modeled to fit the maps in the different structures.
Table 2.
Crystallographic Data Collection and Refinement Statistics
| Protease Mutant | PRI50V | PRI54V | PRI54M | PRG48V | PRI54V | PRI54M | |
|---|---|---|---|---|---|---|---|
| Inhibitors | SQV | SQV | SQV | DRV | DRV | DRV | |
| Space group | P21212 | P21212 | P21212 | P21212 | P21212 | P21212 | |
| Unit cell dimensions (Å) | a | 58.9 | 58.7 | 59.5 | 58.2 | 58.8 | 58.6 |
| b | 86.0 | 85.9 | 85.7 | 86.2 | 86.1 | 85.7 | |
| c | 46.4 | 46.38 | 46.2 | 45.9 | 46.2 | 46.0 | |
| Unique reflections | 69158 | 101144 | 103172 | 40435 | 100265 | 53524 | |
| Rmerge (%) Overall (final shell) | 8.9 (53.7) |
10.0 (42.3) |
8.8 (25.8) |
9.4 (20.3) |
10.8 (41.7) |
8.9 (57.6) |
|
| I/sigma(I) Overall (final shell) | 13.5 (1.8) |
16.8 (2.5) |
21.9 (7.0) |
13.2 (3.0) |
17.8 (1.8) |
24.4 (2.2) |
|
| Resolution range for refinement (Å) | 10–1.20 | 10–1.05 | 10–1.05 | 10–1.40 | 10–1.05 | 10–1.30 | |
| Rwork (%) | 15.0 | 15.0 | 11.9 | 16.0 | 15.9 | 15.1 | |
| Rfree (%) | 19.2 | 17.4 | 14.7 | 22.7 | 17.96 | 19.5 | |
| No. of waters | 200 | 223 | 209 | 174 | 212 | 161 | |
| Completeness (%) Overall (final shell) | 93.1 (79.4) |
92.5 (58.1) |
93.4 (79.3) |
87.5 (55.8) |
91.2 (58.7) |
94.4 (80.8) |
|
| RMS deviation from ideality | |||||||
| Bonds (Å) | 0.014 | 0.017 | 0.017 | 0.011 | 0.017 | 0.013 | |
| Angle distance (Å) | 0.032 | 0.033 | 0.036 | 0.029 | 0.037 | 0.034 | |
| Average B-factors (Å2) | |||||||
| Main chain | 10.2 | 14.0 | 10.1 | 15.7 | 15.5 | 16.9 | |
| Side chain | 18.7 | 19.9 | 19.1 | 25.8 | 21.4 | 26.4 | |
| Inhibitor | 9.4 | 17.0 | 10.8 | 13.8 | 12.8 | 16.8 | |
| Solvent | 26.1 | 33.5 | 26.0 | 29.8 | 32.0 | 19.0 | |
| Inhibitor occupancy (%) | 80/20 | 100 | 75/25 | 60/40 | 60/40 | 70/30 | |
Figure 2.

The Fo-Fc omit maps showing the mutated residues and the flap residues (47–54) contoured at 3.3 sigma. Val 48 is from PRG48V-DRV, Val50 from PRI50V-SQV, Val54 from PRI54V-SQV, Met54 from PRI54M-SQV and flap residues from PRI54M-SQV. The cyan sticks indicate the alternate conformations of main-chain and side-chain atoms in Val50 and Met54.
Figure 3.
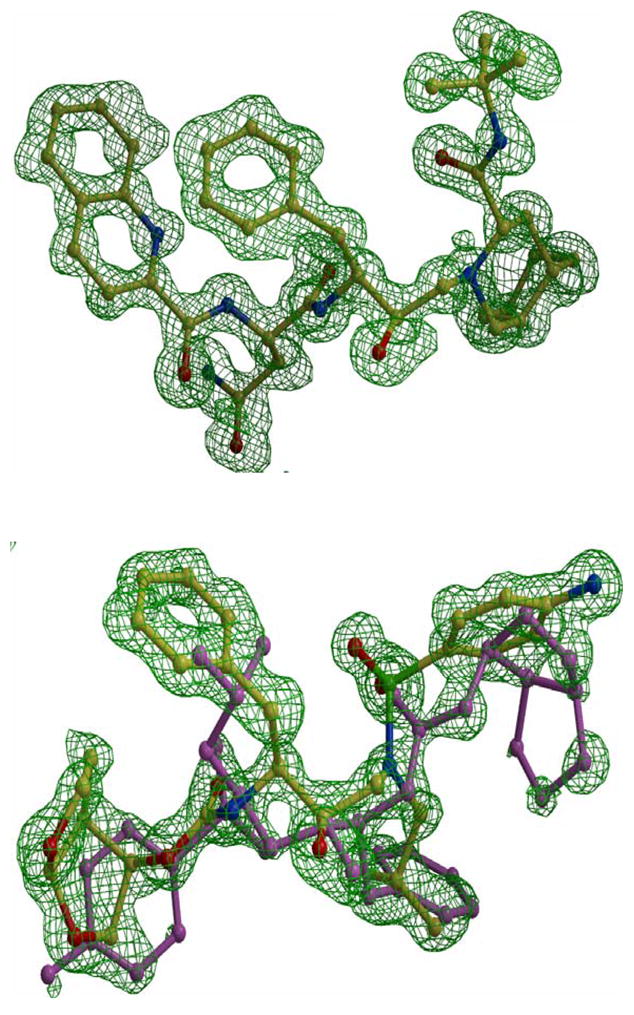
The Fo-Fc omit maps of saquinavir (upper panel) and darunavir (lower panel) contoured at 3.3 sigma. Saquinavir is colored by atom type from complex PRI54V-SQV. Darunavir is from complex PRI54V-DRV showing alternate conformations of 60/40% occupancy. The major conformation is colored by atom type and the minor is colored pink.
