

Abstract
TAB1, a subunit of the kinase TAK1, was phosphorylated by SAPK2a/p38α at Ser423, Thr431 and Ser438 in vitro. TAB1 became phosphorylated at all three sites when cells were exposed to cellular stresses, or stimulated with tumour necrosis factor-α (TNF-α), interleukin-1 (IL-1) or lipopolysaccharide (LPS). The phosphorylation of Ser423 and Thr431 was prevented if cells were pre-incubated with SB 203580, while the phosphorylation of Ser438 was partially inhibited by PD 184352. Ser423 is the first residue phosphorylated by SAPK2a/p38α that is not followed by proline. The activation of TAK1 was enhanced by SB 203580 in LPS-stimulated macrophages, and by proinflammatory cytokines or osmotic shock in epithelial KB cells or embryonic fibroblasts. The activation of TAK1 by TNF-α, IL-1 or osmotic shock was also enhanced in embryonic fibroblasts from SAPK2a/p38α-deficient mice, while incubation of these cells with SB 203580 had no effect. Our results suggest that TAB1 participates in a SAPK2a/p38α-mediated feedback control of TAK1, which not only limits the activation of SAPK2a/p38α but synchronizes its activity with other signalling pathways that lie downstream of TAK1 (JNK and IKK).
Keywords: p38/SB 203580/stress-activated protein kinase/TAB1/TAK1
Introduction
The past decade has seen the elucidation of many of the major protein kinase cascades that allow extracellular signals to control intra- and intercellular communication. One of the major goals in this field is to identify the physiological substrates of each protein kinase and to find out how phosphorylation of these substrates alters cell physiology in response to particular signals. This is a daunting task because, based on the number of protein kinases encoded by the human genome, and the proportion of proteins that are phosphorylated in cells, protein kinases would be expected, on average, to phosphorylate ∼20 substrates. In fact, the number of substrates may be much higher, because so many proteins are phosphorylated at multiple sites by a number of different protein kinases.
One of the major approaches that has been used to identify substrates and activators of protein kinases is the yeast ‘two-hybrid’ system (Fields and Song, 1989). Its success depends on the validity of the assumption that the interaction between these proteins is sufficiently strong to enable their detection by this method. The finding that some protein kinases and their substrates possess specific ‘docking’ sites to allow them to interact specifically with one another suggests that this method is worth pursuing. Indeed, several substrates of the protein kinase PDK1 have been identified using the ‘two-hybrid’ system (Balendran et al., 2000), while MAPKAP-K3 (McLaughlin et al., 1996) and MEF2C (Han et al., 1997) were identified as substrates for stress-activated protein kinase 2a (SAPK2a, also called p38α) (Han et al., 1994; Rouse et al., 1994) by this method.
In this study, we have exploited the ‘two-hybrid’ system to identify further proteins that interact with SAPK2a/p38α. Using this technique, we identified several established substrates of SAPK2a/p38α, as well as several proteins that were not previously known to be substrates. Here we demonstrate that transforming growth factor-β (TGF-β)-activated kinase-1 (TAK1)-binding protein 1 (TAB1) is a physiological substrate of SAPK2a/p38α. TAB1 is an essential activator of TAK1 (Yamaguchi et al., 1995; Shibuya et al., 1996), a kinase that is thought to lie upstream of SAPK2a/p38α, c-jun N-terminal kinase (JNK) and IκB kinase (IKK) in a signalling pathway that is triggered by proinflammatory cytokines or bacterial lipopolysaccharide (LPS) (Ninomiya-Tsuji et al., 1999; Lee et al., 2000; Wang et al., 2001). We present evidence which suggests that the phosphorylation of TAB1 may be involved in a feedback control mechanism for terminating the activation of TAK1.
Results
Identification of TAB1 as a binding partner of SAPK2a/p38α
In order to identify proteins that regulate, or are phosphorylated by SAPK2a/p38α, a yeast two-hybrid screen was carried out with SAPK2a/p38α as bait. Approxim ately 1 × 106 yeast colonies were screened from a human brain cDNA library. Several positive clones that interacted with SAPK2a/p38α were identified as MEF2C (full-length), MNK2 (residues 260–465) (Waskiewicz et al., 1997) and MAPKAP-K2 (residues 308–400) (Rouse et al., 1994), which are all known substrates of SAPK2a/p38α. Another clone was identified as CL100 (also called MAP kinase phosphatase-1), a dual-specificity protein phosphatase that is known to dephosphorylate and inactivate SAPK2a/p38α (Alessi et al., 1993). Several other positive clones were identified that were not known to interact with SAPK2a/p38α, including TAB1 (residues 229–504). This protein was of interest, since it is reported to be required for the activity of TAK1 (Shibuya et al., 1996), a MAP kinase kinase kinase that is implicated in the activation of SAPK2a/p38α and JNK (Lee et al., 2000).
In order to define the region on TAB1 that interacts with SAPK2a/p38α, full-length and truncated mutants were expressed as GAL4 activation domain fusion proteins and tested in the two-hybrid system against the GAL4 DNA-binding domain fused to SAPK2a/p38α. Full-length TAB1 was able to interact with SAPK2a/p38α, as did the construct encoding residues 229–436, which is missing the TAK1-binding domain. The TAK1-binding domain, which is located within residues 437–504 (Shibuya et al., 1996), was unable to bind to SAPK2a/p38α, showing that SAPK2a/p38α and TAK1 bind to distinct regions of TAB1. SAPK2a/p38α was able to bind to a TAB1 construct bearing residues 371–504, but not 391–504. Similarly, SAPK2a/p38α bound to TAB1 constructs encoding residues 350–436 but not 350–400. These studies indicate that the region on TAB1 that interacts with SAPK2a/p38α lies within residues 371–436 (data not shown).
The association between TAB1 and SAPK2a/p38α was demonstrated by co-expression of vectors coding for GST–TAB1 and haemagglutinin (HA)-tagged SAPK2a/p38α in HEK 293 cells, and by showing that the HA-SAPK2a/p38α could be pulled-down with GST–TAB1 on gluthathione–Sepharose (Figure 1). GST–TAB1(229–436) also interacted with HA-tagged SAPK2a/p38α after co-expression in 293 cells (data not shown). The interaction of TAB1 with SAPK2a/p38α was specific, since TAB1 did not associate with the closely related isoforms SAPK2b/p38β2, SAPK3/p38γ or SAPK4/p38δ, nor did GST–TAB1 interact with MKK3 and MKK6 (Figure 1), the ‘upstream’ activators of SAPK2a/p38α (reviewed in Cohen, 1997). The endogenous SAPK2a/p38α in the cell lysates could also be co-precipitated with GST–TAB1 on glutathione beads (data not shown).
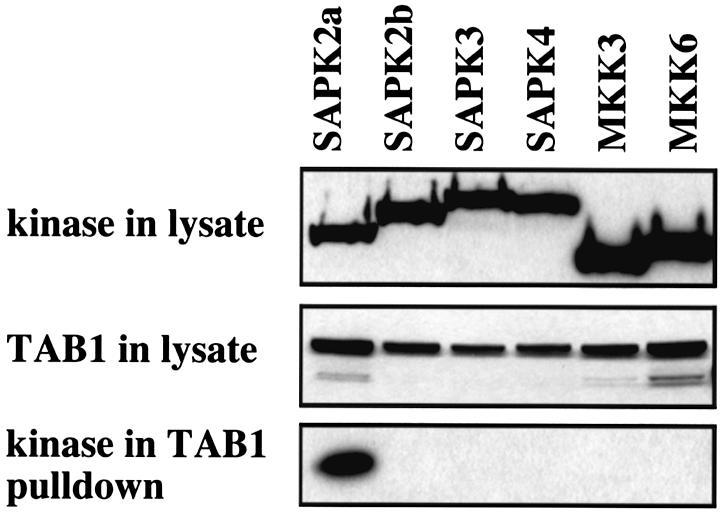
Fig. 1. TAB1 interacts specifically with SAPK2a/p38α. HEK 293 cells were transfected with plasmids coding for GST–TAB1 and SAPK2a/p38α, SAPK2b/p38β2, SAPK3/p38γ, SAPK4/p38δ, MKK3 or MKK6 fused to an HA epitope tag. After 36 h, the cells were lysed and the cell lysate (20 µg of protein) denatured in SDS, subjected to SDS–PAGE, transferred onto nitrocellulose membrane and the membrane probed with an antibody specific for the HA epitope to detect the expression of each protein kinase or with an anti-GST antibody to detect the level of expression of GST–TAB1. The GST–TAB1 in the cell lysates was then affinity purified on glutathione–Sepharose and proteins binding to this support were subjected to SDS–PAGE and immunoblotting with the anti-HA antibody to investigate the binding of each protein kinase to TAB1.
