

Abstract
Secretory and membrane proteins that fail to fold in the endoplasmic reticulum (ER) are retained and may be sorted for ER-associated degradation (ERAD). During ERAD, ER-associated components such as molecular chaperones and lectins recognize folding intermediates and specific oligosaccharyl modifications on ERAD substrates. Substrates selected for ERAD are then targeted for ubiquitin- and proteasome-mediated degradation. Because the catalytic steps of the ubiquitin–proteasome system reside in the cytoplasm, soluble ERAD substrates that reside in the ER lumen must be retrotranslocated back to the cytoplasm prior to degradation. In contrast, it has been less clear how polytopic, integral membrane substrates are delivered to enzymes required for ubiquitin conjugation and to the proteasome. In this review, we discuss recent studies addressing how ERAD substrates are recognized, ubiquitinated and delivered to the proteasome and then survey current views of how soluble and integral membrane substrates may be retrotranslocated.
Keywords: degradation, ER, glycosylation, molecular chaperone, proteasome, proteolysis, transport, ubiquitin
Over one-third of newly synthesized proteins are translocated into the endoplasmic reticulum (ER), which ensures their delivery into the secretory pathway (1). Because of this high flux of proteins through the ER and because the folding of multi-domain proteins may be inefficient, the ER lumen contains a high concentration of molecular chaperones that maintain polypeptide solubility, enzymes that posttranslationally modify proteins, and factors that directly assist in the folding of newly synthesized polypeptides. Proteins that fail to acquire their native conformations, because of genetic error, cellular stress or stochastic events, may harm the cell. Therefore, components within the ER lumen also mediate quality control ‘decisions’ that result in the resolution of terminally misfolded proteins from correctly folded proteins and folding intermediates. This decision-making process is known as ER quality control (ERQC) and may result in the retention of aberrantly folded proteins in the ER (2).
To offset the potentially catastrophic consequences of misfolded protein accumulation, ER-retained species are most commonly destroyed. The primary mechanism of disposal is ER-associated degradation (ERAD) (3–9). During ERAD, misfolded proteins are delivered to the 26S proteasome, which resides in the cytoplasm. Therefore, the destruction of ERAD substrates requires polypeptide recognition, delivery from the ER to the cytoplasm (which has been termed retrotranslocation or dislocation) and in most cases ubiquitination, which ensures efficient delivery to the proteasome. The importance of ERAD is underscored by the fact that a growing number of ERAD substrates are linked to human diseases (10) and that the accumulation of ERAD substrates may induce the unfolded protein response (UPR), which if unresolved will trigger apoptosis (11). Another mechanism utilized by the cell to destroy misfolded proteins, especially those that aggregate in the secretory pathway or in the cytoplasm, is autophagy, and the reader is referred to a recent review on this topic (12). In this article, we will focus on and briefly summarize current views of how substrates are recognized during ERAD. We will then discuss the ER membrane-associated degradation machineries that facilitate ERAD substrate retrotranslocation and ubiquitination.
ERAD Substrate Recognition
Soluble substrates
It is imperative that ERAD substrates, especially those that are aggregation prone, are retained in solution to ensure efficient retrotranslocation. Thus, the simplest mechanism underlying molecular chaperone-mediated selection during ERAD is that early folding intermediates and misfolded proteins remain bound to chaperones. The chaperones would then deliver these species to secondary quality control complexes that escort selected polypeptides from the ER and to the proteasome. In fact, molecular chaperones such as BiP (an Hsp70) and its Hsp40 co-chaperones have been shown to prevent soluble substrate aggregation prior to degradation (13). Protein disulfide isomerase (PDI) and PDI homologues have also been shown to act prior to substrate retrotranslocation (14–16).
Another method of ERAD substrate retention employs ER-resident chaperone-like lectins (2,9, 17). Nearly, all secreted proteins contain a core glycosylation consensus site onto which an N-linked oligosaccharide (NAc-Gln2-Man9-Glc3) is appended during or immediately after translocation. Soon thereafter, two of three glucose residues in the N-glycan are trimmed by glucosidase I and II. This species is a substrate for calnexin and calreticulin, which catalyzes polypeptide folding and prevents aggregation. Indeed, in the absence of calnexin function, BiP provides a backup mechanism to maintain substrate solubility and retain aberrant proteins in the ER (18). In what is most probably a stochastic process, the last glucose residue may be trimmed by glucosidase II, which releases the protein from the lectins. If, however, the substrate is still misfolded, it is recognized by the glucosyl transferase (GT), which preferentially recognizes unfolded or molten globule (19–21) species and reglucosylates the polypeptide. This permits lectin rebinding and prevents the misfolded protein from leaving the ER.
Misfolded proteins in the ER cannot cycle between calnexin/calreticulin and the GT perpetually and thus a folding ‘timer’ aborts this cycle and induces the destruction of dead-end products. The timer is an ER mannosidase that trims mannose residues from the core glycan (22–24). This reduces the efficiency of calnexin/calreticulin rebinding and diverts substrates to a second, putative lectin known as ER degradation enhancing α-mannosidase-like protein (EDEM) (25–27). Three EDEM homologues have been identified in mammals (28,29), and one homologue resides in the yeast ER (30,31). Although the protein was originally thought to simply recognize polypeptides with trimmed mannose residues, recent data suggest that EDEM may be a mannosidase and exhibits chaperone-like activity (32,33). It was also originally thought that EDEM might recognize only modestly mannose-trimmed species, but again more recent data suggest that ERAD substrates may be trimmed more extensively (34). Many of these issues, and the nature of ERAD substrate hand off between ER-resident chaperones, lectins and the retrotranslocation machinery, will clearly be aided by the development of reconstituted systems in which each event can be recapitulated.