Comparison of Protease Structures
The mutant PR complexes were compared to the PR structures with the corresponding inhibitor, which were determined in our previous studies. The same PR sequence is used to construct all mutants so any changes represent the effects of the introduced mutations. The wild type PR-DRV (2IEN) 21 structure has been solved at 1.30 Å resolution in space group P21212, while the PR-SQV structure has been determined to 1.16 Å resolution in P21212122. The mutations in the flap are expected to alter interactions with neighboring residues, the flap conformation and inhibitor binding. The overall structural differences between mutant PRs and the wild type PRs are shown by the RMS deviations, which do not reflect significant local differences, as observed in other PR structures 8; 12. The pairwise overall RMS deviations of Cα were 0.1–0.3 Å for complexes with darunavir with the same unit cell and space group. The complex PRI54V-DRV is the most similar to the wild type PR-DRV, while the other two complexes have similar deviations (0.3 Å) from the wild type PR. The pair-wise overall RMS deviations of Cα for the three complexes with saquinavir were very similar in the range of 0.6–0.7 Å, as observed for comparison of HIV PR structures in different space groups 13; 22; 32. For all complexes, larger deviations were consistently located around the tip of the flap and 80’s loop (residues 78–82) in both subunits. The alternate conformations of the main chain of residues 50/50′ contributed to the higher RMS deviations in the flaps. Structural differences for flap residues 47–54 are shown in Figure 4. PRG48V had the biggest difference from the wild type PR among all the complexes with darunavir. The Cα atom of Ile50 shifted about 0.8 Å towards the active site cavity in one subunit and 0.5 Å at the equivalent position in the other subunit. In high resolution (better than 1.5 Å) structures, the atomic shifts over 0.3 Å are considered to be significant changes, using the estimation of crystallographic errors described in 33.
Figure 4.

The flap regions for superimposed complexes with darunavir. The wild type PR is in green, PRG48V in cyan, PRI54V in magenta and PRI54M in red. The arrows indicate the shifts between Cα atoms of PRG48V and of the wild type PR with distances in Å.
Notably, in all the mutant structures, the main chain atoms of residues 78–82 (the 80’s loop) were shifted relative to their positions in wild type PR. The 80’s loop shows intrinsic flexibility as described previously for atomic resolution structures of saquinavir complexes with wild type PR and mutants V82A and I84V 22 and a 9X mutant with indinavir 34. The shifts appeared in both subunits of the dimer, and were slightly larger in one subunit than the other. The shifts of the 80’s loop were related to the size of the side chain in the mutated residues, independent of the type of bound inhibitor. The 80’s loop shifts away from a longer side chain (as in PRI54M) and towards a smaller side chain (as in PRI54V) to maintain the separation between residues 50, 54 and the 80’s loop. However, the observed conformational changes in the 80’s loop did not produce substantial changes in the interactions with inhibitors. Furthermore, the flap mutants showed varied effects for the interactions of the mutated residues with the bound inhibitors, as discussed for each mutant individually.
Protease Interactions with Saquinavir and Darunavir
The interatomic interactions that contribute to the binding of inhibitor in the active site cavity of PR include: strong N-H…O and O-H…O hydrogen bonds, weaker C-H…O, C-H…πinteractions, and the weakest van der Waals C-H…H-C contacts, as described previously for darunavir 12. The different interactions are illustrated for the complexes of PRI54M with the two inhibitors (Figure 5). Saquinavir with 49 non-hydrogen atoms is larger than darunavir with 38 non-hydrogen atoms. Saquinavir has larger hydrophobic groups at both ends of the molecule and forms more van der Waals interactions with numerous PR residues compared to darunavir 22. Saquinavir forms eight direct hydrogen bonds, three water-mediated hydrogen bonds, and one C-H…O interaction with Gly 49. However, darunavir forms two more direct hydrogen bonds with the main chain atoms of Asp29 and Asp30 and two more C-H…O interactions with main chain atoms than does saquinavir 21; 22. Similar hydrogen bond interactions are retained in the inhibitor complexes with wild type protease and the other mutants. The only exception is the interaction of the aniline NH2 of darunavir with the carboxylate oxygen of Asp30, which is a direct hydrogen bond in the wild type and other mutant complexes, and a water-mediated interaction for the major darunavir conformation in the PRI54M-DRV complex. The van der Waals and C-H…π interactions between PR and hydrophobic groups of saquinavir are likely to be vulnerable to changes due to mutations of hydrophobic residues in the binding site. On the contrary, darunavir was designed to form more hydrogen bond interactions with main-chain atoms of PR, which cannot be directly altered by mutations 23. Therefore, the binding of darunavir can adapt to the changes due to mutations of hydrophobic residues in the active site cavity. Through this strategy darunavir retains its effectiveness for many drug resistant mutants, with the exception of PRI50V due to the loss of hydrogen bond interactions 11; 20; 21. However, altered interactions are observed in some mutant complexes as described in the following sections, which are organized by individual mutants.