We compared the strength of interaction between TAB1 and SAPK2a/p38α and TAB1 and TAK1. When GST–TAB1 was co-expressed with myc-tagged TAK1, the two proteins could be pulled-down as a 1:1 complex from HEK293 cell lysates using either glutathione beads or immunoprecipitating myc antibodies. In contrast, only a small proportion of the HA-SAPK2a/p38α was pulled-down on the glutathione beads (data not shown). Similarly, as shown later, endogenous TAK1 was readily detectable in immunoprecipitates of endogenous TAB1, whereas endogenous SAPK2a/p38α was not. These experiments suggest that, in contrast to the TAB1–TAK1 complex, SAPK2a/p38α and TAB1 do not exist as a stoichiometric complex in 293 cells.
TAB1 is phosphorylated by SAPK2a/p38α
TAB1 expressed as a His-tagged fusion protein in Escherichia coli migrated as a single major band on SDS–PAGE. The protein was phosphorylated rapidly by bacterially expressed SAPK2a/p38α and approached a plateau at nearly 2 mol phosphate incorporated per mol protein, indicating that at least two residues were becoming phosphorylated (Figure 2A). Consistent with this finding, the electrophoretic mobility of TAB1 on SDS–polyacrylamide gels was reduced after phosphorylation for 30 min (Figure 2B), indicating stoichiometric phosphorylation of at least one site. The phosphorylation of TAB1 was prevented by the inclusion of SB 203580 in the assays, which is a relatively specific inhibitor of SAPK2a/p38α (Cuenda et al., 1995). No phosphorylation occurred in the absence of SAPK2a/p38α. These experiments indicate that all three sites are phosphorylated by SAPK2a/p38α in vitro.

Fig. 2. TAB1 is phosphorylated stoichiometrically by SAPK2a/p38α. Bacterially expressed His-TAB1 was phosphorylated at 30°C with 2 U/ml GST–SAPK2a/p38α and Mg [γ-32P]ATP. At the times indicated, an aliquot of the reaction was denatured in SDS and subjected to SDS–PAGE. The gel was stained with Coomassie blue and destained (B). The band corresponding to His-TAB1 was excised and analysed by Cerenkov counting to quantitate the incorporation of phosphate into TAB1 (A).
The 32P-labelled TAB1 that had been maximally phosphorylated by SAPK2a/p38α was excised from the gel, digested with trypsin and the tryptic phosphopeptides resolved by chromatography on a Vydac C18 column (Figure 3A). Nearly all the 32P-labelled material eluted at a high acetonitrile concentration as several overlapping peaks, indicating that the tryptic phosphopeptides were either very large or hydrophobic. All the 32P radioactivity was pooled, further subdigested with Asp-N proteinase and rechromatographed on the C18 column (Figure 3B). The four phosphopeptides D1–D4 were subjected to mass spectrometry and solid phase sequencing. Peptides D2 and D3 were identified as di- and mono-phosphorylated forms of the peptides corresponding to residues 428–462 phosphorylated at Thr431 and/or Ser438 (Figure 3C and D). Peptide D4 comprised residues 403–427 phosphorylated at Ser423 (Figure 3E). The identity of the minor peptide D1 was not determined, but 32P radioactivity was released after the fifth cycle of Edman degradation (Figure 3F). This is consistent with it being the peptide 419–427 phosphorylated at Ser423, cleavage with Asp-N protease occurring as a result of the partial deamidation of Asn419. Thus TAB1 is phosphorylated by SAPK2a/p38α in vitro at three sites, namely Ser423, Thr431 and Ser438.

Fig. 3. Identification of the residues on TAB1 phosphorylated by SAPK2a/p38α. His-TAB1 was phosphorylated with active GST–SAPK2a/p38α and subjected to SDS–PAGE. The band corresponding to 32P-labelled TAB1 was visualized by staining with Coomassie blue, excised and digested with trypsin. (A) The tryptic phosphopeptides were separated by HPLC on a Vydac C18 column equilibrated in 0.1% (v/v) trifluoroacetic acid. The column was developed with an acetonitrile gradient in 0.1% (v/v) trifluoroacetic acid (broken line). Radioactivity is indicated by the full line. (B) All the major 32P-labelled peptides from (A) were pooled, further digested with the protease Asp-N and rechromatographed on the Vydac C18 column as in (A). Peptides D2, D3, D4 and D1 were then subjected to solid phase sequencing in (C), (D), (E) and (F), respectively, to determine the sites of phosphorylation. The methodology has been described previously (Stokoe et al., 1992).
TAB1 is phosphorylated at three residues in vivo in response to lipopolysaccharide, proinflammatory cytokines and cellular stresses
We raised antibodies that immunoprecipitate TAB1 and phospho-specific antibodies that recognize TAB1 phosphorylated at Ser423, Thr431 or Ser438 (see Materials and methods). Recognition by each phospho-specific antibody was abolished by incubation with the relevant phosphopeptide immunogen, but not by the other two phosphopeptide immunogens or by the unphosphorylated form of the immunogen (Figure 4). We then examined the ability of a number of MAP kinase family members to phosphorylate the different sites on TAB1 (Figure 5). Ser423 was phosphorylated specifically by SAPK2a/p38α, with only relatively weak phosphorylation by SAPK2b/p38β2, trace phosphorylation by SAPK4/p38δ and no phosphorylation by any other MAP kinase. Thr431 and Ser438 were phosphorylated in vitro by all the MAP kinases tested.
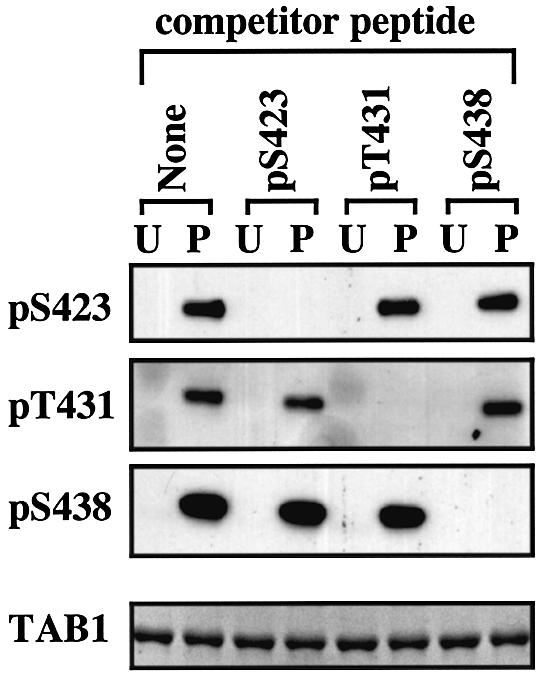
Fig. 4. Development of phospho-specific antibodies recognizing each of the three phosphorylation sites on TAB1. Bacterially expressed His-TAB1 (0.5 µg) was phosphorylated with SAPK2a/p38α (P) or left unphosphorylated (U). The proteins were then separated by SDS–PAGE, transferred to nitrocellulose and probed with each of the three phospho-specific antibodies in the absence (none) or presence of 10 µg/ml of one of the three phosphopeptide immunogens (termed competitor peptide). Each antibody also contained 10 µg/ml of the unphosphorylated form of the relevant peptide immunogen to neutralize any antibodies present that recognize the unphosphorylated species. The bottom panel shows the amount of His-TAB1 in each gel lane stained with Coomassie Blue.
