

Abstract
Plasmodium falciparum erythrocyte invasion is powered by an actin/myosin motor complex that is linked both to the tight junction and to the merozoite cytoskeleton through the Inner Membrane Complex (IMC). The IMC association of the myosin motor, PfMyoA, is maintained by its association with three proteins: PfMTIP, a myosin light chain, PfGAP45, an IMC peripheral membrane protein, and PfGAP50, an integral membrane protein of the IMC. This protein complex is referred to as the glideosome, and given its central role in erythrocyte invasion, this complex is likely the target of several specific regulatory effectors that ensure it is properly localized, assembled, and activated as the merozoite prepares to invade its target cell. However, little is known about how erythrocyte invasion as a whole is regulated, or about how or whether that regulation impacts the glideosome. Here we show that P. falciparum erythrocyte invasion is regulated by the release of intracellular calcium via the cyclic-ADP Ribose (cADPR) pathway, but that inhibition of cADPR-mediated calcium release does not affect PfGAP45 phosphorylation or glideosome association. By contrast, the serine-threonine kinase inhibitor, staurosporine, affects both PfGAP45 isoform distribution and the integrity of the glideosome complex. This data identifies specific regulatory elements involved in controlling P. falciparum erythrocyte invasion and reveals that the assembly status of the merozoite glideosome, which is central to erythrocyte invasion, is surprisingly dynamic.
Keywords: erythrocyte invasion, glideosome, merozoite, calcium signaling, post-translational modification
Introduction
Infection by Plasmodium parasites results in 200 to 500 million cases of malaria each year, with between 1 and 3 million of those cases being fatal [1, 2]. This illness represents a significant burden on global public health—a burden that is becoming more severe as drug-resistance spreads among both parasite and mosquito populations, and as environmental factors converge to increase the number of at-risk individuals [3]. Malaria can result from infection by any of four species of Plasmodium parasite, but almost all malaria mortality is caused by infection with Plasmodium falciparum. Like other Plasmodium parasites, P. falciparum infects through the bite of an infected mosquito and causes disease as a result of its subsequent invasion and occupation of erythrocytes.
As all clinical symptoms of malaria result from the parasite’s intraerythrocytic development, a clear understanding of the molecular mechanisms that underpin the process of erythrocyte invasion could allow the development of invasion-inhibitory therapeutics. Erythrocyte invasion begins when a merozoite comes into contact with a circulating red blood cell (RBC) and forms an initial interaction with the RBC surface. This is followed by a reorientation step that allows the merozoite to bring its apical organelles into direct opposition to the RBC plasma membrane and form the tight junction—a close and irreversible interaction between the merozoite and RBC that is then translocated along the merozoite’s periphery as invasion proceeds. As the tight junction is passed along the merozoite periphery, the RBC plasma membrane is forced to invaginate to form the parasitophorous vacuole (PV), and invasion is complete once the merozoite has passed the tight junction to its posterior and moved completely into the fully formed PV [4].
This tightly coordinated sequence of events relies on a variety of specific molecular interactions. The initial interactions that allow RBC recognition and tight junction formation depend on ligands on the merozoite surface that interact with specific receptors on the RBC surface. These interactions are subject to immune selection pressure and as a result the merozoite proteins responsible for them are highly variable and are often part of multi-member protein families with overlapping and interchangeable functions [5]. However, the process of tight junction translocation depends on a multi-member myosin associated protein complex that is conserved across a number of Apicomplexan species [6-8]. This complex functions by anchoring the motor protein, PfMyoA, to the Inner Membrane Complex (IMC), a set of flattened membrane cisternae that are associated with the merozoite cytoskeleton. The anchoring of PfMyoA to the IMC is essential for the generation of directional force, which occurs when PfMyoA binds and releases F-actin filaments that are associated with specific transmembrane proteins in the tight junction [6, 9, 10]. This myosin associated protein complex was first described in Toxoplasma gondii, but is present in P. falciparum merozoites and in other invasive/motile Plasmodium life cycle stages; it is collectively referred to as the glideosome [6-8]. The glideosome in P. falciparum merozoites consists of PfMyoA, an associated light chain, PfMTIP, an IMC-associated peripheral membrane protein, PfGAP45, and an IMC integral membrane protein, PfGAP50, which is thought to function in anchoring this complex to the IMC [6,8].
As outlined above, erythrocyte invasion involves complex interactions between two eukaryotic cells, the merozoite and the erythrocyte, and must rely on the coordinated action of a tightly controlled regulatory network. This regulatory network is likely active at numerous points in the invasive process, and given its central role in invasion, the glideosome is likely an important regulatory target. In order to function properly, the glideosome must be both fully assembled and activated as the tight junction is translocated rearward, and both assembly and activity of this complex could be regulated. Furthermore, while some of the broad regulatory elements responsible for controlling the overall process of RBC invasion have been defined—calcium signaling and protein phosphorylation are known to play an invasion regulatory role, for example—the specific signaling pathways active in controlling invasion have been poorly characterized, and we know nothing of the subsequent impact of these pathways on the glideosome [11-13]. In order to address these major gaps in our understanding of P. falciparum erythrocyte invasion, we set out to describe the specific signaling elements involved in the regulation of erythrocyte invasion, and to determine the impact of these pathways on the assembly status of the glideosome and on the post-translational modification of one of its constituents.
Materials and Methods
P. falciparum Culture and Invasion Assays
P. falciparum parasite strains were cultured in O+ human erythrocytes at 5% hematocrit and 10% O+ human serum as described previously [14]. Synchronization of parasite cultures was by treatment with 5% sorbitol.