Figure 5.

PRI54M interactions with inhibitor. (a) The major orientation of saquinavir. (b) The major orientation of darunavir. Hydrogen bonds are indicated in red, CH-π interactions in blue and C-H…O in purple. Interatomic distances are shown in Å. Note that the water-mediated interaction of the NH2 of darunavir with the Asp30 side chain is replaced by a direct hydrogen bond in the wild type PR and the other mutant structures.
PRG48V-DRV
Residue Gly48 is located in the active site cavity and its carbonyl O makes C-H…O interactions with darunavir in the wild type PR complexes. Replacing Gly48 by Val in PRG48V is expected to disrupt the interactions with neighboring residues on the flap, and possibly alter the interactions with the inhibitor, thus destabilizing the flap and reducing the affinity for inhibitor. Despite extensive efforts, no crystals were obtained for the single mutant PRG48V in complex with saquinavir. Fortunately, crystals were obtained for PRG48V with darunavir that diffracted to 1.40 Å. The comparison of PRG48V-DRV with the wild type PR-DRV complex showed that the Cα of Val48 in both subunits had lost interactions with the Phe53 aromatic ring, while new hydrophobic interactions were observed for the Val48 side chain (Figure 6a). In one subunit, residues Gly49 and Ile50 have shifted 0.7 Å away from the neighboring residues Gly52 and Phe53, while there is relatively little change in the position of the Phe53 side chain relative to the wild type. As a result, the Cα of Val48 has lost most of the favorable C-H…π interactions with the aromatic ring of Phe53. However, the Cβ and Cγ atoms of Val48 have gained new interactions with the aromatic ring of Phe53. In the other subunit, the side chain of Phe53′ had rotated about 180° relative to its position in the wild type PR, which eliminated C-H…πinteractions with the Cα of Gly48′. However, a new C-H…O interaction was formed between the Cγ of Val48′ and the carbonyl O of Gly52′, which partially compensated for the lost contacts. Thus, the introduction of the larger Val side chain at position 48 has disturbed the interactions with the neighboring residues in the complex of PRG48V-DRV, while structural adjustments partially compensated for lost interactions.
Figure 6.


(a) Comparison of the flaps of PRG48V-DRV and of wild type PR-DRV in the two subunits. (b) Selected PRG48V interactions with darunavir in the minor conformation. The mutant structures are colored by atom type and wild type PR is in grey. Dashed lines indicate van der Waals interactions with interatomic distances shown in Å. The arrows show the shift between the two structures with distances in Å.
Other structural changes in the PRG48V-DRV complex were observed for the 80’s loop. Residue 48 is located near the 80’s loop of the same PR subunit, but no van der Waals contacts shorter than 4.2 Å were observed between the two regions. In the mutant, the 80’s loop has shifted up to 0.7 Å towards Val48 relative to its position in the wild type structure; nevertheless the residues remained too far apart to make favorable interactions.
The main chain carbonyl group of Gly48 makes direct interactions with the major (60%) and minor (40%) orientations of darunavir, as shown in Figure 5b. In the mutant the main chain atoms of Val48 have shifted by as much as 0.9 Å at the carbonyl oxygen relative to their positions in wild type PR-DRV structure. As a result, the carbonyl oxygen of Val48 has gained additional interactions with darunavir, which are not observed in the wild type structure. There are several shorter and stronger C-H…O interactions with the minor orientation of darunavir, and the side chain of Val48 has gained a new van der Waals interaction with the aromatic ring of the inhibitor (Figure 6b). On the other hand, the major orientation has similar contacts to those of the wild-type complex. However, the favorable interactions described above are likely offset by several repulsive short C-H…H-C hydrophobic contacts made with the side chain atoms of Pro81, Pro81′ and Ile84′. The P1 benzene ring of darunavir in both orientations is involved in short 3.1–3.5 Å contacts with residues 81 and 81′, while the aniline part of the minor darunavir orientation makes very short (2.7–3.2 Å) interactions with Cδ of Ile84′. These short contacts were not observed in the wild type PR-DRV structures or other mutant complexes examined 11; 12; 13; 35. Therefore, PRG48V-DRV represents a unique structure with unfavorably close PR-ligand interactions, rather than longer or lost contacts as observed in other mutant complexes. In agreement with the structural changes, the Ki value for the PRG48V inhibition by darunavir increased ~ 30 times relative to the wild type PR, suggesting that the repulsive contacts have a significantly negative effect on the darunavir affinity for the mutant PR.
The crystal structure of PRG48V-SQV is not available for comparison. However, the crystal structure of the double mutant PRG48V/L90M with SQV (1FB7) shows that saquinavir moves to accommodate the bigger side chain of Val48, giving rise to weaker interactions than in the PR-SQV structure 30. The carbonyl O of Val48 loses the hydrogen bond interaction with SQV. In addition, there are fewer van der Waals interactions between saquinavir and the flap (residues 47–50). These structural changes and reduced protease-inhibitor interactions, including loss of a hydrogen bond, explain why G48V is a primary mutation observed in isolates that are resistant to saquinavir. On the other hand, the G48V mutation is not considered to be resistant to darunavir, although potentially repulsive protease-inhibitor contacts were observed in the new structure of PRG48V-DRV.