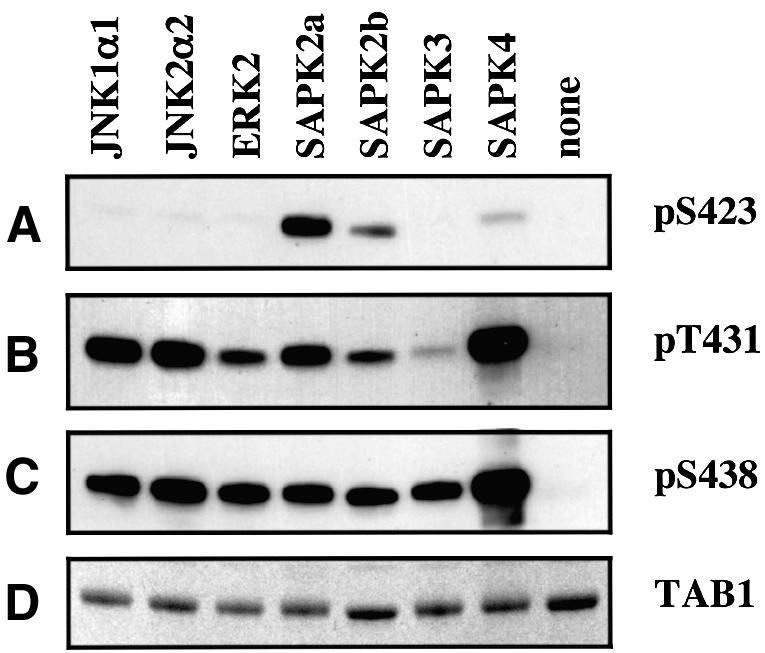
Fig. 5. Phosphorylation of TAB1 in vitro by different MAP kinase family members. Bacterially expressed His-TAB1 (0.5 µg) was phosphorylated with the protein kinases indicated, subjected to SDS-–PAGE and the proteins either transferred onto a nitrocellulose membrane (A–C) or stained with Coomassie blue (D). They were then immunoblotted with antibodies recognizing phosphorylated Ser423, Thr431 or Ser438 as in Figure 6.
The antibodies were then used to examine the phosphorylation state of TAB1 after exposure to different stimuli known to activate SAPK2a/p38α. Exposure of human epithelial KB cells to oxidative stress (hydrogen peroxide), UV-C radiation, osmotic shock (a high concentration of sorbitol) or a protein synthesis inhibitor (anisomycin) induced the phosphorylation of TAB1 at Ser423, Thr431 and Ser438 (Figure 6). The phosphorylation of Ser423 and Thr431 by all four stimuli was completely suppressed by prior incubation of the cells with SB 203580, but not by incubation with PD 184352, a specific inhibitor of the classical MAP kinase cascade (Sebolt-Leopold et al., 1999; Davies et al., 2000). In contrast, the phosphorylation of Ser438 was not prevented by SB 203580, but was largely (hydrogen peroxide) or partially (UV-C and sorbitol) inhibited by PD 184352. However, the anisomycin-induced phosphorylation of Ser438 was unaffected by either SB 203580 or PD 184352.

Fig. 6. TAB1 is phosphorylated on Ser423, Thr431 and Ser438 in response to stressors. KB cells were pre-treated for 1 h with or without 10 µM SB 203580 or 2 µM PD184352 then stimulated with H2O2 (2 mM for 15 min), UV-C (30 min), anisomycin (10 µg/ml for 30 min) or sorbitol (0.5 M for 30 min) and lysed. For anti-phospho-Ser423 (pS423) and anti-phospho-Thr431 (pT431) blots, 2 µg of an antibody that immunoprecipitates TAB1 was incubated for 1 h at 4°C with 250 µg of protein lysate. The immunocomplex was collected and washed as described in Materials and methods. The immunoprecipitated proteins were then denatured in SDS, separated by SDS–PAGE, transferred to a nitrocellulose membrane and blotted with phospho-specific antibodies that recognize TAB1 phosphorylated at Ser423 or Thr431. The antibody that recognizes TAB1 phosphorylated at Ser438 (pS438) was more sensitive and immunoblotting could be carried out using the cell lysate without immunoprecipitation. A protein migrating slightly faster than TAB1 was also recognized non-specifically by the anti-pSer438 antibody.
The stimulation of RAW 264 macrophages with LPS or human epithelial KB cells with tumour necrosis factor-α (TNF-α) or interleukin-1 (IL-1) induced the phosphorylation of TAB1 at Ser423 and Thr431 (Figure 7). As for cellular stresses, the phosphorylation of both sites was completely suppressed by prior incubation of the cells with SB 203580, but not by PD 184352. LPS and IL-1 also induced the phosphorylation of Ser438, which was not prevented by SB 203580, but largely prevented by PD 184352. TNF-α did not induce the phosphorylation of TAB1 at Ser438 in the 5 min stimulation shown in Figure 7, but did induce this phosphorylation after 20 min (data not shown). This reflects the slower activation of the classical MAP kinase cascade by TNF-α, compared with the activation of SAPK2a/p38α (Deak et al., 1998).

Fig. 7. TAB1 is phosphorylated on Ser423, Thr431 and Ser438 in LPS-stimulated RAW macrophages and TNF-α- or IL-1-stimulated KB cells. Cells were pre-treated for 1 h with or without 10 µM SB 203580 or 2 µM PD184352 then stimulated with LPS (100 ng/ml for 15 min), TNF-α (50 ng/ml for 5 min) or IL-1α (20 ng/ml for 20 min), and lysed and analysed as described in the legend to Figure 6.
An improved assay for TAK1
In order to study the effects of different agonists on the activation of TAK1, we developed the improved assay described in Materials and methods. In this assay, the TAK1 present in TAB1 immunoprecipitates is assayed by its ability to activate MKK6, as judged by the activation of SAPK2a/p38α. The active SAPK2a/p38α generated in this first reaction is then quantitated in a second assay by measuring its ability to phosphorylate myelin basic protein (MBP). The assay is analogous to the standard assay for the protein kinase Raf in the classical MAP kinase cascade (Alessi et al., 1995), which occupies a position equivalent to TAK1. The new assay was validated by the finding that no activity was detected if MKK6 or inactive SAPK2a/p38α or MBP were omitted from the assay (Table I). A small amount of activity was observed if the TAK1-containing immunoprecipitates were omitted or replaced by an immunoprecipitate in which the TAB1 antibody was replaced by a control IgG, because the bacterially expressed unphosphorylated form of MKK6 is not totally inactive (Table I). No activity was detected if the anti-TAB1 antibody was replaced by a control IgG. This assay for TAK1 activity in cell extracts is much faster and far more sensitive than measuring TAK1 activity by the direct phosphorylation of MKK6, which requires SDS–PAGE followed by autoradiography.
Table I. Validation of the TAK1 assay in cell extracts.
| Assay component omitted | Unstimulated (c.p.m.) | LPS-stimulated (c.p.m.) | LPS + SB 203580 (c.p.m.) |
|---|---|---|---|
| None | 2023 ± 657 | 5022 ± 1437 | 11 624 ± 787 |
| TAB1 immunoprecipitate | 1546 ± 184 | 1546 ± 184 | 1546 ± 184 |
| MKK6 | 295 ± 61 | 292 ± 40 | 367 ± 106 |
| SAPK2a/p38α | 237 ± 41 | 251 ± 41 | 243 ± 2 |
| Myelin basic protein | 233 ± 16 | 240 ± 23 | 238 ± 40 |
TAK1 was immunoprecipitated from the cell extract using an anti-TAB1 antibody as described in Materials and methods. TAK1 from unstimulated RAW264 cells, LPS-stimulated cells and cells pre-incubated with 10 µM SB 203580 prior to stimulation with LPS was assayed in the presence of every protein component of the assay (none) or with the omission of one protein component as indicated. The results show 32P radioactivity (c.p.m.) bound to phosphocellulose paper in each assay ± SEM for three assays. The significant incorporation of 32P radioactivity into myelin basic protein in the absence of TAB1 immunoprecipitate is caused by the trace activity of the unphosphorylated form of MKK6. This value is subtracted from that obtained in the complete assay to give the activity due to the added TAK1.