For invasion assays, mid-late stage 3D7 schizonts (43-46 hrs post-invasion at 1-2% parasitemia) were incubated with signaling effector compound or compound solvent alone and allowed to re-invade. After 12-16 hrs, assays were smeared and Giemsa-stained and fully formed rings were counted. Each experiment was performed at least three times in duplicate or in triplicate. Invasion in the presence of the compound solvent alone was considered to be 100%, and the influence of each signaling effector compound on ring formation was compared to its own solvent control in order to calculate invasion efficiency.
PfGAP45 immuno-affinity Purification Column Preparation and Usage
To generate an immuno-affinity purification column specific for PfGAP45, affinity purified PfGAP45 rabbit antisera (prepared as previously described, [8]) was coupled to NHS-Sepharose loaded onto a glass Econo-Column (Bio-Rad, 1ml column volume). In order to purify PfGAP45 and the associated glideosome, late schizonts (45-48 hrs post-invasion) were collected and lysed in 1% TX100 IP lysis buffer (1% TX100, 50mM Tris-Cl pH 8.0, 5mM EDTA) with complete protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktail II (Calbiochem) at a concentration of 1-5 × 108 parasites/ml. After lysis on ice for 30min, parasite material was spun at 13000 X G for 10min at 4°C and lysate supernatants were put over the PfGAP45 immuno-affinity column by gravity flow. After washing with 5-10 column volumes of 1% TX100 IP lysis buffer, PfGAP45 and its binding partners were eluted from the column using 3-column volumes elution buffer (.1M Glycine, .5M NaCl, pH 2.5-2.8). Elutions were collected directly into 1/10 volume neutralization buffer (1M Tris-Cl pH 9.0) and were either mixed with an equal volume 2X SDS-Sample Buffer with 10% β-Mercaptoethanol or were chloroform/methanol precipitated and resuspended in 1X SDS-Sample Buffer with 5% β-Mercaptoethanol for analysis by SDS-PAGE.
Experiments examining PfGAP45 and its associated complex members under treatment with invasion-inhibitory compounds were performed exactly as described above except as follows. For each signaling compound used, an invasion-inhibitory concentration of either the compound under study or an equal volume of the compound solvent was added to well synchronized, mid-schizont (42-45 hrs post-invasion) stage parasites and culture was maintained for between 2 and 4 hrs before lysis of parasites in 1% TX100 IP lysis buffer and application to the PfGAP45 immuno-affinity purification column as described above.
SDS PAGE, Western blotting, and Pro-Q analysis
PfGAP45 column elutions were loaded onto either 10% or 12% polyacrylamide Tris-Glycine gels for SDS-PAGE. For Western blot analysis, SDS-PAGE gels were transferred to either nitrocellulose or PVDF membranes and blocked in 2% Milk/PBS solution overnight. Membranes were probed with anti-glideosome sera and then with HRP-conjugated secondary sera and visualized using enhanced chemoluminescence (ECL, Amersham Biosciences). Densitometry was performed using ImageJ analysis, which is freely available at (http://rsbweb.nih.gov/ij/). For Pro-Q analysis, SDS-PAGE gels were transferred to PVDF membranes and treated with Pro-Q membrane stain per manufacturer’s instruction (Molecular Probes). Following Pro-Q staining, PVDF membranes were also subjected to Western blot analysis as described above.
PfMTIP Immunodepletion
Immunodepletion experiments were basically immunoprecipitation (IP) experiments performed repeatedly with the same lysate. Briefly, 600ul of 1% TX100-IP lysate was pre-adsorbed against Protein G/Sepharose for 1hr at 4°C and then added to 20ul of either rabbit pre-immune sera or rabbit anti-MTIP. This initial IP was left overnight at 4 degrees with rotation. The resulting immune-complexes were precipitated by incubation with Protein G/Sepharose for 1hr at 4°C, after which IP supernatants were again added to 20ul of rabbit pre-immune or MTIP sera and incubated with rotation at 4°C for 2hrs. Protein G/Sepharose was again added to pull-down the subsequent immune-complexes, and this process was repeated for three rounds of 2hr incubation/1hr immunoprecipitation. Complete protease and phosphatase inhibitor cocktails were added with each addition of antisera. After immuno-depletion, the resulting IP lysate was split into 200ul aliquots for IP with MTIP, GAP45, or GAP50 rabbit antisera as described above. Subsequent immune complexes were separated by SDS-PAGE and transferred to nitrocellulose membranes for Western blotting as described above.
Results
Signaling pathways regulating P. falciparum erythrocyte invasion
Erythrocyte invasion is dynamic and must rely on a sophisticated regulatory network that organizes the molecular events driving the process, but little is known about the specific signaling elements involved in this regulatory network. In order to address this significant gap in our understanding of merozoite biology, we set out to describe several signaling elements that are required for successful erythrocyte invasion. Previous work has indicated that calcium signaling is potentially involved in regulating P. falciparum erythrocyte invasion, though whether this involvement is intra or extracellular in relation to the merozoite has been unclear [11, 13]. We performed erythrocyte invasion assays in the presence of the extracellular calcium chelator, BAPTA, or its membrane permeable counterpart, BAPTA AM, in order to determine whether intra or extracellular calcium signaling is required for successful P. falciparum erythrocyte invasion. Treatment with BAPTA AM had a significant and dose-dependent inhibitory effect on invasion, whereas BAPTA had no such effect (fig. 1A), indicating the release of intracellular calcium is required for successful invasion. As there are no intracellular calcium stores within erythrocytes, it is likely that the effect of BAPTA AM on invasion is directly related to the chelation of calcium released within the merozoite.
Figure 1.