Another intermediary that appears to act between ERAD substrate selection and retrotranslocation is Yos9, which resides in the yeast ER (35–37). Yos9 is also a lectin that appears to act as a component of a ‘gate-keeping’ complex through which ERAD substrates must pass prior to retro-translocation (38). Other components of the Yos9 complex include BiP and Hrd3 (39), which is a partner of the Hrd1 E3 ubiquitin ligase (see below). Because Hrd3 recognizes misfolded proteins independent of Yos9, and BiP might as well, these data suggest that a multiple-step recognition mechanism may be employed prior to the final decision-making process that targets some ERAD substrates for degradation. In mammals, two homologues of Yos9, OS-9 and XTP3-B, have been noted. Similar to yeast Yos9, OS-9 and XTP3B bind to misfolded proteins and to SEL1L, a homologue of Hrd3, suggesting that these two proteins link ERAD substrates to the membrane-associated ubiquitination machinery. OS-9 also associates with Grp94, which is required for misfolded protein degradation. Therefore, Grp94 may contribute to substrate recognition and/or to the regulation of the assembly and disassembly of the OS-9–SEL1L–Hrd complex (40).
Although an N-glycan seems to be a critical element for the ERAD of misfolded glycoproteins (see above), it is less clear how nonglycosylated misfolded proteins are recognized and targeted for degradation. The importance of this pathway is underscored by the fact that treatment of cells with tunicamycin, an inhibitor for N-glycosylation, results in the accumulation of misfolded proteins in the ER and UPR induction. A recent study demonstrates that HERP, a membrane-associated cytoplasmic protein in mammals, binds to nonglycosylated BiP substrates and to the 26S proteasome but not to substrates whose degradation depends on calnexin function. Moreover, reduction of HERP levels inhibits the degradation of nonglycosylated BiP substrates but has no effect on calnexin substrates. These results suggest that there is some distinction in the degradation pathways for glycosylated and nonglycosylated proteins (41).
Integral membrane substrates
Far less is known about how misfolded membrane proteins are recognized and sorted to the ERAD pathway, although the cytoplasmic Hsp70/Hsp40 system and other chaperones clearly help maintain the solubility of large, cytoplasmic loops in selective ERAD substrates (42–44). We recently reported that ER-associated, cytoplasmic Hsp70–Hsp40 chaperones assist the interaction of this class of ERAD substrates with E3 ubiquitin ligases and suggested that a chaperone complex may play an active role during degradation rather than simply retaining polypeptides in solution (45). This view is consistent with other recent reports suggesting that a cytoplasmic chaperone network may lead substrates to a folding and/or degradation pathway (46,47). Interestingly, defects in cytoplasmic Hsp90 function accelerate the degradation of integral membrane proteins with misfolded cytoplasmic domains, implying that selective chaperones are ‘prodegradative’, whereas others are primarily involved in folding (48–50). In any event, integral membrane ERAD substrates with misfolded cytoplasmic domains have been proposed to utilize the ‘ERAD-C’ (cytoplasmic) pathway, and ongoing work has defined many of the requirements for the degradation of proteins in this class (Table 1 and Figure 1) (51,52).
Table 1.
Selective components required for ERAD in yeasta
| Cytosol- and membrance-associated | Merribrane-associated | ER lumen- and membrane-associated | ||||
|---|---|---|---|---|---|---|
| ERAD-C | Degradation | 26S proteasome | E3 | Doa10 | ||
| Retrotranslocation | Cdc48–Ufd1–Npl4–Ubx2 | E2 | Ubc6 and Ubc7–Cue1 | |||
| Chaperones | Ssa1, Ydj1 and HIj1 | |||||
| ERAD-M | Degradation | 26S proteasome | E3 | Hrd1–Hrd3 | ||
| Retrotranslocation | Cdc48–Ufd1–Npl4–Ubx2 | E2 | Ubc7–Cue1 | |||
| ERAD-L | Degradation | 26S proteasome | E3 | Hrd1–Hrd3 | Putative lectins | Yos9 and Htm1/Mnl1 |
| Retrotranslocation | Cdc48–Ufd1–Npl4–Ubx2 | E2 | Ubc7–Cue1 | Chaperones | BiP (Kar2), Scj1, Jem1 and PDI | |
| Putative retrotranslocon components | Der1, Sec61, Ssh1 and Usa1 | |||||
| Other components | Polyubiquitination | Ufd2 | ||||
| Deubiquitination | Rpn11 and Otu1 | |||||
| Escort | Rad23–Dsk2 | |||||
| Deglycosylation | Png1 | |||||
Components involved in the ERAD-C, -M or -L pathways as listed by their locations. Note that the functions of Ufd2 and Rad23-Dsk2 have not been well characterized for multiple ERAD substrates, and the direct involvement of Rpn11, Otu1 and Png1 during ERAD is still being elucidated. Ubx2 and Cue1 are membrane proteins that recruit the Cdc48 complex and Ubc7 to the ER membrane, respectively. Hrd3 is a membrane protein that stabilizes Hrd1 and recognizes lumenal substrates through its lumenal domain. Usa1 is also a membrane protein that appears to help link the Hrd1 E3 ligase complex and Der1 and is a specific component of the ERAD-L pathway (52).
Figure 1. A current working model of the ERAD pathway in yeast.