Role of SAPK2a/p38α in the regulation of TAK1 activity
The experiments presented in Figures 6 and 7 indicated that SAPK2a/p38α activity was rate limiting for the phosphorylation of TAB1 at Ser423 and Thr431 in cells. Since TAB1 is essential for TAK1 activity, we studied the effects of all seven stimuli on the activity of the TAB1–TAK1 complex in cells. After exposure to each stimulus, TAK1 was immunoprecipitated from the cell lysates using an anti-TAB1 antibody and assayed for TAK1 activity (see Materials and methods). LPS, TNF-α and IL-1 all caused a striking increase in TAK1 activity, while exposure to sorbitol induced a small transient activation of TAK1 (Figure 8A). In contrast, three other stressors (UV-C, hydrogen peroxide and anisomycin) were unable to activate TAK1 (data not shown).

Fig. 8. SB 203580 enhances the activity of TAK1 in LPS-stimulated RAW macrophages and TNF-α-, IL-1- or sorbitol-stimulated KB cells. (A) Cells were pre-treated for 1 h with or without 10 µM SB 203580 then stimulated with LPS (RAW 264.7 macrophages), TNF-α, IL-1 or sorbitol (KB cells). The cells were lysed at the times indicated and the TAK1 complexed to TAB1 was immunoprecipitated from 150 µg of protein lysate using 2 µg of a TAB1-specific antibody bound to protein G–Sepharose. The immune complex was then assayed for TAK1 activity as described in Materials and methods. The results are expressed as the mean ± SEM for three separate experiments with all assays performed in duplicate. Results are shown as a percentage of the activity of TAK1 measured at the time at which activation was maximal in the absence of SB 203580. (B) Same as (A), except that the TAB1 immunoprecipitates were assayed for TAK1 activity by the phosphorylation of MKK6 as described in Materials and methods. The figure shows the incorporation of 32P into the MKK6 substrate as revealed by autoradiography.
The failure of UV-C, hydrogen peroxide and anisomycin to induce any activation of TAK1, although they are even stronger inducers of the phosphorylation of TAB1 than proinflammatory cytokines or LPS, indicated that the phosphorylation of Ser423, Thr431 and Ser438 was insufficient for the activation of TAK1. In order to investigate different roles for the phosphorylation of Ser423 and Thr431, we therefore studied the effect of SB 203580 on the activation of TAK1 by LPS, TNF-α, IL-1 and sorbitol. These experiments revealed that SB 203580 enhanced by ∼2-fold the maximal activation attained in response to LPS, TNF-α and IL-1, and caused an even larger enhancement of sorbitol-induced TAK1 activation (Figure 8A). In contrast, no activation of TAK1 was observed after exposure of KB cells to hydrogen peroxide, UV-C or anisomycin, even in the presence of SB 203580 (data not shown).
In order to confirm these results, we repeated the experiments, but this time assayed TAK1 by the direct phosphorylation of MKK6 (see Materials and methods). These studies confirmed the results obtained by the new assay method; SB 203580 enhanced the LPS-induced activation of TAK1 several fold and greatly enhanced the sorbitol-induced activation of TAK1 (Figure 8B).
The results presented in Figure 8 suggested that SAPK2a/p38α is involved in a feedback control mechanism for limiting the activation of TAK1, perhaps by phosphorylating TAB1. Consistent with this hypothesis, the SB 203580-sensitive phosphorylation of TAB1 was clearly detectable (Figure 7) at the time at which downregulation of TAK1 was observed (Figure 8A), as was the phosphorylation of MAPKAP-K2 (data not shown), which is a well established substrate of SAPK2a/p38α. In order to investigate this idea further, we compared the activation of TAK1 in immortalized fibroblasts from wild-type mice and mice deficient in SAPK2a/p38α (Adams et al., 2000). These experiments revealed a striking increase in the TNF-α-, IL-1- and sorbitol-induced activation of TAK1 in the SAPK2a/p38α knockout fibroblasts compared with wild-type fibroblasts (Figure 9A), consistent with the proposed feedback control by SAPK2a/p38α. Moreover, incubation of the SAPK2a/p38α-deficient fibroblasts with SB 203580 did not increase the activity of TAK1 significantly in SAPK2a/p38α-deficient fibroblasts that had been stimulated with TNF-α, IL-1 or sorbitol, indicating that the effect of SB 203580 in wild-type cells did not result from a non-specific effect of this compound.

Fig. 9. The TNF-α-induced activation of TAK1 is enhanced in embryonic mouse fibroblasts deficient in SAPK2a/p38α. Immortalized embryonic fibroblasts from wild-type (WT, open bars) and SAPK2a/p38α knockout (KO, black bars) mice were treated for 1 h with or without 10 µM SB 203580, then stimulated for 10 min with 10 ng/ml of mouse TNF-α, for 20 min with 20 ng/ml of mouse IL-1 or for 30 min with 0.5 M sorbitol, and the cells lysed. (A) The complexes containing TAK1 were immunoprecipitated from the cell lysates (0.15 mg of protein) using a TAB1-specific antibody, and the activity of TAK1 in the cell lysates determined (see Materials and methods). The results are expressed as the mean ± SEM for three separate experiments with all assays performed in duplicate. Results are shown as a percentage of the activity of TAK1 in stimulated wild-type cells measured without pre-incubation with SB 203580. (B) The TAK1 complexes immunoprecipitated from the lysates of cells stimulated with TNF-α were prepared as in (A), then subjected to SDS–PAGE followed by immunoblotting with antibodies that recognize TAK1, TAB1 and TAB2. Cell lysate (20 µg of protein) was also subjected to SDS–PAGE followed by immunoblotting for SAPK2a/p38α (C). TAK1 complexes immunoprecipitated from the lysates of cells stimulated with TNF-α or IL-1 were prepared as in (A), and then analysed as in (B), except that the gels were immunoblotted with antibodies that recognize TAB1 phosphorylated at Ser423, Thr431 or Ser438.
TNF-α, IL-1 and sorbitol induced the phosphorylation of TAB1 at Ser423 and Thr431 in wild-type fibroblasts, but not in SAPK2a/p38α-deficient fibroblasts. In contrast, the phosphorylation of Ser438 was not abolished in the SAPK2a/p38α-deficient fibroblasts (Figure 9C). The results also show that TAK1, TAB1 and TAB2 are always present as a complex in both stimulated and unstimulated cells and expressed at similar levels in either the wild-type or SAPK2a/p38α ‘knockout’ cells after stimulation with TNF-α (Figure 9B), IL-1 or sorbitol (data not shown).
SB 203580 enhances the activation of JNK isoforms and the phosphorylation of IκB
Since TAK1 is reported to mediate the activation of JNK and IKK, as well as the activation of SAPK2a/p38α (see Introduction), the enhanced activation of TAK1 in the presence of SB 203580 (Figure 8) would be predicted to enhance the phosphorylation/activation of JNK isoforms and the phosphorylation of IκB. That this is indeed the case is demonstrated in Figure 10. After stimulation with IL-1, LPS or sorbitol, there was a striking increase in the level of phosphorylated JNK, provided the cells were first pre-incubated with SB 203580 prior to exposure to each agonist. SB 203580 enhanced the TNF-α-induced phosphorylation of JNK to a lesser extent, consistent with the smaller effect of SB 203580 to increase the TNF-α-induced activation of TAK1 (Figure 8). SB 203580 also greatly enhanced the sorbitol-induced phosphorylation of IκB (Figure 10). However, the effect of SB 203580 on the phosphorylation of IκB induced by LPS or proinflammatory cytokines could not be easily assessed because, as is well known, these agonists trigger an immmediate degradation of IκB as soon as it is phosphorylated (data not shown). SB 203580 did not enhance the IL-1- or LPS-induced phosphorylation of ERK1/ERK2 and only slightly increased the sorbitol-induced phosphorylation of ERK1/ERK2, which are not thought to be mediated by TAK1. TNF-α does not activate ERK1/ERK2 significantly in KB cells after 5 min, but only after 15 min.