Signaling pathways involved in the regulation of P. falciparum erythrocyte invasion: (A) The release of intracellular calcium is required for efficient invasion to proceed. BAPTA (an extracellular calcium chelator) has no effect on invasion, while BAPTA A/M (a membrane-permeable calcium chelator) shows a dose-dependent inhibition of invasion. (B) The release of intracellular calcium required for invasion is not via the Phospholipase-C/IP3 dependent pathway. U73122 (a PLC-specific inhibitor) has no specific effect on invasion, as the decrease in invasion efficiency using this compound is less significant than that seen using a non-specific analog of this compound, U73343. 2-APB, an inhibitor of IP3-dendent calcium release, also has no effect on invasion. (C) The cADP-Ribose dependent pathway for calcium release is involved in the regulation of invasion. 8Br-cADP-Ribose and Dantrolene are inhibitors of cADP-Ribose dependent calcium release, and each of these compounds causes a significant decrease in RBC invasion. (D) The release of intracellular calcium that is required for invasion must itself be well regulated. The calcium ionophore, A23187, artificially induces intracellular calcium release and causes a significant reduction in invasion efficiency at low concentrations.
In most eukaryotic systems, release of intracellular calcium occurs in response to one or both of two different second messenger signaling pathways. The first pathway depends upon Phospholipase C (PLC) generated IP3 and IP3-responsive ER calcium channels. The second pathway depends upon cyclic-ADP Ribose (cADPR) dependent stimulation of calcium release from ryanodine-responsive ER calcium channels. We performed invasion assays with inhibitors specific for each pathway to establish whether one or both were involved in regulating P. falciparum erythrocyte invasion. Treatment with the PLC-specific inhibitor, U73122, did prevent invasion at relatively high concentrations, but the effect was less significant than that of its non-active analog, U73343, which suggests that the effect was non-specific (fig. 1B). Furthermore, treatment with 2-APB, which specifically prevents IP3 induced release of calcium from IP3-responsive calcium channels had no impact on invasion (fig. 1B). These two results suggest that PLC dependent IP3-induced calcium release is not required for invasion. By contrast, invasion assays performed with both dantrolene, which prevents cADPR binding to ryanodine-responsive Ca2+ channels, and 8Br-cADPR, a competitive inhibitor of cADPR, showed a significant reduction in P. falciparum erythrocyte invasion in a dose dependent fashion (fig. 1C). These data clearly indicate that the release of intracellular calcium in response to cADPR-dependent signaling is required for invasion. Furthermore, it is likely that release of calcium through the cADPR-dependent pathway must be tightly regulated, as using the calcium ionophore, A23187, to artificially stimulate calcium release in an unregulated manner results in a marked inhibition of invasion (fig. 1D). The invasion-inhibitory effect of A23187 in merozoites is reminiscent of ionophore-induced death as reported in T. gondii, where continuous incubation of tachyzoites with this compound results in parasite death as a result of calcium signaling hyperstimulation. Although the regulatory target(s) of cADPR-regulated calcium release are not known, calcium-responsive kinases (such as the CDPKs) are at least one likely target, as treatment with the broad spectrum serine/threonine kinase inhibitor staurosporine inhibits invasion in a dose-dependent manner (fig. 1E), as has been previously shown in P. knowlesi [12].
PfGAP45 is a phospho-protein and calcium signaling effectors do not inhibit PfGAP45 phosphorylation or glideosome complex association
Having identified several signaling effector molecules that interfere with the process of invasion, we next tested whether these effectors impacted the P. falciparum merozoite glideosome in terms of post-translational modification and complex association. The peripheral IMC protein, PfGAP45, which runs as a triplet of bands by SDS-PAGE [8], has been shown by others to be both acylated and phosphorylated [15-17]. In order to follow the post-translational modification of PfGAP45, as well as the other members of the merozoite glideosome in vivo, we generated a PfGAP45 specific immuno-affinity column and used it to immuno-precipitate the glideosome complex from P. falciparum extracts. Silver staining and Western blotting of the eluate from the PfGAP45-specific immuno-affinity column revealed quantitative pull down of all four glideosome components (fig. 2A and B). We then probed column eluates with a phospho-specific membrane stain, Pro-Q (Molecular Probes). As expected, PfGAP45 was labeled by Pro-Q (fig. 2C), confirming that at least one of the three PfGAP45 isoforms detectable by SDS-PAGE is phosphorylated. As Pro-Q treatment to identify phospho-proteins must be performed separately from Western blotting to identify the glideosome members present, it is difficult to unequivocally identify which PfGAP45 isoform is the major target of phosphorylation, but careful alignment of each membrane suggests that the highest MW PfGAP45 isoform is the major Pro-Q binding band, as previously reported [16].
Figure 2.
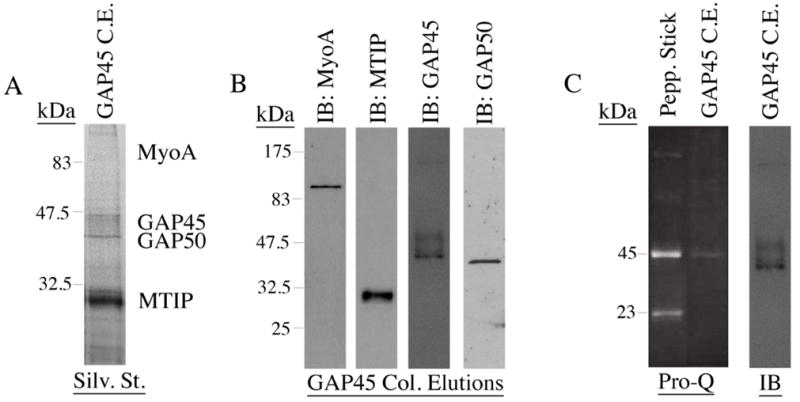
PfGAP45 is a phospho-protein: (A) The fully assembled merozoite glideosome is precipitated by the PfGAP45-specific immuno-affinity column. A silver stained SDS-PAGE gel of a PfGAP45 immuno-affinity column elution shows that each member of the invasion motor complex is precipitated. (B) The presence of each glideosome complex member in PfGAP45 column elutions is further confirmed by Western blotting with sera specific for each complex member. (C) PfGAP45 is phosphorylated in vivo. After labeling with the phospho-protein specific membrane stain, Pro-Q, a major phosphorylated band is present that corresponds to the highest MW PfGAP45 isoform. Peppermint Stick is a positive control for Pro-Q staining. The right hand image is a blot of the Pro-Q stained PVDF membrane using anti-PfGAP45 sera.