PRI50V Complexes
The new structure of PRI50V-SQV is described and compared with the structure of PRI50V- DRV (2FGG). Residue 50 lies at the tip of the flap and the carbonyl oxygen forms a hydrogen bond interaction with the main chain amide of Gly51′ from the other subunit. The role of Ile/Val50 in binding of indinavir or darunavir has been described previously 11; 13. In the wild type PR, the side chain of Ile50 forms van der Waals interactions with Thr80′ and Pro81′. Thr80 has been proposed to maintain the mobility of the flap tips by pulling Ile50′ of the other subunit out of a hydrophobic pocket 36. In PRI50V-SQV, the smaller side chain in Val50 has induced a shift towards the 80’s loop to maintain favorable contacts (Figure 7a). Moreover, the side chain of Val50 has rotated to gain van der Waals interactions with Ile54′. Therefore, the PRI50V-SQV structure shows adaptations to improve intersubunit contacts, unlike the PRI50V-indinavir complex that showed reduced contacts between the two subunits at the flap tip 13.
Figure 7.


The interactions of residues 50, 51, 54 and 79–81 in (a) PRI50V-SQV, (b) PRI54M-DRV, and (c) PRI54V-DRV. The structures of mutants are colored by atom types and the corresponding structures of wild type PR are grey. Dashed lines indicate van der Waals interactions with interatomic distances shown in Å. The arrows show the shift between the two structures with distances in Å.
Overall, the interactions of PRI50V and saquinavir show little change relative to those of wild type PR-SQV, even for residue 50. Val50 in one subunit has lost two van der Waals interactions with saquinavir compared with those of Ile50, while in the other subunit this residue has gained a new van der Waals interaction with saquinavir. In contrast, our previous studies showed that several attractive interactions between the side chain of Ile50 and inhibitor were lost in the complexes of PRI50V with darunavir and indinavir 11; 13. Moreover, the aniline of darunavir had lost two hydrogen bonds with the main chain of Asp30, in agreement with the higher relative Ki for the mutant. All three inhibitors show reduced interactions and higher relative Ki values forPRI50V compared to wild type enzyme. However, the loss of hydrogen bonds in the mutant PRI50V-DRV complex is consistent with the presence of this mutation in isolates resistant to darunavir. Also, the absence of large structural changes in the saquinavir complex agrees with the rarity of the I50V mutation during resistance to saquinavir.
PRI54M and PRI54V complexes with DRV and SQV
Residue 54 is located on the opposite side of the flap from residue 48, and close to residue 50 and the 80’s loop (Figure 1). Significant structural changes were observed at the 80’s loop in the complexes of mutants with saquinavir and darunavir in comparison with the corresponding wild type PR complexes (Figure 7). The 80’s loop was shifted away from Met54 in PRI54M and towards Val54 in PRI54V to compensate for the different sizes of the side chains of residue 54. In PRI54M-DRV, the carbonyl O of Pro79 has shifted away from the longer Met54 by 0.7 Å and 1.4 Å in the two subunits, respectively, in order to accommodate the longer Met side chain. Despite this movement, Met54 has gained more van der Waals contacts with Pro79 and the side chain of Ile50′ than are observed for wild type Ile54. Moreover, the flap tip has shifted towards Met54 by 0.5 Å at the carbonyl O of Gly51, because there is more space due to the absence of the β-branched methyl group of Ile in Met54. On the other hand, the mutation I54V introduces a smaller side chain, so the 80’s loop has shifted towards Val54 by 0.6 Å at the carbonyl O of Pro79′ in only one subunit of PRI54V-DRV. However, no new interactions are formed between Val54 and the 80’s loop (Figure 7c). Unlike in PRI54M-DRV, the tip of the flap had no significant change in PRI54V–DRV relative to the wild type PR structure.
The shift of the 80’s loop caused by the mutation of residue 54 has modified the interactions with inhibitor in PRI54MDRV, but not in the other complexes. In PRI54M-DRV, residues 78–82 were further away from 54 in the flap, so the interactions between Pro81-Val82 and the major conformation of darunavir were a little weaker (the interatomic distances are 0.3–0.4 Å longer) than in the wild type complex. For the major conformation of darunavir, the direct hydrogen bond interaction between the aniline NH2 of darunavir and OD2 of Asp30 in the wild type PR was replaced by a water-mediated interaction in PRI54M-DRV (Figure 5). For the minor conformation of darunavir, however, the direct hydrogen bond interaction was maintained, while the hydrogen bond interaction between the NH2 of darunavir and the carbonyl O of Asp30 was lost (interatomic distance is 4.5 Å). On the other hand, no significant structural changes in the PR-inhibitor interactions were observed in the PRI54V-DRV, PRI54V-SQV and PRI54M-SQV complexes relative to the corresponding wild type PR complexes. In summary, the I54M mutation caused larger structural changes than I54V in the flap and in the interactions with darunavir, which is consistent with the more frequent occurrence of mutation I54M compared to I54V in the treatment with amprenavir and darunavir 28.