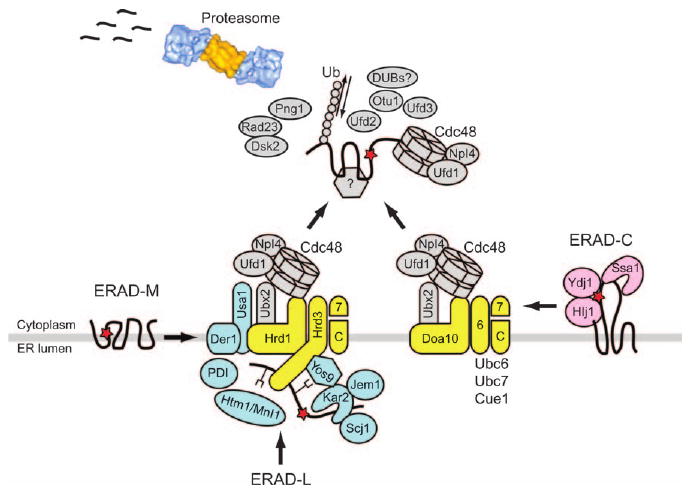
ERAD-L substrates are recognized by Hsp70/Hsp40 chaperones (Kar2, Jem1 and Scj1), PDI, putative lectins (Yos9 and Htm1/Mnl1) and the lumenal domain of Hrd3. These substrates are then retrotranslocated to the cytoplasm. In contrast, ERAD-M substrates may be directly recognized by the Hrd1 E3 ligase. ERAD-C substrates are recognized by cytoplasmic Hsp70/Hsp40 chaperones (Ssa1, Ydj1 and Hlj1) and by the Doa10 E3 ligase. The ubiquitinated substrates are delivered to the proteasome core for degradation by a series of escort factors, including the Cdc48 complex and the 19S proteasome cap. Steps following substrate ubiquitination are not yet clear for ERAD substrates. However, the polyubiquitin chain may be further remodeled by Ufd2 and by deubiquitinating enzymes (Rpn1, and/or DUBs such as Otu1). N-glycans may be cleaved by Png1, and some substrates are escorted to the proteasome by Rad23 and Dsk2. Membrane-associated E2–E3 enzymes, ERAD-L-specific components and retrotranslocation components are colored in yellow, blue and gray, respectively. The red star depicts a mutation that leads to protein misfolding, and the proteasome image was adopted from Voges et al. (126).
At this time, it is unclear how misfolded, integral membrane proteins that lack prominent soluble domains are recognized for destruction, and only a few members of this ‘ERAD-M’ class of species have been studied (5,52). It is possible that these substrates are recognized directly by E3 ubiquitin ligases; it is worth noting that most of the E3s known to be required for ERAD substrate ubiquitination contain multiple transmembrane-spanning domains (53–55), suggesting that intramembrane substrate recognition may occur. E3-encoded transmembrane domains may also constitute a component of the long-sought retrotranslocation channel (see below).
Do ERAD substrates possess a common recognition motif?
Because secretory proteins are diverse, there are potentially an infinite number of states at which proteins may misfold, depending on the positions of the mutation, environmental and cellular stress, or expression level. As noted above, the accumulation of misfolded and/or non-native proteins in the ER can trigger the UPR, which leads to the synthesis of ER chaperones and enzymes involved in secretory protein maturation and to the synthesis of components required to divert misfolded proteins to the ERAD and other degradation pathways (11). UPR induction appears to be enhanced by both a chaperone-dependent mechanism (i.e. BiP) and an UPR-inducing sensor in the ER (56). Not surprisingly, the ERAD and UPR pathways are linked such that the simultaneous disruption of genes required for each pathway results in synthetic phenotypes (57–59). This suggests that diverse misfolded proteins may be targeted to ERAD.
Although ERAD seems to be a major mechanism to dispose misfolded secretory proteins, some misfolded proteins in the ER are degraded through different pathways. In fact, a quality control system exists in subsequent secretory organelles (60). In addition, highly accumulated misfolded proteins may aggregate and can be delivered to the autophagic pathway (61). Therefore, at this time, the existence of a common biophysical feature of ERAD substrates cannot be assumed. Notably, a recent study has suggested that ERAD efficiency does not simply correlate with a protein's thermodynamic instability or with its rate of folding (62). Moreover, there seems to be a complex interplay between ERAD, the ER-associated folding machinery, secretion efficiency and a substrate's innate biophysical properties (63).
One attribute of secreted proteins that might confound attempts to generalize mechanisms of ERAD regards the number and placement of appended oligosaccharides. Several recent reports indicate that the specific position of the N-glycan controls ERAD substrate selection (64,65). In mammalian cells, a C-terminal 19 amino acid cassette with one N-glycan is sufficient to mediate the entry of cyclooxygenase-2 into the ERAD pathway (66). In the future, it will be vital to extend these important studies to include more diverse substrates and to examine whether the embedded polypeptide sequence plays a contributing role in glycan position-dependent selection.
ERAD Substrate Retrotranslocation
Nearly, all ERAD substrates are ubiquitinated prior to their degradation, a process that requires the sequential action of an E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases, each of whose catalytic sites are located in the cytoplasm. Therefore, ER lumenal substrates must first gain access to the cytoplasm before they can be modified. It remains unknown how this occurs. Next, a critical, minimal length of the polyubiquitin chain is imperative for retrotranslocation (67), and it was shown that ubiquitinated and/or unmodified substrates can be detected in the cytosol or on the cytosolic face of the ER membrane when the proteasome is inhibited (68–72). The extraction of ubiquitinated substrates from the ER is driven by the Cdc48/p97 complex, an ATP-requiring hexameric AAA adenosine triphosphatase (ATPase) that is thought to couple ATP hydrolysis with polypeptide retrotranslocation (67,73,74). However, a selective group of soluble ERAD substrates retrotranslocate in an ubiquitin-independent manner, and these use a different energy-requiring complex, the 19S particle of the proteasome (see below). These substrates also accumulate in the cytosol when the proteolytic activity of the proteasome is inhibited (75–77). Together, these data imply that a protein-conducting channel facilitates the retrotranslocation of substrates to the cytoplasm (8).
Guilt by association: which protein – if any – constitutes the ‘retrotranslocon’?
The identity of the retrotranslocation channel and mechanism of retrotranslocation remain contentious, even though defined integral membrane proteins in the ER interact with ERAD substrates en route to their degradation and interact with components of the ubiquitination/degradation machineries (Table 1 and Figure 1). In this section, we summarize evidence that implicates the involvement of each candidate as the retrotranslocon.