Pro-Q staining, while not as sensitive as P32-labeling, detects phosphoproteins in the single nanogram range, which is roughly equivalent to the detection limit of silver stain. PfGAP45 was the only Pro-Q stained band in our column eluates, suggesting that PfGAP45 is the only significant target of phosphorylation in vivo when the full glideosome is assembled. Recent experiments have shown that PfMTIP can be phosphorylated in vitro by PfCDPK1 [16, 18], but we were not able to detect phosphorylated PfMTIP in vivo, nor has it been reported by others. It is however possible that PfMTIP and/or other members of this complex are phosphorylated when not in association with PfGAP45 (as we are precipitating the glideosome through PfGAP45), or are phosphorylated within the fully assembled complex but at a significantly different level to PfGAP45.
This PfGAP45 column/Pro-Q strategy gave us the ability to repeatedly precipitate significant quantities of the merozoite glideosome and to determine the phosphorylation status of PfGAP45 under several conditions while avoiding the use of copious amounts of radioactivity. Having established that cADPR-responsive release of intracellular calcium is involved in the regulation of erythrocyte invasion, we next wanted to use this strategy to determine what role calcium-regulated signaling plays in regulating PfGAP45 phosphorylation and glideosome association. We therefore treated mid-late schizont stage cultures with invasion-inhibitory amounts of either A23187, BAPTA AM, or dantrolene and precipitated PfGAP45 from these treated cultures using our PfGAP45-specific immuno-affinity column. Column elutions were then treated with Pro-Q membrane stain to determine whether any treatment prevents PfGAP45 phosphorylation. Surprisingly, the gross level of PfGAP45 phosphorylation, as detected by Pro-Q staining, was not affected by any single treatment, and there was no reproducible impact on the distribution of PfGAP45 isoforms (Fig. 3A-C). Again, it should be mentioned that Pro-Q analysis is not as sensitive as P32-labeling, but it does clearly allow the detection of PfGAP45 phosphorylation (fig. 2C). Furthermore, neither dantrolene (fig. 3D), BAPTA AM, nor A23187 (data not shown) had any discernible effect on the assembly of the glideosome complex, as the full glideosome complex co-precipitated with PfGAP45 from extracts made from parasites treated with each inhibitor at a concentration that strongly inhibited erythrocyte invasion. It is possible that the concentrations of inhibitor used in these experiments, while clearly sufficient to disrupt erythrocyte invasion (see fig. 1), were not adequate for interfering with PfGAP45 phosphorylation or glideosome complex association. However, an alternative, and we think more likely explanation, is that PfGAP45 is phosphorylated by multiple kinases, some of which are not regulated by calcium-responsive signaling elements.
Figure 3.

Influence of signaling effector compounds versus their vehicle solvent on PfGAP45 phosphorylation and glideosome association: Invasion-inhibitory amounts of (A) A23187, (B) BAPTA AM, and (C) Dantrolene (Dan.) do not inhibit PfGAP45 phosphorylation as determined by Pro-Q staining. Phosphorylated Peppermint Stick (P.S.) markers serve both as a positive control for Pro-Q staining and for size verification. (D) Treatment with invasion-inhibitory amounts of Dantrolene (Dan.) has no significant effect on glideosome association as judged by co-immunoprecipitation. Extracts from parasites treated with vehicle solvents at the same concentration as was present in the inhibitors were used as negative controls. Solvents were chloroform (Chlor.) for A23187 and DMSO for BAPTA AM and Dantrolene.
Phosphorylation is linked to PfGAP45 isoform distribution and glideosome association
In an attempt to overcome the possibility that PfGAP45 is phosphorylated by multiple kinases, some of which are calcium-insensitive, we tested the effect of the broader range serine/threonine kinase inhibitor, staurosporine. This compound has been shown to inhibit RBC invasion by Plasmodium knowlesi merozoites at low concentrations [12], and we established that it also inhibits P. falciparum erythrocyte invasion at similar concentrations (fig. 1E). We then treated late schizont stage P. falciparum cultures with an invasion-inhibitory amount of this compound (5uM) and immuno-precipitated the glideosome using the anti-PfGAP45 column/ProQ strategy described above. Treatment with staurosporine again did not completely prevent PfGAP45 phosphorylation; however, staurosporine treatment did have a significant impact on PfGAP45 isoform distribution, as this distribution was clearly shifted toward a predominance of the higher MW PfGAP45 isoform (fig. 4A). Furthermore, staurosporine treatment resulted in a marked reduction in the amount of PfMyoA and PfMTIP that co-precipitated with PfGAP45 from the anti-PfGAP45 column (9.9% and 36.3% of control by densitometry respectively, fig. 4B). By contrast, the amount of PfGAP50 co-precipitated with PfGAP45 remained unchanged between control and staurosporine-treated extracts (94.1% by densitometry, fig. 4B). This data suggests that a previously unrecognized glideosome sub-complex consisting of only PfGAP45 and PfGAP50 exists, at least in staurosporine treated extracts, apart from PfMyoA and PfMTIP.