Structural and kinetic changes and drug resistance
These new crystal structures of the drug resistant mutants revealed changes in PR conformation and interaction with inhibitor relative to the equivalent wild type PR complexes. The introduction of mutations in the flap caused conformational modifications of the flap in all the complexes, with the biggest deviations observed in the PRG48V-DRV complex. In all the complexes, the 80’s loop has flexibly adjusted to accommodate the size of neighboring residues 50 and 54. It shifted towards smaller residues like Val50 and Val54, and away from the longer residue Met54. However, the shifts of the 80’s loop did not significantly alter the interactions between inhibitor and PR, except in the cases of PRG48V-DRV and PRI54M-DRV. The van der Waals interactions with darunavir were enhanced in PRG48V-DRV but were weakened slightly in PRI54M-DRV in correlation with the shift of the 80’s loop.
Analysis of the available structures of these flap mutants with saquinavir or darunavir showed the largest changes in PRG48V/L90M-SQV 30 and PRI50V–DRV relative to the corresponding wild type complexes 11. In both cases, at least one PR-inhibitor hydrogen bond interaction is eliminated in the mutant complex, and the differences relate to the chemical structures of the two drugs. One notable difference is that saquinavir forms one hydrogen bond with the carbonyl O of Gly48, while this interaction is replaced by two weaker CH…O interactions in the darunavir complexes. The hydrogen bond between Gly48 and saquinavir was absent in the structure of PRG48V/L90M-SQV 30, consistent with the 90-fold reduced inhibition. However, the CH…O interactions between Val48 and darunavir in PRG48V-DRV were even tighter than in the wild type PR-DRV. This difference in polar interactions may explain why PRG48V provides resistance to saquinavir yet maintains susceptibility to darunavir. The opposite occurs with the mutant PRI50V; the darunavir complex has lost hydrogen bond, as well as van der Waals interactions, while only a few changes in hydrophobic interactions were seen in the saquinavir complex. The loss of hydrogen bond interactions with inhibitor in PRI50V appears to provide resistance to darunavir, and in PRG48V/L90M to provide resistance to saquinavir, as suggested in previous studies 11; 30.
Saquinavir appeared to adapt better than darunavir to the changes caused by I54M and I54V, which is consistent with the increased prevalence of both mutations in HIV isolates showing resistance to darunavir. PRI54M showed reduced van der Waals interactions with darunavir in correlation with the shift of the 80’s loop, and both darunavir conformations had lost a direct hydrogen bond with Asp30. However, no significant reduction in inhibitor interactions was observed in the structures of PRI54V-DRV, PRI54V-SQV and PRI54M-SQV.
The observed changes in PR structure and activity in the flap mutants PRG48V, PRI50V, PRI54V, and PRI54M are in good agreement with the occurrence of these mutations in drug resistance to darunavir and saquinavir. In addition to these structural changes, several other factors must be considered since drug resistance to PR inhibitors can arise by various molecular mechanisms, including reduced inhibition, altered PR activity and stability 8; 13; 14. The structure and activity of PR is especially sensitive to mutations in the flap region, consistent with their frequent appearance in drug resistant HIV 6. In fact, PRG48V, PRI50V and PRI54V showed reduced catalytic efficiency and PRI50V had reduced stability, and these mutations are likely to be deleterious for correct polyprotein processing and viral replication. Such unfavorable mutations will only be selected under the severe pressure of drug treatment. The Ile54 mutants showed relatively small changes in inhibition by saquinavir and darunavir, and the mutants are similar to wild type in catalytic activity and stability. However, PRI54M showed significant structural changes and reduced polar interactions with darunavir consistent with clinical resistance data. Moreover, an unusual mechanism for resistance was proposed for mutant PRI54V, which showed fewer hydrogen bonds with a peptide reaction intermediate 37. The interactions with substrates or reaction intermediates have not been investigated for other mutants.
These new structures revealed that saquinavir and darunavir differ in their interactions with specific mutants. Importantly, saquinavir showed reduced inhibition and hydrogen bond interactions for mutants containing G48V and less change with PRI50V, while the opposite is seen for darunavir, in excellent agreement with the resistance data and despite the reduced activity of these mutants. Overall, the changes in polar interactions between PR mutants and inhibitor have the best correlation with observed resistance mutations. Therefore, this structural analysis will assist with predictive genotyping of clinical isolates and in the development of new protease inhibitors to combat HIV drug resistance.
MATERIALS AND METHODS
Preparation of HIV-1 Protease Mutants
The optimized HIV-1 PR clone with mutations Q7K, L33I, and L63I to diminish the autoproteolysis of the PR, as well as mutations C67A and C95A to prevent cysteine-thiol oxidation was used as the initial template for adding drug resistant mutations 31. This optimized PR had almost identical kinetic parameters and stability with the mature PR (Genbank HIVHXB2CG) 31. Plasmid DNA (pET11a, Novagen) encoding PR was utilized to construct mutant PR by the Quick-Change mutagenesis kit (Stratagene). The PR mutants were expressed in Escherichia coli BL21 (DE3) and the protein was purified from inclusion bodies as described 38. The presence of the appropriate mutations was confirmed by DNA sequencing.