Several lines of evidence have suggested that the retrotranslocon may be Sec61, which is the major component of the translocation channel that imports polypeptides into the ER (78,79). Some ERAD substrates, including major histocompatibility complex class I (MHCI) in mammals (whose degradation is induced by a viral gene product) and a nonglycosylated yeast mating pheromone (pαF) in yeast, coprecipitate with or cross-link to Sec61 (80,81). Consistent with these data, sec61 mutant yeast exhibit a pαF degradation defect in vitro and CPY* degradation defect in vivo (81,82). Also, the prebinding of ribosomes to ER-derived microsomes, and most likely Sec61, abolishes the retrotranslocation of a glycopeptide, which exhibits some properties in common with ERAD substrates (83). Furthermore, Sec61 depletion in reconstituted mammalian vesicles or Sec61 blockage with ribosome–nascent chain complexes prevents the export of cholera toxin and the amyloid beta-peptide from the ER to the cytosol (84,85). Less direct – but in further support of Sec61-mediated retrotranslocation – is the reported association of yeast Sec61 to the 19S subunit of the proteasome (86), which suggests a mechanism for the proteasome-mediated extraction of pαF from the ER (76). But, perhaps the best evidence that at least a small subset of ERAD substrates utilizes Sec61 derives from studies on the regulated degradation of apolipoprotein B (apoB) in mammals; in the lipid-deficient state, apoB translocation into the ER is halted, and the protein is then cotranslocationally degraded by the proteasome (87). It has been proposed that the regulated degradation of apoB requires an ER and Sec61-associated chaperone-like protein, p58, which may ‘shift’ Sec61 from acting as a translocon to a retrotranslocon (88).
Together, the data presented above suggest that Sec61 can directly retrotranslocate proteins or may serve as a component of the retrotranslocon, particularly those that are stalled during translation and translocation (i.e. apoB). Nevertheless, accumulating data conflict with the assumption that Sec61 is the retrotranslocon for all ERAD substrates. First, complexes containing the principle E3s utilized during ERAD (Hrd1 and Doa10) lack Sec61 (38,52,89), even though another putative retrotranslocon, known as Der1, associates with Hrd1 (see below). Second, sec61 mutants degrade integral membrane ERAD substrates proficiently, suggesting that the channel – if it is involved in retrotranslocation – is only co-opted for the degradation of soluble substrates (50,90). Third, the realtime retrotranslocation of pαF from mammalian microsomes was unaffected by the addition of anti-Sec61 antibody, but the retrotranslocation of paF was significantly inhibited by anti-Der1 antibody. Furthermore, pαF cross-linked to this protein but not to Sec61 (77).
As noted above, the Hrd1 and Doa10 E3 ligases seem to be central components of the membrane-associated ubiquitination machineries in the yeast Saccharomyces cerevisiae (Table 1 and Figure 1). Hrd1 can be immunoprecipitated in complex with components in the lumen, in the ER membrane and in the cytoplasm. Each of these factors is required for the degradation of lumenal substrates or more formally substrates that harbor misfolded domains that reside in the lumen (ERAD-L) (51,52). In contrast, Doa10 mediates the turnover of ERAD-C substrates (see above). Doa10p can be immunoprecipitated in complex with a more specific group of ERAD-requiring proteins in the membrane and in the cytoplasm (52). The catalytic RING domain in both Hrd1 and Doa10 is situated on the cytoplasmic face of the ER membrane, and the proteins are estimated to span the lipid bilayer 6 times and 14 times, respectively (53–55). Based on this fact, and the fact that Hrd1 and Doa10 co-ordinate so many ERAD contributors, one view is that the enzymes function as both retrotranslocation channels and E3 ligases. Therefore, retrotranslocation and substrate modification are effectively coupled. This hypothesis has also been forwarded for gp78, a polytopic, integral membrane E3 in the mammalian ER (91).
Another protein that may constitute or be a part of the retrotranslocon is Der1, which was introduced above and was first isolated as a protein required for ‘degradation in the ER’ of two different soluble substrates (92). While Der1 is only required for the degradation of lumenal proteins in yeast, accumulating evidence has suggested that Derlin1 – one of three homologues in the mammalian ER – is a critical component that links lumenal and cytoplasmic components during ERAD. Specifically, Derlin1 can be isolated in complex with factors in the membrane and cytoplasm and with ERAD substrates (41,93–99) (Table 2). Consistent with a central role in ERAD, Derlin1 depletion induces the UPR (97) and retards the degradation of select substrates, and – as mentioned above – Derlin1 is a central player during pαF retrotranslocation from mammalian vesicles (77). Interestingly, Derlin proteins have also recently been implicated in the liberation of polyomaviral-encoded proteins from the ER after infection (16,100) and in the retrotranslocation of cholera toxin from the ER (101). These processes require other factors required for ERQC, indicating that the ERAD pathway has been co-opted by opportunistic pathogens (102).
Table 2.
Selective components required for ERAD in mammalsa
| Mammals | Saccharomyces cerevisiae |
|---|---|
| Cytosol- and membrane-associated | |
| p97-UFD1-NPL4 | Cdc48–Ufd1–Npl4 |
| Carboxyl terminus of Hsc70-interacting protein (CHIP) | |
| FBX2 | |
| Parkin | |
| ATX3 | |
| Rad23 | Rad23 |
| N-glycanase | Png1 |
| Membrane-associated | |
| gp78 | |
| HRD1–SEL1L | Hrd1–Hrd3 |
| RMA1 | |
| TEB4 | Doa10 |
| Ubc6e | Ubc6 |
| Ubc7 | Ubc7 |
| HERP | Usa1 |
| VCP-interacting membrane protein (VIMP) | |
| Derlin-1, -2 and -3 | Der1 |
| Sec61 | Sec61 |
| ER lumen- and membrane-associated | |
| OS-9 and XTP3-B | Yos9 |
| EDEM-1, -2 and -3 | Htm1/Mnl1 |
| BiP | BiP (Kar2) |
| PDI | PDI |
Selective conserved components are listed by their locations. Proteins whose names are underlined can be coimmunoprecipitated with Derlin-1 (see text). Note that the functions of many of the components in mammals have not been fully delineated.