Figure 4.
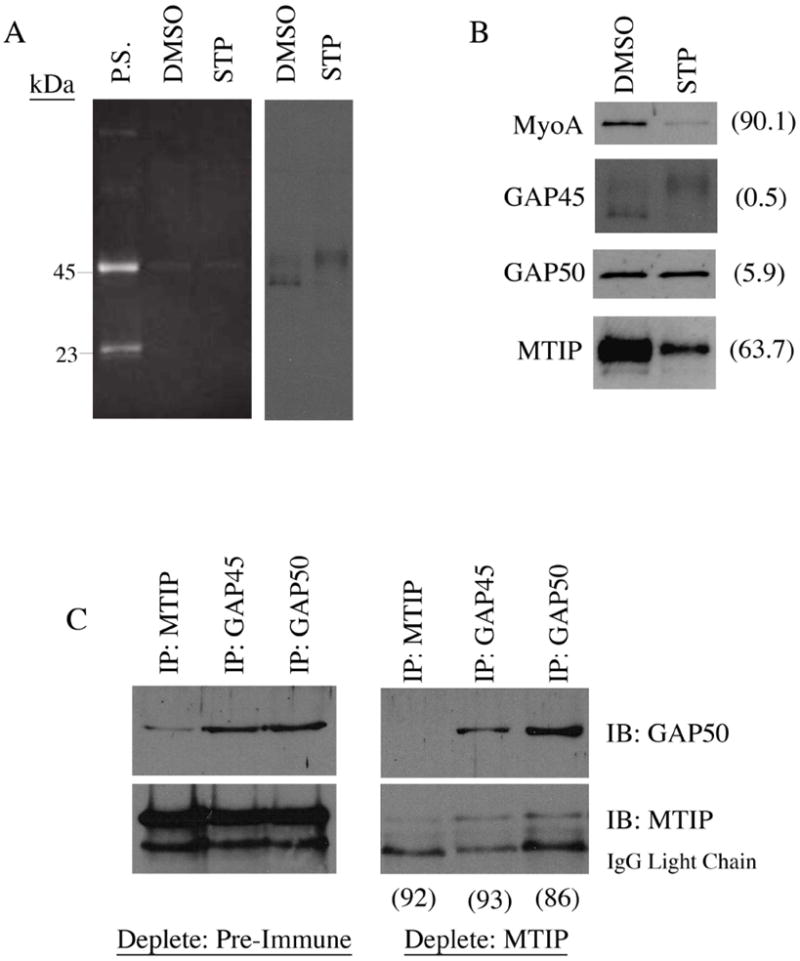
Phosphorylation is linked to PfGAP45 isoform distribution and glideosome association: (A) Treatment of late blood-stage parasites with 5uM staurosporine (STP) does not completely inhibit PfGAP45 phosphorylation, but affects PfGAP45 isoform distribution. Treatment of PfGAP45 column elutions with the phospho-specific stain, Pro-Q, shows that PfGAP45 phosphorylation is not inhibited by treatment with 5uM STP. Western blotting of PfGAP45 column elutions shows that 5uM STP treatment results in a shift in PfGAP45 isoform distribution toward the highest MW isoform. (B) STP treatment affects glideosome complex association. Western blotting of STP-treated PfGAP45 column elutions shows a significant reduction in the amount of PfMyoA and PfMTIP precipitated with PfGAP45 compared to DMSO-treated control elutions. The percentage of each glideosome complex member precipitated in STP-treated parasites compared to DMSO-treated parasites as measured by densitometry is in parentheses. (C) Specific depletion of PfMTIP confirms that a sub-complex of PfGAP45 and PfGAP50 is present in late schizont stage parasites. Results show that PfGAP45 and PfGAP50 can be co-precipitated from PfMTIP-depleted lysates. Densitometry analysis of MTIP depletion and amounts of PfGAP50 precipitated from Pre-Immune vs. MTIP depleted lysates is in parentheses.
To test whether such a sub-complex could be detected in P. falciparum schizonts and merozoites in the absence of any drug treatment, we used an immuno-depletion approach. By performing repeated immunoprecipitations with anti-PfMTIP specific antibodies, we were able to remove the majority of PfMTIP from TX-100 extracts (by densitometry, the level of PfMTIP remaining in PfMTIP-depleted extract is 8% of that that remains after depletion with pre-immune sera; fig. 4C). In this PfMTIP-depleted extract, immunoprecipitation of PfGAP45 still resulted in the co-immunoprecipitation of PfGAP50 at a level comparable to that of pre-immune sera-depleted control extract (74% by densitometry; fig. 4C). By contrast, PfMTIP co-immunoprecipitation with PfGAP45 was reduced to only 7% of control after PfMTIP depletion (fig. 4C). A sub-complex of PfGAP45 and PfGAP50 can therefore be detected in untreated P. falciparum schizonts after the near complete depletion of PfMTIP (fig. 4C). Taken together, the effect of staurosporine treatment and PfMTIP immunodepletion on the glideosome complex suggests that PfGAP45 and PfGAP50 form a sub-complex in vivo and that the association of the fully assembled glideosome complex is affected by a staurosporine-sensitive kinase.
Discussion
Erythrocyte invasion by P. falciparum merozoites is the initiating step in the pathogenic phase of this parasite’s life cycle. Invasion is a complex biological process, and the mechanisms underpinning it remain poorly defined, particularly in regards to the signaling network controlling the process and the effects of this network on specific proteins involved. Here we have shown that erythrocyte invasion depends upon calcium signaling and that this signaling is predominantly through the cADPR second messenger pathway. We have also shown that PfGAP45 phosphorylation is not completely dependent upon calcium signaling, but that phosphorylation does play a role in regulating the merozoite glideosome. The precise invasion-regulatory role of second messenger induced calcium release, and of the phosphorylation events described here await further study, but our data suggest several important points.