Enzyme Kinetic Assays
Kinetic parameters were determined by a fluorescence assay. The substrate was Abz-Thr-Ile-Nle-pNO2Phe-Gln-Arg-NH2, where Abz is anthranylic acid, Nle is norleucine, based on the p2-NC cleavage site of the natural polyprotein substrate. Protease (10 μl, final concentration of ~20–70 nM) was mixed with 98 μl reaction buffer (100 mM MES, pH=5.6, 400 mM NaCl, 1 mM EDTA, 5% glycerol) and 2 μl of DMSO or inhibitor in DMSO, and the mixture was preincubated at 26°C for 5 min. The reaction was initiated by adding 90 μl of substrate from stock solution of 240 μM (final concentration of 10–70 μM), due to the solubility limit of substrate. The resulting mixture was assayed over 5 min for the increase in fluorescence using 340 nM and 420 nM bandpass filters for the excitation and emission. Data analysis was performed with the program SigmaPlot 8.02. kcat and Km values were obtained by standard data-fitting techniques for a reaction obeying Michaelis-Menten kinetics. Ki values were obtained from the IC50 values estimated from an inhibitor dose-response curve with the fluorescent assay using the equation Ki = (IC50 − [E]/2)/(1 + [S]/Km), where [E] and [S] are the protease and substrate concentrations, respectively 39.
Urea Denaturation Assay
The effect of urea denaturation was measured using the spectroscopic assay 14. The decrease of absorbance of chromogenic substrate (The Lys-Ala-Arg-Val-Nle-p-nitroPhe-Glu-Ala-Nle-amide, where Nle is norleucine) was monitored at 310 nm by a PerkinElmer Lambda 35 UV/Vis spectrophotometer. The substrate was concentrated at 400 μM and PR activity was measured at urea concentration of 0–3.0 M with a final enzyme concentration of 300–500 nM. The UC50 values were determined by plotting the initial velocities against increasing urea concentration using SigmaPlot 8.02.
Crystallographic Analysis
PRI50V, PRI54V and PRI54M were cocrystallized with saquinavir, while PRG48V, PRI54V and PRI54M were cocrystallized with darunavir at room temperature by the hanging drop method. The protein (2–5 mg/ml) was preincubated with the inhibitor in a molar ratio of 1:5–10. The crystallization drops had a 1:1 ratio by volume of reservoir solution and protein. For PRI50V -SQV, the reservoir contained 0.2 M sodium citrate, phosphate buffer, pH 6.0–6.2 and 30–35% saturated ammonium sulfate as precipitant; For PRI54V-SQV, 0.1 M sodium acetate buffer, pH 5.4, 1M NaCl as precipitant; For PRI54M-SQV, 0.2 M sodium acetate buffer, pH 4.8–5.2 and 10–15% NaCl as precipitant; For PRI50V-DRV, 0.2M sodium citrate, phosphate buffer, pH 6.0–6.2 and 30–35% saturated (NH4)2SO4 as precipitant; For PRG48V-DRV, 0.1 M citrate/0.2 M phosphate buffer, pH 5.6–5.8, 1.3M KCl as precipitant and 7–10% MPD; For PRI54V-DRV, 0.1 M sodium acetate buffer, pH 4.4, 1M NaCl as precipitant; For PRI54M-DRV, 0.2 M sodium acetate buffer, pH 4.4–4.6, 25–30% NaCl as precipitant. The crystals grew from overnight to 10 days into bricks, plates, and rod shapes. Some of them formed crystal clusters and were cut into individual crystals before mounting. Crystals were frozen in liquid nitrogen after soaking in 20–35% glycerol as a cryoprotectant.
X-ray diffraction data for all the complexes were collected on the SER-CAT ID beamline of the Advanced Photon Source, Argonne National Laboratory. Data were processed using HKL200040, and the structures were solved by molecular replacement using CCP4i 41; 42, refined using SHELX 43 and refitted using O 44. Alternate conformations for residues were modeled according to the electron density maps. Anisotropic B factors were refined for all the structures. Hydrogen atom positions were included in the last stage of refinement using all data.
The mutant crystal structures were compared with the wild type by superimposing their Cα atoms on each other and structural figures were made using Bobscript 45; 46 and PyMOL 47.
Protein Data Bank Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession code 3CYX for PRI50V-SQV, 3D1Y for PRI54V-SQV, 3D1X for PRI54M-SQV, 3CYW for PRG48V-DRV, 3D20 for PRI54V-DRV and 3D1Z for PRI54M-DRV, respectively.
Acknowledgments
We thank the staff at the SER-CAT beamline at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02–06CH11357. The research was supported in part by the Georgia State University Molecular Basis of Disease Program, the Georgia Research Alliance, the Georgia Cancer Coalition, and the National Institute of Health grants GM062920 and GM053386.