Another scenario, which will surely muddy future attempts to define the retrotranslocon, is that the channel forms only transiently and/or is a composite of several candidates. In other intracellular organelles, such as the peroxisome, the transient pore model has been postulated, which could explain the fact that peroxisomes import folded, even oligomeric, proteins (103). Yet another scenario is that retrotranslocation requires the formation of a lipid bi-cell in the ER membrane (104). This model has been invoked to explain data indicating that protein complexes and large folded domains and N-glycans on ERAD substrates can be retrotranslocated (105–107). It is possible that a proteinaceous channel would be unable to expand to accommodate these large cargo molecules.
Could there be as-yet undiscovered or other candidate proteins that constitute the retrotranslocon? One factor proposed to fulfill this role is Ssh1, a Sec61 homologue in yeast (108). However, other data suggest that the yeast genetic background may dictate the Ssh1 dependence on ERAD (50) and thus it remains unclear how this protein contributes to ERQC.
Substrate Delivery to the Proteasome
After retrotranslocation, a series of ubiquitin-binding proteins escort modified substrates from the ER membrane to the proteasome (109,110). One of these factors is the AAA ATPase, Cdc48 (p97 in mammal), that in conjugation with two cofactors (Ufd1 and Npl4) contributes to ERAD after substrate ubiquitination but prior to degradation (67,73,74,111,112). Biochemical studies have shown that Cdc48/p97 extracts ubiquitinated substrates to the cytosol (45,73,113) either by actively pulling a polypeptide through the retrotranslocon and/or lipid membrane or by segregating a polypeptide that has already been liberated from the ER membrane. Cdc48/p97 might then deliver substrates to the proteasome by virtue of its interaction with ubiquitin-binding and ubiquitin-like domain-containing molecules such as Ufd2 (which has been termed an E4), Ufd3 (a protein of unknown function), Otu1 (a deubiquitinating enzyme) and Rad23/Dsk2 (109,114–118).
Accumulating evidence indicates that Cdc48/p97 is not required for the destruction of all ERAD substrates. For example, the Cdc48/p97 requirement during the extraction of a membrane protein might depend on the overall hydrophobicity of the substrate. Membrane proteins possessing multiple transmembrane-spanning segments might have a heightened requirement for the enzyme, whereas substrates containing fewer transmembrane segments or transmembrane segments with charged residues appear to be less dependent on Cdc48/p97 function (119). Second, Cdc48/p97 activity is dispensable for the retrotranslocation of pαF from yeast or mammalian microsomes (76,77). Third, the retrotranslocation of cholera toxin from the ER is p97 independent (120). For pαF and cholera toxin, the lack of Cdc48/p97-dependent export may arise because the substrates are not polyubiquitinated. Instead, the 19S particle of the proteasome drives the retrotranslocation of these substrates. Of note, the base of the 19S particle, like Cdc48/p97, harbors a ring of AAA ATPases (109), and some recent studies have attempted to define which subunit(s) of the base are directly involved in catalyzing retrotranslocation (67,121).
How are Integral Membrane Substrates Delivered to the Proteasome?
By definition, integral membrane substrates expose one or more regions to the cytoplasm, which could be ubiquitinated before retrotranslocation. The subsequent degradation of these substrates could start from either end of the polypeptide after dislocation through the retrotranslocon or through direct membrane extraction (Figure 2). Alternatively, degradation could start from an internal site on a cytoplasmically exposed loop after an endoproteolytic clip by the proteasome. In these models, it is assumed that degradation and retrotranslocation are tightly coupled and occur at the ER membrane. On the other hand, some integral membrane ERAD substrates, such as MHCI (70) and cystic fibrosis transmembrane conductance regulator (CFTR) (122), have been observed to reside in the cytoplasm when proteasome function is compromised. These data suggested that membrane-spanning segments might be solubilized from the lipid bilayer of the ER prior to proteasome-mediated degradation. By employing a reconstituted system, we were able to demonstrate directly that Ste6p*, another 12 transmembrane-spanning ERAD substrate, is released into the cytosol in a Cdc48/p97- and ATP-dependent manner (45). The electrophoretic profiles of these species suggest that the transmembrane domain of Ste6p* became solvent exposed. Therefore, a future research challenge is to determine how transmembrane segments are maintained in solution.
Figure 2. Models for membrane substrate delivery to the proteasome.
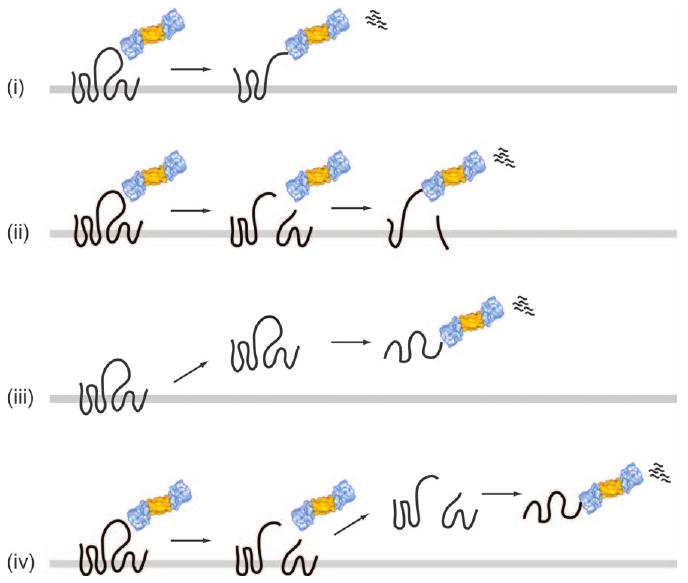
Polytopic membrane proteins may be processively dislocated and degraded from either the N- or the C-terminus by the proteasome (i), as was proposed for the FtsH proteolytic system in bacteria (127). Alternatively, polytopic membrane proteins may be extracted to the cytoplasm and then degraded by the proteasome (iii). In this case, the solubility of hydrophobic transmembrane segments may be maintained by the Cdc48 complex, the 19S cap of the proteasome and/or other components including chaperones and proteasome-escorting factors. Because the proteasome has an endoproteolytic activity (128), cytoplasmic loop(s) of substrates may first be ‘clipped’ by the proteasome and then dislocated or extracted (ii or iv). Not shown in these models is the potential role of a retrotranslocon in substrate degradation. The proteasome image was adopted from Voges et al. (126).