First, our studies of the signaling pathways that influence erythrocyte invasion have revealed a previously unrecognized role for cADPR-dependent calcium release in regulating P. falciparum invasion. These results are largely reflective of what seem to be broadly conserved invasion/motility regulatory mechanisms active in both Plasmodium and Toxoplasma parasites, but with some interesting and significant differences. In T. gondii tachyzoites, transient fluctuations in intracellular calcium concentrations are involved in regulating various aspects of gliding motility, host-cell invasion, and host-cell egress [19-21]. These transient fluctuations in the intracellular calcium concentration depend on the action of PLC generated IP3 and IP3-responsive calcium channels, and also on cADPR-stimulated calcium release from ryanodine-responsive calcium channels [22, 23]. Here we have shown that, like T. gondii, P. falciparum erythrocyte invasion does require the release of calcium from internal stores, and that inhibitors of cADPR signaling block invasion. However, we have found no clear evidence that PLC generated IP3 is involved in the regulation of erythrocyte invasion. The conclusion that PLC-dependent calcium release is not involved in the regulation of RBC invasion is in contrast to that of a recently published report where it was shown that treatment with the PLC inhibitor, U73122, blocks invasion [17]. We question that conclusion for several reasons. First, the invasion-inhibitory concentration of U73122 used (30uM) was fifteen times higher than the concentration required to block motility and invasion in T. gondii tachyzoites (2uM)—a difference that is surprising given that Plasmodium and Toxoplasma are related parasites [24]. Second, while we also observed that U73122 affects invasion at a concentration of 20uM or greater, an inactive analog of this compound, U73343, which was not tested in the previously published work, had a more significant impact on invasion (fig. 1B), calling into question whether the effect of U73122 on invasion is through specific disruption of PLC-dependent signaling. Lastly, we have shown that the IP3-receptor antagonist, 2-APB, has no effect on P. falciparum erythrocyte invasion, which provides independent support for our conclusion that PLC dependent IP3-stimulation of calcium release is likely not involved in the regulation of P. falciparum erythrocyte invasion. While we have taken care in these studies to use inhibitor concentrations similar to those shown to be effective in T. gondii or near to their I.C.50 in eukaryotic cells, in each case it should be noted that the targets of these respective inhibitors are not well characterized in Plasmodium parasites, and that the conclusions drawn from both this and other signaling-inhibitor studies will require further experimental confirmation.
Second, while we found evidence for the involvement of cADPR-stimulated calcium release in the regulation of P. falciparum erythrocyte invasion, the impact of this pathway on the glideosome is clearly far from simple and may involve multiple kinases. Treatment of blood-stage parasites with BAPTA AM, dantrolene, or A23187—all of which have a significant inhibitory effect on erythrocyte invasion—had no detectable effect on PfGAP45 phosphorylation in vivo, or on complex assembly. This could reflect differences between the inhibitor concentration needed to effectively prevent PfGAP45 phosphorylation by a specific kinase and that needed to prevent invasion, which likely occurs as a result of the interruption of several invasion-related processes—protease activation, or microneme and rhoptry discharge, for example. While this is possible, we think a more likely explanation is that PfGAP45 is phosphorylated by multiple kinases, some of which are not regulated by calcium. Two recent reports have shown that both PfCDPK1 and PfPKB will phosphorylate PfGAP45 in vitro, though in each of these instances, the kinase in question is regulated by calcium or calmodulin and should therefore be affected by treatment with BAPTA AM or dantrolene [16, 17]. However, we have shown that even treatment with staurosporine does not completely prevent PfGAP45 phosphorylation in vivo, and while concerns regarding the inhibitor concentration required to inhibit specific kinases in vivo persist, our work and that of others suggest that PfGAP45 phosphorylation is regulated by multiple kinases, and that inhibition of only one kinase or class of kinase is not sufficient to completely inhibit its phosphorylation [16, 17].
Phosphorylation site prediction programs identify several potential phosphorylation sites in the PfGAP45 sequence and a number of kinases that potentially phosphorylate this protein. Among these, Casein Kinase II (CKII) is of particular interest, as this kinase is not sensitive to staurosporine, does not depend on second-messenger activation, and has been shown to be active in the regulation of invasion by T. gondii tachyzoites [25, 26]. Protein Kinase A is also identified as a potential regulator of PfGAP45, and this kinase has recently been shown to regulate microneme discharge in Plasmodium sporozoites [27]. We attempted to address the role of these kinases in regulating erythrocyte invasion and PfGAP45 phosphorylation using the CKII inhibitor, DRB, and the PKA inhibitor, H89, and saw no effect (data not shown), though in the case of H89, this inhibitor has recently been shown to have little effect on P. falciparum PKA [28]. Further experiments will clearly be required to determine the exact number of times PfGAP45 is phosphorylated and exactly which kinases are responsible for effecting this regulation. These experiments will also need to be performed specifically with P. falciparum merozoites as it appears that PfGAP45 is likely phosphorylated at fewer sites than TgGAP45 in T. gondii tachyzoites [16, 29].