Fig. 10. SB 203580 enhances the activation of JNK isoforms and phosphorylation of IκB. KB cells were stimulated with 50 ng/ml TNF-α (5 min), 20 ng/ml IL-1 (20 min) or 0.5 M sorbitol (30 min), and RAW264 macrophages were stimulated with 100 ng/ml LPS (15 min). The cells were incubated for 1 h without (–) or with (+) 2 µM PD 184352 or 10 µM SB 203580 prior to stimulation with each agonist. The cells were lysed and lysate protein (30 µg) was subjected to SDS–PAGE and immunoblotted with phospho-specific antibodies that recognize the active forms of JNK isoforms, the active forms of ERK1 and ERK2, IκB phosphorylated at Ser32, and an antibody that recognizes all forms of IκB.
Discussion
In this study, we have demonstrated that SAPK2a/p38α interacts with TAB1 at a region towards the C-terminus of the latter protein, but N-terminal to the region that binds TAK1. Interestingly, this region has no cluster of basic residues, which is the hallmark of docking sites for other substrates of SAPK2a/p38α so far identified.
The interaction between TAB1 and SAPK2a/p38α was also reported recently by others (Ge et al., 2002) while our study was in progress. However, a major conclusion reached by these investigators was that the TAB1–SAPK2a/p38α interaction allows SAPK2a/p38α to autophosphorylate and activate itself (Ge et al., 2002). This was suggested to provide a mechanism for activation that is independent of MKK3 and MKK6, the protein kinases thought to mediate the activation of SAPK2a/p38α by extracellular signals. In our hands, the incubation of GST–SAPK2a/p38α with MgATP and TAB1 in vitro failed to stimulate any autophosphorylation or activation of SAPK2/p38α. The TAB1 used in our experiments was expressed either in E.coli with six additional histidine residues at the C-terminus or in insect Sf21 cells with six additional histidine residues at the N-terminus. Both preparations were >90% pure as judged by SDS–PAGE. The same GST–SAPK2a/p38α preparation could be activated several hundred fold by MKK3 or MKK6 in parallel experiments (data not shown).
The relatively weak interaction of SAPK2a/p38α with TAB1, compared with the interaction of TAB1 with TAK1, suggested that the interaction of TAB1 with SAPK2a/p38α might have another function. This led us to find that TAB1 can be phosphorylated by SAPK2a/p38α at three sites in vitro and that the residues phosphorylated in vitro are also phosphorylated in vivo in response to cellular stresses, proinflammatory cytokines and LPS. Moreover, the phosphorylation of two of the three residues, Ser423 and Thr431, was prevented by the SAPK2a/p38α inhibitor SB 203580 and did not occur in fibroblasts deficient in SAPK2a/p38α activity. These studies establish that TAB1 is a physiological substrate of SAPK2a/p38α.
Ser423 is followed by alanine in murine as well as human TAB1. This is highly unusual because MAP kinases almost invariably phosphorylate serine or threonine residues that are followed by a proline. There have been two reports that SAPK2a/p38α phosphorylates serine and threonine residues of the microtubule-associated protein Tau (Reynolds et al., 2000) and the phosphatase cdc25 (Bulavin et al., 2001) in vitro that are not followed by proline. However, to our knowledge, Ser423 of TAB1 is the first such residue phosphorylated by SAPK2a/p38α that has been shown to be an in vivo phosphorylation site for this protein kinase. Phosphorylation of Ser423 in vitro was prevented by SB 203580, demonstrating that it is catalysed by SAPK2a/p38α. It could be argued that the phosphorylation of Ser423 is not catalysed by SAPK2a/p38α, but by another protein kinase, activated by SAPK2a/38α, present as a trace contaminant in the preparation of SAPK2a/p38α or its activator MKK6 or its substrate TAB1. However, this seems inconceivable, since all three proteins were expressed in E.coli, and not in eukaryotic cells, and were purified to near homogeneity by affinity chromatography. This finding was so unexpected that we resequenced the DNA several times to check that in the expressed protein the residue C-terminal to Ser423 would really be alanine. Thus potential residues in substrates phosphorylated by SAPK2a/p38α cannot always be predicted by inspecting the amino acid sequence of a protein.
The results presented here also show that SAPK2a/p38α suppresses the activity of TAK1 in cells, because the activation of TAK1 by proinflammatory cytokines and LPS is enhanced if cells are first pre-incubated with SB 203580 or in cells that do not express SAPK2a/p38α. Moreover, SB 203580 has no further effect in SAPK2a/p38α–/– fibroblasts, indicating that the effect of this compound in wild-type cells results from its inhibition of SAPK2a/p38α and not a non-specific effect.
The sites on TAB1 phosphorylated by SAPK2a/p38α in vivo lie at the interface of the SAPK2a/p38α- and TAK1-binding regions of TAB1. Since TAK1 is not a substrate for SAPK2a/p38α (data not shown), it seems likely that the phosphorylation of TAB1 induces a conformational change that suppresses either TAK1 activity or the ability of TAK1 to be activated. Alternatively, the phosphorylation of TAB1 may stimulate the rate of deactivation of TAK1. However, it cannot be excluded that SAPK2a/p38α downregulates TAK1 by phosphorylating another protein in the complex, such as TAB2 (Takaesu et al., 2000). Indeeed, we have noticed that the same agonists consistently induce a slight decrease in the electrophoretic mobility of TAB2 in wild-type fibroblasts, which is not observed in SAPK2a/p38α-deficient cells (Figure 9). The sites on TAB2 that become phosphorylated under these conditions have yet to be identified.
The mechanism by which TAK1 is activated by proinflammatory cytokines or LPS has not yet been fully elucidated but, once activated, TAK1 is thought to switch on MKK3 and/or MKK6 and hence SAPK2a/p38α (Lee et al., 2000). The downregulation of TAK1 by SAPK2a/p38α reported here may underlie the finding that incubation of RAW macrophages with SB 203580 enhances the activation of MKK3 by LPS (Xiao et al., 2002).
TAK1 appears to be essential for the LPS-induced activation of JNK in Drosophila cells (Chen et al., 2002). If this is also true in mammalian cells, then elimination of the feedback inhibition of TAK1 by SB 203580 should enhance the activation of JNK by agonists that activate TAK1. The results presented in Figure 10 show that this is indeed the case for four such agonists. SB 203580 has also been reported by others to enhance the LPS-induced activation of JNK in RAW macrophages (Hall and Davis, 2002).
Another target of TAK1 is reported to be IKK, leading to the phosphorylation of IκB, which is the trigger for its proteolytic degradation and hence the activation of NF-κB (Wang et al., 2001). The downregulation of TAK1 by SAPK2a/p38α may therefore not just be a feedback control device for limiting the activation of SAPK2a/p38α and JNK, but may also limit the activation of IKK, thereby synchronizing three signalling pathways that play key roles in the inflammatory response. Consistent with this idea, the sorbitol-induced phosphorylation of IκB, like the activation of TAK1 (Figure 9), was greatly enhanced in cells that had been pre-incubated with SB 203580 (Figure 10). However, LPS, TNF-α and IL-1 triggered such a rapid degradation of IκB that it was not possible to assess whether SB 203580 enhanced the phosphorylation of IκB in the presence of these agonists. Why osmotic shock induces the phosphorylation of IκB without triggering its degradation is unclear. Osmotic shock may inhibit the proteolytic degradation of IκB by an unknown mechanism. Alternatively, LPS and proinflammatory cytokines may activate the proteolytic degradation of IκB by a second mechanism that is unrelated to the phosphorylation of IκB.