Conclusions and Perspectives
As described in this review, many open questions remain on the mechanistic details by which different ERAD substrates are degraded. As is often the case in ‘new’ fields, early attempts to generalize accumulated phenomenology have invariably been met with exceptions to the rule. Indeed, we are continually humbled by the growing number of factors that impact ERAD efficiency and diverse mechanisms employed during ERAD. Thus, it is clear that the degradation pathways of new and noncanonical ERAD substrates must be defined.
Nevertheless, specific, general questions in the field that remain to be answered include the following: Are substrates actively released from the ERQC machinery, or is the final decision to degrade an ERAD substrate simply a stochastic event? What is the nature of the retrotranslocon, or in the end, will there be a variety of means by which the retro-translocation reaction can be engineered? Are there other specialized ERAD machineries, and how is ERAD regulated, as evidenced by the examination of apoB degradation (88) or cells under stress (123–125)? How are substrates, such as 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase, converted from functional enzymes in the ER membrane to ERAD substrates (6)? Because proteasome substrates must be deubiquitinated, how is ERAD substrate deubiquitination and degradation coupled, and are specific deubiquitinating enzymes required for ERAD? What is the rate-limiting step during ERAD, or does this depend on the nature of the substrate? And finally, how is ERAD efficiency controlled during development, disease and stress? Needless to say, answers to each of these questions will keep many of us busy for some time.
Acknowledgments
Studies on ERAD in the Brodsky laboratory are supported by grants from the National Institutes of Health (GM075061 and HL58541), the Cystic Fibrosis Foundation (FRIZZE05XO) and the Alpha One Foundation. K. N. acknowledges the support of the Uehara Memorial Foundation and the American Heart Association, and we also thank K. A. Hecht, S. Nishikawa, T. Sato, Y. Tamura and J. Tran for their helpful discussions.
References
- 1.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 2.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 3.Brodsky JL. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation) Biochem J. 2007;404:353–363. doi: 10.1042/BJ20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 5.Sayeed A, Ng DT. Search and destroy: ER quality control and ER-associated protein degradation. Crit Rev Biochem Mol Biol. 2005;40:75–91. doi: 10.1080/10409230590918685. [DOI] [PubMed] [Google Scholar]
- 6.Hampton RY. Proteolysis and sterol regulation. Annu Rev Cell Dev Biol. 2002;18:345–378. doi: 10.1146/annurev.cellbio.18.032002.131219. [DOI] [PubMed] [Google Scholar]
- 7.Kostova Z, Wolf DH. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003;22:2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romisch K. Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol. 2005;21:435–456. doi: 10.1146/annurev.cellbio.21.012704.133250. [DOI] [PubMed] [Google Scholar]
- 9.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 10.Aridor M. Visiting the ER: the endoplasmic reticulum as a target for therapeutics in traffic related diseases. Adv Drug Deliv Rev. 2007;59:759–781. doi: 10.1016/j.addr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 12.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 13.Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gillece P, Luz JM, Lennarz WJ, de La Cruz FJ, Romisch K. Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J Cell Biol. 1999;147:1443–1456. doi: 10.1083/jcb.147.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3:246–255. doi: 10.1038/nrm780. [DOI] [PubMed] [Google Scholar]
- 16.Schelhaas M, Malmstrom J, Pelkmans L, Haugstetter J, Ellgaard L, Grunewald K, Helenius A. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell. 2007;131:516–529. doi: 10.1016/j.cell.2007.09.038. [DOI] [PubMed] [Google Scholar]
- 17.Caramelo JJ, Parodi AJ. How sugars convey information on protein conformation in the endoplasmic reticulum. Semin Cell Dev Biol. 2007;18:732–742. doi: 10.1016/j.semcdb.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang JX, Braakman I, Matlack KE, Helenius A. Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Mol Biol Cell. 1997;8:1943–1954. doi: 10.1091/mbc.8.10.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor SC, Ferguson AD, Bergeron JJ, Thomas DY. The ER protein folding sensor UDP-glucose glycoprotein-glucosyltransferase modifies substrates distant to local changes in glycoprotein conformation. Nat Struct Mol Biol. 2004;11:128–134. doi: 10.1038/nsmb715. [DOI] [PubMed] [Google Scholar]
- 20.Caramelo JJ, Castro OA, Alonso LG, De Prat-Gay G, Parodi AJ. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc Natl Acad Sci U S A. 2003;100:86–91. doi: 10.1073/pnas.262661199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritter C, Quirin K, Kowarik M, Helenius A. Minor folding defects trigger local modification of glycoproteins by the ER folding sensor GT. EMBO J. 2005;24:1730–1738. doi: 10.1038/sj.emboj.7600645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Swulius MT, Moremen KW, Sifers RN. Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc Natl Acad Sci U S A. 2003;100:8229–8234. doi: 10.1073/pnas.1430537100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jakob CA, Burda P, Roth J, Aebi M. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J Cell Biol. 1998;142:1223–1233. doi: 10.1083/jcb.142.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosokawa N, Tremblay LO, You Z, Herscovics A, Wada I, Nagata K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong alpha1-antitrypsin by human ER mannosidase I. J Biol Chem. 2003;278:26287–26294. doi: 10.1074/jbc.M303395200. [DOI] [PubMed] [Google Scholar]
- 25.Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, Nagata K. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- 27.Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science. 2003;299:1397–1400. doi: 10.1126/science.1079474. [DOI] [PubMed] [Google Scholar]