While we were unable to completely inhibit PfGAP45 phosphorylation in vivo, treatment with staurosporine did have a significant impact on PfGAP45 isoform distribution and glideosome complex association. These data have two important implications. First, that PfGAP45 post-translational modification is dynamic and is regulated directly or indirectly by phosphorylation events. The shift in PfGAP45 isoform distribution toward the highest MW isoform suggests that some PfGAP45 post-translational modifications are reversible and that removal of specific modifications depends upon either direct phosphorylation of PfGAP45 or of an upstream regulator that is itself responsible for affecting the post-translational modification of PfGAP45. Given that others have shown that PfGAP45 is multiply acylated and that the lowest mobility PfGAP45 isoform is likely both dually acylated and phosphorylated, one possible hypothesis is that the acylation status of PfGAP45 is regulated by a staurosporine-sensitive kinase, with a palmitoyl group being removed in a phosphorylation dependent manner to give rise to the higher mobility PfGAP45 isoforms [15, 16]. However, it is clearly important to directly determine the identity of each PfGAP45 isoform and account for precisely which post-translational modifications are present at each MW.
The second major implication of the staurosporine treatment data presented here is that associations between members of the glideosome are dynamic and are regulated by specific phosphorylation events. Previous work has shown that the glideosome, in T. gondii, is assembled in two stages with TgMyoA, TgMLC1 (the orthologue of PfMTIP), and TgGAP45 being assembled in the cytoplasm and subsequently relocated to the IMC where an association with TgGAP50 is established [7]. More recent work by the same group has also shown that TgGAP45 phosphorylation is responsible for regulating the full assembly of the tachyzoites glideosome, with a requirement that TgGAP45 be dephosphorylated at two specific residues before it can associate with TgGAP50 to form the mature glideosome [29]. A similar two-step process of glideosome assembly is thought to occur in Plasmodium, though the data presented here suggest that while this model likely represents the process of glideosome assembly quite well, it does not incorporate the complete range of glideosome dynamics, which may involve the formation of both assembly and disassembly intermediate sub-complexes. The negative effect of staurosporine treatment on the association of PfGAP45/PfGAP50 with PfMyoA and PfMTIP suggests that the continued association of the complete glideosome complex is regulated by a staurosporine-sensitivie kinase. The results presented here and outlined above raise the possibility that the glideosome is assembled and disassembled in response to specific stimuli, creating specific sub-complexes depending upon various signaling cues and suggesting a degree of regulation—potentially at the tight junction—that is surprisingly complex.
The data presented here suggests for the first time that cADPR-regulated calcium release plays a significant role in P. falciparum erythrocyte invasion, and also illustrates several important points related to PfGAP45 phosphorylation and glideosome dynamics. However, several significant questions remain unanswered. First, the precise number of times that of PfGAP45 is phosphorylated is not known, and, with the exception of CDPK1, the kinases responsible for phosphorylating PfGAP45 have not been identified. Furthermore, whether some relationship exists between PfGAP45 phosphorylation and acylation, as is suggested by the change in PfGAP45 isoform distribution after staurosporine treatment, requires further study. Finally, the precise role of phosphorylation in regulating the assembly and/or disassembly of the merozoite glideosome, and the nature of each putative intermediate glideosome assembly/disassembly sub-complex remain to be defined. While these questions present significant technical challenges, a more complete understanding of the precise mechanisms involved in P. falciparum erythrocyte invasion will nevertheless provide many new therapeutic targets and perhaps will allow the development of new and effective malaria treatments.
Acknowledgments
We would like to thank Oliver Billker, PhD., for careful reading of the manuscript. M.L.J was supported by an NIH training grant (T32 AI055438).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Breman JG, Alilio MS, Mills A. Conquering the Intolerable Burden of Malaria: What’s New, What’s Needed: A Summary. Am J Trop Med Hyg. 2004;(Supplement 2):1–15. [PubMed] [Google Scholar]
- 2.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The Global Distribution of Plasmodium falciparumi Malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller LH, Baruch DI, Marsh K, Doumbo OK. The Pathogenic Basis of Malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 4.Cowman AF, Crabb BS. Invasion of Red Blood Cells by Malaria Parasites. Cell. 2006;124:755–766. doi: 10.1016/j.cell.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Gaur D, Mayer G, Miller LH. Parasite ligand-host receptor interactions during invasion of erythrocytes by Plasmodium merozoites. Int J Parasitol. 2004;34:1413–1429. doi: 10.1016/j.ijpara.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Baum J, Richard D, Healer J, Rug M, Krnajski Z, Gilberger TW, Green JL, Holder AA, Cowman AF. A conserved molecular motor drives cell invasion and gliding motility across malaria lifecycle stages and other apicomplexan parasites. J Biol Chem. 2006;281:5197–5208. doi: 10.1074/jbc.M509807200. [DOI] [PubMed] [Google Scholar]