Cellular stresses also trigger the activation of SAPK2a/p38α and JNK and the phosphorylation of TAB1 at Ser423 and Thr431. However, with the exception of sorbitol, these stimuli do not activate TAK1, whether or not the cells are pre-incubated with SB 203580 (data not shown), and they presumably activate these MAP kinase cascades via one or more of the other protein kinases that switch on MKK3 and MKK6 (reviewed in Garrington and Johnson, 1999). The phosphorylation of TAB1 by SAPK2a/p38α may therefore also be a device for limiting further activation of SAPK2a/p38α and JNK by proinflammatory cytokines, under conditions where these enzymes have already been activated by another stimulus.
The downregulation of TAK1 by SAPK2a/p38α, which is illustrated schematically in Figure 11, may have important implications for the development of anti-inflammatory drugs. Several compounds that are more potent and specific inhibitors of SAPK2a/p38α than SB 203580 currently are undergoing human clinical trials for the treatment of rheumatoid arthritis and other chronic inflammatory diseases. However, in view of the results obtained in the present study, it seems important to examine whether these compounds also upregulate the JNK and NF-κB pathways which are themselves proinflammatory and may contribute to unwanted side effects. Finally, we should mention that the downregulation of TAK1 by SAPK2a/p38α is not the only feedback control mechanism that limits the activation of the pathway. For example, it has been shown recently that SAPK2a/p38α negatively regulates the stability of the mRNA encoding its upstream activator MKK6 (Ambrosino et al, 2002).

Fig. 11. Schematic representation of the feedback control of TAK1 activity by SAPK2a/p38α and its implication for the regulation of JNK and NF-κB. (A) SAPK2a/p38α downregulates TAK1, probably via the phosphorylation of TAB1. (B) The inhibition of SAPK2a/p38α by SB 203580 (or other inhibitors of this protein kinase) abolishes this feedback control of TAK1, causing upregulation of the JNK and IKK pathways (illustrated by the thicker arrows).
Materials and methods
Materials
The SAPK2a/p38α inhibitor SB 203580 was obtained from Calbiochem (La Jolla, CA), pre-cast SDS–polyacrylamide gels and MBP were from Invitrogen (Groningen. The Netherlands), protein G–Sepharose was from Amersham Pharmacia Biotech (Little Chalfont, UK), LPS and human and mouse TNF-α and IL-1α were from Sigma, Pfu Turbo DNA polymerase used for all PCRs was from Stratagene (La Jolla, CA) and restriction enzymes were purchased from New England Biolabs (Beverly, MA). All other chemicals were obtained from Sigma, BDH (Poole, Dorset, UK). Peptides were synthesized by Dr Graham Bloomberg (University of Bristol, UK).
Protein kinase preparations
All protein kinases were expressed from human cDNAs and purified by affinity chromatography on glutathione–Sepharose (GST fusions), and/or nickel–nitrilotriacetate agarose (His6 fusions) or on amylose resin (MBP fusions). The MAP kinase family members SAPK2a/p38α, SAPK2b/p38β2, SAPK3/p38γ, SAPK4/p38δ and ERK2, and the MAP kinase kinase family members MKK4 and MKK7 were expressed as inactive GST fusion proteins in E.coli. MKK1 was expressed as a fusion protein with GST at the N-terminus and a His6 tag at the C-terminus. A truncated form of MEKK1 [MEKK1(2–317)] was also expressed as a GST fusion protein and used to activate GST–MKK4 and GST–MKK7. Wild-type MKK6 and an active mutant in which Ser210 and Thr214 were mutated to aspartate were expressed as MBP fusions in E.coli. MBP–MKK6[D210,D214] was used to activate all the p38 isoforms, and then removed by passage through amylose resin. The wild-type inactive MBP–MKK6 and inactive GST–SAPK2a/p38α were used to assay TAK1. JNK1α1 and JNK2α2 were expressed as His6-tagged proteins, and an active form of Raf (Raf[307–649][D340,D341] in which Tyr340 and Tyr341 were changed to aspartate) was expressed in insect Sf21 cells. The JNK isoforms were activated with MKK4 and MKK7, the activating enzymes being removed subsequently by passage through glutathione–Sepharose. The MKK1 was activated with Raf, and then used to activate the ERK2. The MKK1 was then removed on nickel–nitriliotriacetate agarose.
Two-hybrid screen with SAPK2a/p38α
A two-hybrid screen was carried out using the protein kinase SAPK2a/p38α as bait. Full-length SAPK2a/p38α[T180A, Y182F] with Thr180 mutated to alanine and Tyr182 mutated to phenylalanine was subcloned into the EcoRI and BamHI sites of pAS2-1 (Clontech, Palo Alto, CA) and in-frame with the GAL4 DNA-binding domain. The plasmid pAS2-1 was transformed into Y190 yeasts with a human brain cDNA library (Clontech), subcloned into the pACT2 vector and expressed as GAL4 activation domain fusions. The transformed yeast were grown on Synthetic Dropout medium supplemented with 25 mM 3-aminotriazole and 2% (w/v) glucose, but lacking histidine, leucine and tryptophan. The plates were incubated for 10 days at 30°C and colonies which grew in the absence of histidine were assayed for β-galactosidase activity and taken for further analysis.
Two-hybrid analysis of interactions between SAPK2a/p38α and TAB1
Various deletion mutants of TAB1 were constructed in the pACT2 vector and tested against pAS-SAPK2a/p38α[T180A, Y182F]. Y190 strain yeasts were co-transformed with the combinations of vectors indicated in the Results and grown on SD agar lacking tryptophan and leucine at 30°C until the appearance of colonies. Yeast colonies were picked and resuspended in 50 µl of sterile water and 1 µl dropped onto SD agar lacking leucine and tryptophan or SD agar supplemented with 25 mM 3-aminotriazole and lacking histidine, leucine and tryptophan. The yeast patches were incubated for 2 days at 30°C and filter lifts taken of yeast grown on SD agar lacking leucine and tryptophan. Reporter β-galactosidase activity of the transformants was tested by incubating filters for 4 h in X-Gal at 30°C.
Bacterial expression of His-TAB1
An expressed sequence tag (EST) cDNA clone (IMAGE 3506362) encoding TAB1 was obtained from the IMAGE consortium (HGMP, Hinxton Hall, Cambridge, UK) and sequenced. Full-length TAB1 cDNA was amplified by PCR and subcloned into the bacterial expression vector pET21a (Novagen, Madison, WI) by NheI–XhoI. The plasmid construct was transformed into BL21 E.coli, grown to A600 nm of 0.6 and induced for 1 h at 37°C with 200 µM isopropyl-β-d-thiogalactopyranoside. The cells were harvested by centrifugation and resuspended in extraction buffer [50 mM Tris–HCl pH 7.5, 0.1 mM EGTA, 150 mM NaCl, 0.03% (w/v) Brij-35, 5% (v/v) glycerol, 0.1% (v/v) 2-mercaptoethanol, 0.1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 1 mM dithiothreitol]. The cells were freeze-thawed once and sonicated for 2 min on ice. Triton X-100 was added to a final concentration of 1% (v/v) and the suspension left on ice for 30 min. The cell debris was cleared by centrifugation for 30 min at 75 000 g and the supernatant (termed lysate) decanted. The lysate (30 ml) was incubated for 1 h at 4°C with 3 ml of a 50% slurry of nickel–agarose (Qiagen) and the pellet washed with 50 mM Tris–HCl pH 7.5, 0.27 M sucrose, 0.5 M NaCl, 0.1% (v/v) 2-mercaptoethanol, 10 mM imidazole, followed by 50 mM Tris–HCl pH 7.5, 10 mM NaCl, 0.27 M sucrose, 0.1% (v/v) 2-mercaptoethanol, and eluted with 200 mM imidazole in the same buffer. Imidazole was removed by dialysis in 50 mM Tris pH 7.5, 10 mM NaCl, 0.27 M sucrose, 0.1% (v/v) 2-mercaptoethanol.