Abbreviations
- HIV-1
human immunodeficiency virus type 1
- PR
HIV-1 protease
- PRG48V
PR with G48V mutation
- PRI50V
PR with I50V mutation
- PRI54V
PR with I54V mutation
- PRI54M
PR with I54M mutation
- RMS
root mean square
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB, Wlodawer A. Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 A resolution. Science. 1989;246:1149–52. doi: 10.1126/science.2686029. [DOI] [PubMed] [Google Scholar]
- 2.Gustchina A, Weber IT. Comparison of inhibitor binding in HIV-1 protease and in non-viral aspartic proteases: the role of the flap. FEBS. 1990;269:269–272. doi: 10.1016/0014-5793(90)81171-j. [DOI] [PubMed] [Google Scholar]
- 3.Ishima R, Freedberg DI, Wang YX, Louis JM, Torchia DA. Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease. Structure. 1999;7:1047–55. doi: 10.1016/s0969-2126(99)80172-5. [DOI] [PubMed] [Google Scholar]
- 4.Shao W, Everitt L, Manchester M, Loeb DD, Hutchison CA, 3rd, Swanstrom R. Sequence requirements of the HIV-1 protease flap region determined by saturation mutagenesis and kinetic analysis of flap mutants. Proc Natl Acad Sci. 1997;94:2243–8. doi: 10.1073/pnas.94.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shafer RW. Genotypic testing for human immunodeficiency virus type 1 drug resistance. Clin Microbiol Rev. 2002;15:247–77. doi: 10.1128/CMR.15.2.247-277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shafer RW, Rhee SY, Pillay D, Miller V, Sandstrom P, Schapiro JM, Kuritzkes DR, Bennett D. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. Aids. 2007;21:215–23. doi: 10.1097/QAD.0b013e328011e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heaslet H, Rosenfeld R, Giffin M, Lin YC, Tam K, Torbett BE, Elder JH, McRee DE, Stout CD. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr D Biol Crystallogr. 2007;63:866–75. doi: 10.1107/S0907444907029125. [DOI] [PubMed] [Google Scholar]
- 8.Liu F, Kovalevsky AY, Louis JM, Boross PI, Wang YF, Harrison RW, Weber IT. Mechanism of Drug Resistance Revealed by the Crystal Structure of the Unliganded HIV-1 Protease with F53L Mutation. J Mol Biol. 2006;358:1191–9. doi: 10.1016/j.jmb.2006.02.076. [DOI] [PubMed] [Google Scholar]
- 9.Logsdon BC, Vickrey JF, Martin P, Proteasa G, Koepke JI, Terlecky SR, Wawrzak Z, Winters MA, Merigan TC, Kovari LC. Crystal structures of a multidrug-resistant human immunodeficiency virus type 1 protease reveal an expanded active-site cavity. J Virol. 2004;78:3123–32. doi: 10.1128/JVI.78.6.3123-3132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prabu-Jeyabalan M, Nalivaika EA, Romano K, Schiffer CA. Mechanism of substrate recognition by drug-resistant human immunodeficiency virus type 1 protease variants revealed by a novel structural intermediate. J Virol. 2006;80:3607–16. doi: 10.1128/JVI.80.7.3607-3616.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovalevsky AY, Tie Y, Liu F, Boross PI, Wang YF, Leshchenko S, Ghosh AK, Harrison RW, Weber IT. Effectiveness of nonpeptide clinical inhibitor TMC-114 on HIV-1 protease with highly drug resistant mutations D30N, I50V, and L90M. J Med Chem. 2006;49:1379–87. doi: 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. Ultra-high Resolution Crystal Structure of HIV-1Protease Mutant Reveals Two Binding Sites for Clinical Inhibitor TMC114. J Mol Biol. 2006;363:161–173. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu F, Boross PI, Wang YF, Tozser J, Louis JM, Harrison RW, Weber IT. Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L24I, I50V, and G73S. J Mol Biol. 2005;354:789–800. doi: 10.1016/j.jmb.2005.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahalingam B, Louis JM, Reed CC, Adomat JM, Krouse J, Wang YF, Harrison RW, Weber IT. Structural and kinetic analysis of drug resistant mutants of HIV-1 protease. Eur J Biochem. 1999;263:238–245. doi: 10.1046/j.1432-1327.1999.00514.x. [DOI] [PubMed] [Google Scholar]
- 15.Wang YF, Tie Y, Boross PI, Tozser J, Ghosh AK, Harrison RW, Weber IT. Potent new antiviral compound shows similar inhibition and structural interactions with drug resistant mutants and wild type HIV-1 protease. J Med Chem. 2007;50:4509–15. doi: 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buonaguro L, Tornesello ML, Buonaguro FM. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J Virol. 2007;81:10209–19. doi: 10.1128/JVI.00872-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coman RM, Robbins AH, Fernandez MA, Gilliland CT, Sochet AA, Goodenow MM, McKenna R, Dunn BM. The contribution of naturally occurring polymorphisms in altering the biochemical and structural characteristics of HIV-1 subtype C protease. Biochemistry. 2008;47:731–43. doi: 10.1021/bi7018332. [DOI] [PubMed] [Google Scholar]
- 18.Sanches M, Krauchenco S, Martins NH, Gustchina A, Wlodawer A, Polikarpov I. Structural characterization of B and non-B subtypes of HIV-protease: insights into the natural susceptibility to drug resistance development. J Mol Biol. 2007;369:1029–40. doi: 10.1016/j.jmb.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 19.De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune MP. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob Agents Chemother. 2005;49:2314–21. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitors (PI) UIC-94017 (TMC114) potent against multi-PI-resistant HIV in vitro. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J Mol Biol. 2004;338:341–52. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 22.Tie Y, Kovalevsky AY, Boross PI, Wang YF, Ghosh AK, Tozser J, Harrison RW, Weber IT. High Resolution Crystal Structures of HIV-1 Protease and Mutants V82A and I84V with Saquinavir. Proteins: Structure, Function, and Bioinformatics. 2006 doi: 10.1002/prot.21304. in press. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh AK, Dawson ZL, Mitsuya H. Darunavir, a conceptually new HIV-1 protease inhibitor for the treatment of drug-resistant HIV. Bioorg Med Chem. 2007;15:7576–80. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noble S, Foulds D. Saquinavir: a review of its pharmacology and clinical potential in the management of HIV infection. Drugs. 1996;52:93–112. doi: 10.2165/00003495-199652010-00007. [DOI] [PubMed] [Google Scholar]