- 7.Gaskins E, Gilk S, DeVore N, Mann T, Ward G, Beckers C. Identification of the membrane receptor of a class XIV myosin in Toxoplasma gondii. J Cell Biol. 2004;165:383–393. doi: 10.1083/jcb.200311137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones ML, Kitson EL, Rayner JC. Plasmodium falciparum erythrocyte invasion: A conserved myosin associated complex. Mol Biochem Parasitol. 2006;147:74–84. doi: 10.1016/j.molbiopara.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Buscaglia CA, Coppens I, Hol WGJ, Nussenzweig V. Sites of interaction between aldolase and Thrombospondin-related Anonymous Protein in Plasmodium. Mol Biol Cell. 2003;14:4947–4957. doi: 10.1091/mbc.E03-06-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jewett TJ, Sibley LD. Aldolase forms a bridge between cell surface adhesins and the actin cytoskeleton in apicomplexan parasites. Mol Cell. 2003;11:885–894. doi: 10.1016/s1097-2765(03)00113-8. [DOI] [PubMed] [Google Scholar]
- 11.McCallum-Deighton N, Holder AA. The role of calcium in the invasion of human erythrocytes by Plasmodium falciparum. Mol Biochem Parasitol. 1992;50:317–324. doi: 10.1016/0166-6851(92)90229-d. [DOI] [PubMed] [Google Scholar]
- 12.Ward GE, Fujioka H, Aikawa M, Miller LH. Staurosporine inhibits invasion of erythrocytes by malarial merozoites. Exp Parasitol. 1994;79:480–487. doi: 10.1006/expr.1994.1109. [DOI] [PubMed] [Google Scholar]
- 13.Wasserman M, Chaparro J. Intraerythrocytic calcium chelators inhibit the invasion of Plasmodium falciparum. Parasitol Res. 1996;82:102–107. doi: 10.1007/s004360050078. [DOI] [PubMed] [Google Scholar]
- 14.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1979;193:673–676. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 15.Rees-Channer RR, Martin SR, Green JL, Bowyer PW, Grainger M, Molloy JE, Holder AA. Dual acylation of the 45 kDA gliding-associated protein (GAP45) in Plasmodium falciparum merozoites. Mol Biochem Parasitol. 2006;149:113–116. doi: 10.1016/j.molbiopara.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Green JL, Rees-Channer RR, Howell SA, Martin SR, Knuepfer E, Taylor HM, Grainger M, Holder AA. The motor complex of Plasmodium falciparum: phosphorylation by a calcium-dependent protein kinase. J Biol Chem. 2008;283:30980–30989. doi: 10.1074/jbc.M803129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaid A, Thomas DC, Sharma P. Role of Ca2+/Calmodulin-PfPKB signaling pathway in erythrocyte invasion by Plasmodium falciparum. J Biol Chem. 2008;283:5589–5597. doi: 10.1074/jbc.M708465200. [DOI] [PubMed] [Google Scholar]
- 18.Kato N, Sakata T, Breton G, Le Roch KG, Nagle A, Andersen C, Bursulaya B, Henson K, Johnson J, Kumar KA, Marr F, Mason D, McNamara C, Plouffe D, Ramachandran V, Spooner M, Tuntland T, Zhou Y, Peters EC, Chatterjee A, Schultz PG, Ward GE, Gray N, Harper J, Winzeler EA. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nature Chem Biol. 2008;4:347–356. doi: 10.1038/nchembio.87. [DOI] [PubMed] [Google Scholar]
- 19.Carruthers VB, Sibley LD. Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma gondii. Mol Microbiol. 1999;31:421–428. doi: 10.1046/j.1365-2958.1999.01174.x. [DOI] [PubMed] [Google Scholar]
- 20.Lovett JL, Sibley LD. Intracellular calcium stores in Toxoplasma gondii govern invasion of host cells. J Cell Sci. 2003;116:3009–3016. doi: 10.1242/jcs.00596. [DOI] [PubMed] [Google Scholar]
- 21.Wetzel DM, Chen LA, Ruiz FA, Moreno SNJ, Sibley LD. Calcium-mediated protein secretion potentiates motility in Toxoplasma gondii. J Cell Sci. 2004;117:5739–5748. doi: 10.1242/jcs.01495. [DOI] [PubMed] [Google Scholar]
- 22.Chini EN, Nagamune K, Wetzel DM, Sibley LD. Evidence that the cADPR signaling pathway controls calcium-mediated microneme secretion in Toxoplasma gondii. Biochem J. 2005;389:269–277. doi: 10.1042/BJ20041971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovett JL, Marchesin N, Moreno SNJ, Sibley LD. Toxoplasma gondii microneme secretion involves intracellular Ca2+ release from inositol 1,4,5-triphosphate (IP3)/ryanodine-sensitive stores. J Biol Chem. 2002;277:25870–25876. doi: 10.1074/jbc.M202553200. [DOI] [PubMed] [Google Scholar]
- 24.Moudy R, Manning TJ, Beckers CJ. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. J Biol Chem. 2001;276:41492–41501. doi: 10.1074/jbc.M106154200. [DOI] [PubMed] [Google Scholar]
- 25.Delorme V, Cayla X, Faure G, Garcia A, Tardieux I. Actin dynamics is controlled by a Casein Kinase II and Phosphatase 2C interplay on Toxoplasma gondii Toxofilin. Mol Biol Cell. 2003;14:1900–1912. doi: 10.1091/mbc.E02-08-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meggio F, Donella Deana A, Ruzzene M, Brunati AM, Cesaro L, Guerra B, Meyer T, Mett H, Fabbro D, Furet P, Dobrowolska G, Pinna LA. Different susceptibility of protein kinases to staurosporine inhibition: kinetic studies and molecular bases for the resistance of protein kinase CK2. Eur J Biochem. 1995;234:317–322. doi: 10.1111/j.1432-1033.1995.317_c.x. [DOI] [PubMed] [Google Scholar]
- 27.Ono T, Cabrita-Santos L, Leitao R, Bettiol E, Purcell LA, Diaz-Pulido O, Andrews LB, Tadakuma T, Bhanot P, Mota MM, Rodriguez A. Adenylyl Cyclase α and cAMP signaling mediate Plasmodium sporozoite apical regulated exocytosis and hepatocyte infection. PLOS Path. 2008;4(2) doi: 10.1371/journal.ppat.1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sudo A, Kato K, Kobayashi K, Tohya Y, Akashi H. Susceptibility of Plasmodium falciparum cyclic AMP-dependent protein kinase and its mammalian homologue to the inhibitors. Mol Biochem Parasitol. 2008;160:138–142. doi: 10.1016/j.molbiopara.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Gilk S, Gaskins E, Ward GE, Beckers CJ. GAP45 phosphorylation controls assembly of the Toxoplasma myosin XIV complex. Eukaryot Cell. 2009;8:190–196. doi: 10.1128/EC.00201-08. [DOI] [PMC free article] [PubMed] [Google Scholar]