Phosphorylation of TAB1 by SAPK2a/p38α
A 1 µg aliquot of bacterially expressed His-TAB1 was incubated in a 0.12 ml reaction with 2 U/ml of activated GST–SAPK2a/p38α (Davies et al., 2000), 10 mM magnesium acetate and 0.1 mM [γ-32P]ATP (106 c.p.m./nmol) in 50 mM Tris–HCl pH 7.5, 1 µM microcystin, 0.1 mM sodium orthovanadate, 0.1 mM EGTA and 0.1% (v/v) 2-mercaptoethanol. At various times, 20 µl aliquots were denatured in SDS, and subjected to SDS–PAGE. After staining with Coomassie blue and destaining, the band corresponding to His-TAB1 was excised and digested with trypsin. The resulting phosphopeptides were then chromatographed on a Vydac-C18 column as described in the Results
Antibodies
Polyclonal antibodies that recognize specific phosphorylation sites in human TAB1 were raised against the following peptides: CMVNGAHpSASTLDE (Ser423), CSTLDEApTPTLTNQ (Thr431) and CPTLTNQpSPT LTLQ (Ser438), where pS and pT denotes a phosphorylated serine and threonine residues, respectively. Antibodies that recognize TAK1 were raised against the peptide CKKQLE VIRSQQQKRQGTS (residues 561–579). Each peptide was coupled separately to keyhole limpet haemocyanin and bovine serum albumin (BSA), mixed and injected into a sheep at Diagnostics Scotland (Edinburgh, UK). A second injection was made 6 weeks later. Polyclonal antibodies against TAB1 were raised in sheep against bacterially expressed His-TAB1. Antibodies that recognize TAB2 were obtained from Santa Cruz (Santa Cruz, CA) and antibodies that recognize all forms of SAPK2a/p38α or IκB, an antibody that recognizes IκB phosphorylated at Ser32 and an antibody that recognizes the active phosphorylated forms of ERK1 and ERK2 were purchased from Cell Signalling (Beverly, MA). Antibodies that recognize the active phosphorylated forms of JNK were obtained from Biosource (Nivelles, Belgium). Sheep-, rabbit- or mouse-specific secondary antibodies conjugated to horseradish peroxidase were from Pierce (Rockford, IL).
Cell culture, stimulation and lysis
Human epithelial KB cells and immortalized mouse embryonic fibroblasts were cultured in 10 cm dishes in 10 ml of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal calf serum (FCS). At 16 h prior to stimulation with human TNF-α or IL-1α for human cells or murine TNF-α or IL-1-α for mouse cells, the medium was removed and replaced with DMEM from which FCS had been omitted. Where indicated, 10 mM SB 203580 or 10 mM PD 184352 dissolved in dimethylsulfoxide (DMSO) was added to the medium to final concentrations of 10 and 2 µM, respectively, 1 h prior to stimulation with agonists. The equivalent volume of DMSO was added as a control. RAW macrophages were cultured in a similar manner except that the FCS was heat inactivated. Cells were harvested by the removal of the medium followed by lysis in ice cold buffer A [50 mM Tris–HCl pH 7.5, 0.1 mM EGTA, 1 mM EDTA, 1% (w/w) Triton X-100, 1 mM Na3VO4, 50 mM NaF, 5 mM sodium pyrophosphate, 0.27 M sucrose, 1 µM microcystin-LR, 0.1% (v/v) 2-mercaptoethanol and ‘complete’ proteinase inhibitor cocktail (one tablet/50 ml; Roche, Lewes, UK)]. Protein concentrations of the lysates were determined using the Bradford method.
Immortalized mouse embryonic fibroblasts
Embryonic mouse fibroblasts were isolated from wild-type or SAPK2a/p38α-deficient E9.5 embryos (Adams et al., 2000) and immortalized by infection with SV40 large T-expressing retrovirus (Ramhauser et al., 2002).
Transfection of HEK 293 cells
293 cells were transiently transfected with 1 µg/ml of DNA encoding each construct according to a modified calcium phosphate method (Chen and Okayama, 1988). The cells were harvested 36 h post-transfection by lysis with ice-cold buffer A.
Immunoprecipitation of TAB1 and associated TAK1
Aliquots of cell lysates (amounts specified in the figure legends) were incubated with 2 µg of affinity-purified TAB1-specific antibodies coupled to 10 µl of protein G–Sepharose for 1 h at 4°C. The immunoprecipitates were washed twice with 1 ml of buffer A containing 0.5 M NaCl followed by two washes with 1 ml of 50 mM Tris–HCl pH 7.5, 0.27 M sucrose and 0.1% (v/v) 2-mercaptoethanol.
Assay of TAK1
The TAK1 associated with TAB1 immunoprecipitates was assayed by its ability to activate MKK6 as judged by the activation of SAPK2a/p38α. The active SAPK2a/p38α generated in this reaction was then quantitated in a second assay by measuring its ability to phosphorylate MBP.
The TAB1 immunoprecipitates obtained as described above were incubated at 30°C with 40 µl of 50 mM Tris–HCl pH 7.5, 0.1% (v/v) 2-mercaptoethanol, 1 µM microcystin, 0.1 mM sodium orthovanadate, 0.1 mM EGTA, 0.1 mg/ml BSA, 50 µg/ml of unactivated MBP–MKK6 (Alonso et al, 2000), 0.1 mg/ml unactivated GST–SAPK2a/p38α, 10 mM magnesium acetate and 0.1 mM unlabelled ATP. After 30 min, 5 µl was withdrawn and added to 45 µl of 50 mM Tris–HCl pH 7.5, 0.1% (v/v) 2-mercaptoethanol, 1 µM microcystin, 0.1 mM sodium orthovanadate, 0.1 mM EGTA, 0.1 mg/ml BSA, containing 0.33 mg/ml of MBP, 10 mM magnesium acetate and 0.1 mM [γ-32P]ATP (106 c.p.m./nmol). After 10 min at 30°C, the reaction was stopped by pipetting 40 µl onto phosphocellulose P81 paper. The P81 papers were washed several times in 75 mM phosphoric acid, once in acetone, dried and the radioactivity incorporated into MBP bound to the papers quantified by Cerenkov counting. The values obtained were corrected for those obtained in control incubations in which TAK1 was omitted to allow for the trace activity of the unphosphorylated form of MKK6 (see Results). TAK1 activity was linear with respect to time and TAK1 concentration in the above assay.
We also assayed TAK1 by its ability to phosphorylate inactive MKK6. The assay was identical to the first step described above except that inactive SAPK2a/p38α was omitted and unlabelled ATP was replaced by [γ-32P]ATP (2 × 106 c.p.m./nmol). After 30 min, the reaction was terminated by the addition of SDS and half the reaction subjected to SDS–PAGE, transferred to nitrocellulose membranes and subjected to autoradiography overnight.
After immunoprecipitation of the TAK1–TAB1 complex by anti-TAB1, no further TAK1 activity could be immunoprecipitated by either a second round of immunoprecipitation with anti-TAB1 or by immunoprecipitation with the anti-TAK1 antibody. In contrast, the anti-TAK1 antibody used by itself immunoprecipitated ∼60% of the TAK1 activity that could be immunoprecipitated with the anti-TAB1 antibody. These experiments indicate that the assays of the anti-TAB1 immunoprecipitates are measuring most of the TAK1 activity in cells.
Acknowledgments
Acknowledgements
We thank Dr Jane Leitch, Division of Signal Transduction Therapy (DSTT), University of Dundee for antibody production, the Protein Production Team of the DSTT coordinated by Dr Hilary McLaughlan and Dr James Hastie for protein kinases, and the DNA Sequencing Service of the DSTT coordinated by Dr Nick Helps for sequencing constructs. This study was supported by the UK Medical Research Council, The Royal Society, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, NovoNordisk and Pfizer.
Note added in proof
Brancho et al. (2003) have just reported that no activation of SAPK2a/p38α is detectable in fibroblasts from mice deficient in both MKK3 and MKK6, also arguing against a role for TAB1 in activating SAPK2a/p38α in cells.