- 28.Mast SW, Diekman K, Karaveg K, Davis A, Sifers RN, Moremen KW. Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology. 2005;15:421–436. doi: 10.1093/glycob/cwi014. [DOI] [PubMed] [Google Scholar]
- 29.Hirao K, Natsuka Y, Tamura T, Wada I, Morito D, Natsuka S, Romero P, Sleno B, Tremblay LO, Herscovics A, Nagata K, Hosokawa N. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J Biol Chem. 2006;281:9650–9658. doi: 10.1074/jbc.M512191200. [DOI] [PubMed] [Google Scholar]
- 30.Jakob CA, Bodmer D, Spirig U, Battig P, Marcil A, Dignard D, Bergeron JJ, Thomas DY, Aebi M. Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2001;2:423–430. doi: 10.1093/embo-reports/kve089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakatsukasa K, Nishikawa S, Hosokawa N, Nagata K, Endo T. Mnl1p, an alpha-mannosidase-like protein in yeast Saccharomyces cerevisiae, is required for endoplasmic reticulum-associated degradation of glycoproteins. J Biol Chem. 2001;276:8635–8638. doi: 10.1074/jbc.C100023200. [DOI] [PubMed] [Google Scholar]
- 32.Olivari S, Cali T, Salo KE, Paganetti P, Ruddock LW, Molinari M. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem Biophys Res Commun. 2006;349:1278–1284. doi: 10.1016/j.bbrc.2006.08.186. [DOI] [PubMed] [Google Scholar]
- 33.Hosokawa N, Wada I, Natsuka Y, Nagata K. EDEM accelerates ERAD by preventing aberrant dimer formation of misfolded alpha1-antitrypsin. Genes Cells. 2006;11:465–476. doi: 10.1111/j.1365-2443.2006.00957.x. [DOI] [PubMed] [Google Scholar]
- 34.Avezov E, Frenkel Z, Ehrlich M, Herscovics A, Lederkremer GZ. Endoplasmic reticulum (ER) mannosidase I is compartmentalized and required for N-Glycan trimming to man5 6GlcNAc2 in glycoprotein ER-associated degradation. Mol Biol Cell. 2008;19:216–225. doi: 10.1091/mbc.E07-05-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim W, Spear ED, Ng DT. Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol Cell. 2005;19:753–764. doi: 10.1016/j.molcel.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 36.Bhamidipati A, Denic V, Quan EM, Weissman JS. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell. 2005;19:741–751. doi: 10.1016/j.molcel.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 37.Szathmary R, Bielmann R, Nita-Lazar M, Burda P, Jakob CA. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol Cell. 2005;19:765–775. doi: 10.1016/j.molcel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 38.Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 39.Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999;18:1492–1505. doi: 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahner A, Nakatsukasa K, Zhang H, Frizzell RA, Brodsky JL. Small heat-shock proteins select deltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol Biol Cell. 2007;18:806–814. doi: 10.1091/mbc.E06-05-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strickland E, Qu BH, Millen L, Thomas PJ. The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1997;272:25421–25424. doi: 10.1074/jbc.272.41.25421. [DOI] [PubMed] [Google Scholar]
- 45.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McClellan AJ, Scott MD, Frydman J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 2005;121:739–748. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, Riordan JR, Kelly JW, Yates JR, III, Balch WE. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 48.Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loo MA, Jensen TJ, Cui L, Hou Y, Chang XB, Riordan JR. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998;17:6879–6887. doi: 10.1093/emboj/17.23.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- 51.Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 53.Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deak PM, Wolf DH. Membrane topology and function of Der3/Hrd1p as a ubiquitin-protein ligase (E3) involved in endoplasmic reticulum degradation. J Biol Chem. 2001;276:10663–10669. doi: 10.1074/jbc.M008608200. [DOI] [PubMed] [Google Scholar]
- 55.Kreft SG, Wang L, Hochstrasser M. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI) J Biol Chem. 2006;281:4646–4653. doi: 10.1074/jbc.M512215200. [DOI] [PubMed] [Google Scholar]
- 56.Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ng DT, Spear ED, Walter P. The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J Cell Biol. 2000;150:77–88. doi: 10.1083/jcb.150.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2:379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- 59.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 60.Arvan P, Zhao X, Ramos-Castaneda J, Chang A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic. 2002;3:771–780. doi: 10.1034/j.1600-0854.2002.31102.x. [DOI] [PubMed] [Google Scholar]
- 61.Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 63.Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. An adaptable standard for protein export from the endoplasmic reticulum. Cell. 2007;131:809–821. doi: 10.1016/j.cell.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 64.Spear ED, Ng DT. Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J Cell Biol. 2005;169:73–82. doi: 10.1083/jcb.200411136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kostova Z, Wolf DH. Importance of carbohydrate positioning in the recognition of mutated CPY for ER-associated degradation. J Cell Sci. 2005;118:1485–1492. doi: 10.1242/jcs.01740. [DOI] [PubMed] [Google Scholar]
- 66.Mbonye UR, Wada M, Rieke CJ, Tang HY, Dewitt DL, Smith WL. The 19-amino acid cassette of cyclooxygenase-2 mediates entry of the protein into the endoplasmic reticulum-associated degradation system. J Biol Chem. 2006;281:35770–35778. doi: 10.1074/jbc.M608281200. [DOI] [PubMed] [Google Scholar]
- 67.Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- 68.Yu H, Kaung G, Kobayashi S, Kopito RR. Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J Biol Chem. 1997;272:20800–20804. doi: 10.1074/jbc.272.33.20800. [DOI] [PubMed] [Google Scholar]