- 25.Shapiro JM, Winters MA, Lawrence J, Merigan TC. Clinical cross-resistance between the HIV-1 protease inhibitors saquinavir and indinavir and correlations with genotypic mutations. AIDS. 1999;13:359–65. doi: 10.1097/00002030-199902250-00008. [DOI] [PubMed] [Google Scholar]
- 26.Condra JH, Petropoulos CJ, Ziermann R, Schleif WA, Shivaprakash M, Emini EA. Drug resistance and predicted virologic responses to human immunodeficiency virus type 1 protease inhibitor therapy. J Infect Dis. 2000;182:758–765. doi: 10.1086/315782. [DOI] [PubMed] [Google Scholar]
- 27.Molla A, Korneyeva M, Gao Q, Vasavanonda S, Schipper PJ, Mo HM, Markowitz M, Chernyavskiy T, Niu P, Lyons N, Hsu A, Granneman GR, Ho DD, Boucher CA, Leonard JM, Norbeck DW, Kempf DJ. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat Med. 1996;2:760–6. doi: 10.1038/nm0796-760. [DOI] [PubMed] [Google Scholar]
- 28.Murphy MD, Marousek GI, Chou S. HIV protease mutations associated with amprenavir resistance during salvage therapy: importance of I54M. J Clin Virol. 2004;30:62–7. doi: 10.1016/j.jcv.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 29.Lambert-Niclot S, Flandre P, Canestri A, Peytavin G, Blanc C, Agher R, Soulie C, Wirden M, Katlama C, Calvez V, Marcelin AG. Factors Associated with the Selection of Mutations Conferring Resistance to Protease Inhibitors (PIs) in PI-Experienced Patients Displaying Treatment Failure on Darunavir. Antimicrob Agents Chemother. 2008;52:491–6. doi: 10.1128/AAC.00909-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong L, Zhang XC, Hartsuck JA, Tang J. Crystal structure of an in vivo HIV-1 protease mutant in complex with saquinavir: insights into the mechanisms of drug resistance. Protein Sci. 2000;9:1898–904. doi: 10.1110/ps.9.10.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 32.Zoete V, Michielin O, Karplus M. Relation between sequence and structure of HIV-1 protease inhibitor complexes: a model system for the analysis of protein flexibility. J Mol Biol. 2002;315:21–52. doi: 10.1006/jmbi.2001.5173. [DOI] [PubMed] [Google Scholar]
- 33.Weber IT, Kovalevsky AY, Harrison RW. Structures Of HIV Protease Guide Inhibitor Design To Overcome Drug Resistance. In: Caldwell GW, Atta-ur-Rahman, Player MR, Choudhary MI, editors. Frontiers in Drug Design and Discovery. Vol. 3. Bentham Science Publishers; 2007. pp. 45–62. [Google Scholar]
- 34.Munshi S, Chen Z, Yan Y, Li Y, Olsen DB, Schock HB, Galvin BB, Dorsey B, Kuo LC. An alternate binding site for the P1-P3 group of a class of potent HIV-1 protease inhibitors as a result of concerted structural change in the 80s loop of the protease. Acta Crystallogr D Biol Crystallogr. 2000;56:381–8. doi: 10.1107/s0907444900000469. [DOI] [PubMed] [Google Scholar]
- 35.Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 angstroms resolution crystal structures of HIV-1 protease mutants with substrate analogs. FEBS J. 2005;272:5265–77. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foulkes JE, Prabu-Jeyabalan M, Cooper D, Henderson GJ, Harris J, Swanstrom R, Schiffer CA. Role of invariant Thr80 in human immunodeficiency virus type 1 protease structure, function, and viral infectivity. J Virol. 2006;80:6906–16. doi: 10.1128/JVI.01900-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovalevsky AY, Chumanevich AA, Liu F, Louis JM, Weber IT. Caught in the Act: the 1.5 A resolution crystal structures of the HIV-1 protease and the I54V mutant reveal a tetrahedral reaction intermediate. Biochemistry. 2007;46:14854–64. doi: 10.1021/bi700822g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug resistant mutants of HIV-1 protease: High resolution crystal structures of the mutant protease/substrate analog complexes. Proteins: Structure, Function, and Genetics. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 39.Maibaum J, Rich DH. Inhibition of porcine pepsin by two substrate analogues containing statine: the effect of histidine at the P2 subsite on the inhibition of aspartic proteinases. J Med Chem. 1988;31:625–629. doi: 10.1021/jm00398a022. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.Collaborative Computational Project N. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst D. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 42.Potterton E, Briggs P, Turkenburg M, Dodson EA. Graphical user interface to the CCP4 program suite. Acta Cryst D. 2003;59:1131–7. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 43.Sheldrick GM, Schneider TR. High resolution refinement. Methods in Enzymology. 1997;277:319–343. [PubMed] [Google Scholar]
- 44.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 45.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graph Model. 1997;15:132–4. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 46.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Cryst. 1999;D55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 47.DeLano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.