Brancho,D., Tanaka,N., Jaeschke,A., Ventura,J.J., Kelkar,N., Tanaka,Y., Kyuuma,M., Takashita,T., Flavell,R.A. and Davis,R.J. (2003) Mechanism of p38 MAP kinase activation in vivo. Genes Dev., 17, 1969–1978.
References
- Adams R.H. et al. (2000) Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell, 6, 109–116. [PubMed] [Google Scholar]
- Alessi D.R., Smythe,C. and Keyse,S.M. (1993) The human CL100 gene encodes a Tyr/Thr-protein phosphatase which potently and specifically inactivates MAP kinase and suppresses its activation by oncogenic ras in Xenopus oocyte extracts. Oncogene, 8, 2015–2020, [PubMed] [Google Scholar]
- Alessi D.R., Cohen,P., Ashworth,A., Cowley,S., Leevers,S.J. and Marshall,C.J. (1995) Assay and expression of mitogen-activated protein kinase, MAP kinase kinase and Raf. Methods Enzymol., 255, 279–290. [DOI] [PubMed] [Google Scholar]
- Alonso G., Ambrosino,C., Jones,M. and Nebreda,A.R. (2000) Differential activation of p38 mitogen-activated protein kinase isoforms depending on signal strength. J. Biol. Chem., 275, 40641–40648. [DOI] [PubMed] [Google Scholar]
- Ambrosino C., Mace,G., Galban,S., Fritsch,C., Vintersten,K., Black,E., Gorospe,M. and Nebreda,A.R. (2002) Negative feedback regulation of MKK6 mRNA stability by p38α mitogen-activated protein kinase. Mol. Cell. Biol., 23, 370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balendran A., Biondi,R.M., Cheung,P.C.F., Casamayor,A., Deak,M. and Alessi,D.R. (2000) A 3-Phosphoinositide-dependent protein kinase-1 (PDK1) docking site is required for the phosphorylation of protein kinase Cζ and PKC-related kinase 2 by PDK1. J. Biol. Chem., 275, 20806–20813. [DOI] [PubMed] [Google Scholar]
- Bulavin D.V., Higashimoto,Y., Popoff,I.J., Gaarde,W.A., Basrur,V., Potapova,O., Appella,E. and Fornace,A.J. (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature, 411, 102–107. [DOI] [PubMed] [Google Scholar]
- Chen C.A. and Okayama,H. (1988) Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques, 6, 632–638. [PubMed] [Google Scholar]
- Chen W., White. M.A and Cobb,M.H. (2002) Stimulus-specific requirements for MAP3 kinases in activating the JNK pathway. J. Biol. Chem., 277, 49105–49110. [DOI] [PubMed] [Google Scholar]
- Cohen P. (1997) The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends Cell Biol., 7, 353–361. [DOI] [PubMed] [Google Scholar]
- Cuenda A., Rouse,J., Doza,Y.N., Meier,R., Cohen,P., Gallagher,T.F., Young,P.R. and Lee,J.C. (1995) SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett., 364, 229–233. [DOI] [PubMed] [Google Scholar]
- Davies S.P., Reddy,H., Caivano,M. and Cohen,P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J., 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M., Clifton,A.D., Lucocq,J.M. and Alessi,D.R. (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38 and may mediate activation of CREB. EMBO J., 17, 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S. and Song,O. (1989) A novel genetic system to detect protein–protein interactions. Nature, 340, 245–246. [DOI] [PubMed] [Google Scholar]
- Garrington T.P. and Johnson,G.L. (1999) Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol., 11, 211–218. [DOI] [PubMed] [Google Scholar]
- Ge B., Gram,H., Di Padova,F., Huang,B., New,L., Ulevitch,R.J., Luo,Y. and Han,J. (2002) MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science, 295, 1291–1294. [DOI] [PubMed] [Google Scholar]
- Hall J.P., Davis,R.J. (2002) Inhibition of the p38 pathway upregulates macrophage JNK and ERK activities and the ERK, JNK and p38 MAP kinase pathways are reprogrammed during differentiation of the murine myeloid M1 cell line. J. Cell. Biochem., 86, 1–11. [DOI] [PubMed] [Google Scholar]
- Han J., Lee,J.D., Bibbs,L. and Ulevitch,R.J. (1994) A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science, 265, 808–811. [DOI] [PubMed] [Google Scholar]
- Han J., Jiang,Y., Li,Z., Kravchenko,V.V. and Ulevitch,R.J. (1997) Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature, 386, 296–299. [DOI] [PubMed] [Google Scholar]
- Lee J., Mira-Arbibe,L. and Ulevitch,R.J. (2000) TAK1 regulates multiple protein kinase cascades activated by bacterial lipopoly saccharide. J. Leukoc. Biol., 68, 909–915. [PubMed] [Google Scholar]
- McLaughlin M.M., Kumar,S., McDonnell,P.C., Van Horn,S., Lee,J.C., Livi,G.P. and Young,P.R. (1996) Identification of mitogen-activated protein (MAP) kinase-activated protein kinase-3, a novel substrate of CSBP p38 MAP kinase. J. Biol. Chem., 271, 8488–8492. [DOI] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J., Kishimoto,K., Hiyama,A., Inoue,J., Cao,Z. and Matsumoto,K. (1999) The kinase TAK1 can activate NIK-IκB as well as the MAP kinase pathway in the IL-1 signalling pathway. Nature, 398, 252–256. [DOI] [PubMed] [Google Scholar]
- Ramhauser K., Sadzak,I., Porras,A., Pilz,A., Nebreda,A.R., Decker,T. and Kovarik,P. (2002) p38 MAPK enhances STAT1-dependent transcription independently of Ser727 phosphorylation. Proc. Natl Acad. Sci. USA, 99, 12859–12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds C.H., Betts,J.C., Blackstock,W.P., Nebreda,A.R. and Anderton,B.H. (2000) Phosphorylation sites on Tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and p38 and glycogen synthase kinase-3β. J. Neurochem., 74, 1587–1595. [DOI] [PubMed] [Google Scholar]
- Rouse J., Cohen,P., Trigon,S., Morange,M., Alonso-Llamazares,A., Zamanillo,D., Hunt,T. and Nebreda,A.R. (1994) A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell, 78, 1027–3107. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold J.S. et al. (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nature Med., 5, 810–816. [DOI] [PubMed] [Google Scholar]
- Shibuya H., Yamaguchi,K., Shirakabe,K., Tonegawa,A., Gotoh,Y., Ueno,N., Irie,K., Nishida,E. and Matsumoto,K. (1996) TAB1: an activator of the TAK1 MAPKKK in TGF-β signal transduction. Science, 272, 1179–1182. [DOI] [PubMed] [Google Scholar]
- Stokoe D., Campbell,D.G., Nakielny,S., Hidadka,H., Leevers,S.J., Marshall,C.J. and Cohen,P. (1992) MAPKAP-kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. EMBO J., 11, 3985–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaesu G., Kishida,S., Hiyama,A., Yamaguchi,K., Shibuya,H., Irie,K., Ninomiya-Tsuji,J. and Matsumoto,K. (2000) TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell, 5, 649–658. [DOI] [PubMed] [Google Scholar]
- Wang C., Deng,L., Hong,M., Akkaraju,G.R., Inoue,J. and Chen,Z.J. (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature, 412, 346–351. [DOI] [PubMed] [Google Scholar]
- Waskiewicz A.J., Flynn,A., Proud,C.G. and Cooper,J.A. (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J., 16, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.Q., Malcolm,K., Worthen,G.S., Gardai,S., Schiemann,W.P., Fadok,V.A., Bratton,D.L. and Henson,P.M. (2002) Crosstalk between ERK and p38 MAPK mediates selective suppresssion of proinflammatory cytokines by transforming growth factor β. J. Biol. Chem., 277, 14884–14893. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K., Shirakabe,K., Shibuya,H., Irie,K., Oishi,I., Ueno,N., Taniguchi,T., Nishida,E. and Matsumoto,K. (1995) Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science, 270, 2008–2011. [DOI] [PubMed] [Google Scholar]