- 69.Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- 70.Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 71.Biederer T, Volkwein C, Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278:1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- 72.Forster ML, Sivick K, Park YN, Arvan P, Lencer WI, Tsai B. Protein disulfide isomerase-like proteins play opposing roles during retro-translocation. J Cell Biol. 2006;173:853–859. doi: 10.1083/jcb.200602046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 74.Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci U S A. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee RJ, Liu CW, Harty C, McCracken AA, Latterich M, Romisch K, DeMartino GN, Thomas PJ, Brodsky JL. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J. 2004;23:2206–2215. doi: 10.1038/sj.emboj.7600232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wahlman J, DeMartino GN, Skach WR, Bulleid NJ, Brodsky JL, Johnson AE. Real-time fluorescence detection of ERAD substrate retro-translocation in a mammalian in vitro system. Cell. 2007;129:943–955. doi: 10.1016/j.cell.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 79.Johnson AE, Haigh NG. The ER translocon and retrotranslocation: is the shift into reverse manual or automatic? Cell. 2000;102:709–712. doi: 10.1016/s0092-8674(00)00059-3. [DOI] [PubMed] [Google Scholar]
- 80.Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 81.Pilon M, Schekman R, Romisch K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 1997;16:4540–4548. doi: 10.1093/emboj/16.15.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- 83.Gillece P, Pilon M, Romisch K. The protein translocation channel mediates glycopeptide export across the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A. 2000;97:4609–4614. doi: 10.1073/pnas.090083497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schmitz A, Herrgen H, Winkeler A, Herzog V. Cholera toxin is exported from microsomes by the Sec61p complex. J Cell Biol. 2000;148:1203–1212. doi: 10.1083/jcb.148.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schmitz A, Schneider A, Kummer MP, Herzog V. Endoplasmic reticulum-localized amyloid beta-peptide is degraded in the cytosol by two distinct degradation pathways. Traffic. 2004;5:89–101. doi: 10.1111/j.1600-0854.2004.00159.x. [DOI] [PubMed] [Google Scholar]
- 86.Kalies KU, Allan S, Sergeyenko T, Kroger H, Romisch K. The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 2005;24:2284–2293. doi: 10.1038/sj.emboj.7600731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fisher EA, Ginsberg HN. Complexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteins. J Biol Chem. 2002;277:17377–17380. doi: 10.1074/jbc.R100068200. [DOI] [PubMed] [Google Scholar]
- 88.Oyadomari S, Yun C, Fisher EA, Kreglinger N, Kreibich G, Oyadomari M, Harding HP, Goodman AG, Harant H, Garrison JL, Taunton J, Katze MG, Ron D. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126:727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 89.Gauss R, Sommer T, Jarosch E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006;25:1827–1835. doi: 10.1038/sj.emboj.7601088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sato BK, Hampton RY. Yeast Derlin Dfm1 interacts with Cdc48 and functions in ER homeostasis. Yeast. 2006;23:1053–1064. doi: 10.1002/yea.1407. [DOI] [PubMed] [Google Scholar]
- 91.Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem. 2004;279:45676–45684. doi: 10.1074/jbc.M409034200. [DOI] [PubMed] [Google Scholar]
- 92.Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- 93.Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 94.Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. Inaugural article: recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A. 2005;102:14132–14138. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lilley BN, Ploegh HL. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A. 2005;102:14296–14301. doi: 10.1073/pnas.0505014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Katiyar S, Joshi S, Lennarz WJ. The retrotranslocation protein Derlin-1 binds peptide:N-glycanase to the endoplasmic reticulum. Mol Biol Cell. 2005;16:4584–4594. doi: 10.1091/mbc.E05-04-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 98.Schulze A, Standera S, Buerger E, Kikkert M, van Voorden S, Wiertz E, Koning F, Kloetzel PM, Seeger M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol. 2005;354:1021–1027. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 99.Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 100.Lilley BN, Gilbert JM, Ploegh HL, Benjamin TL. Murine polyomavirus requires the endoplasmic reticulum protein Derlin-2 to initiate infection. J Virol. 2006;80:8739–8744. doi: 10.1128/JVI.00791-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bernardi KM, Forster ML, Lencer WI, Tsai B. Derlin-1 facilitates the retro-translocation of cholera toxin. Mol Biol Cell. 2007;19:877–884. doi: 10.1091/mbc.E07-08-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van der Wal FJ, Kikkert M, Wiertz E. The HCMV gene products US2 and US11 target MHC class I molecules for degradation in the cytosol. Curr Top Microbiol Immunol. 2002;269:37–55. doi: 10.1007/978-3-642-59421-2_3. [DOI] [PubMed] [Google Scholar]
- 103.Erdmann R, Schliebs W. Peroxisomal matrix protein import: the transient pore model. Nat Rev Mol Cell Biol. 2005;6:738–742. doi: 10.1038/nrm1710. [DOI] [PubMed] [Google Scholar]
- 104.Ploegh HL. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature. 2007;448:435–438. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- 105.Rudd PM, Woods RJ, Wormald MR, Opdenakker G, Downing AK, Campbell ID, Dwek RA. The effects of variable glycosylation on the functional activities of ribonuclease, plasminogen and tissue plasminogen activator. Biochim Biophys Acta. 1995;1248:1–10. doi: 10.1016/0167-4838(94)00230-e. [DOI] [PubMed] [Google Scholar]
- 106.Fiebiger E, Story C, Ploegh HL, Tortorella D. Visualization of the ER-to-cytosol dislocation reaction of a type I membrane protein. EMBO J. 2002;21:1041–1053. doi: 10.1093/emboj/21.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tirosh B, Furman MH, Tortorella D, Ploegh HL. Protein unfolding is not a prerequisite for endoplasmic reticulum-to-cytosol dislocation. J Biol Chem. 2003;278:6664–6672. doi: 10.1074/jbc.M210158200. [DOI] [PubMed] [Google Scholar]
- 108.Wilkinson BM, Tyson JR, Stirling CJ. Ssh1p determines the translocation and dislocation capacities of the yeast endoplasmic reticulum. Dev Cell. 2001;1:401–409. doi: 10.1016/s1534-5807(01)00043-0. [DOI] [PubMed] [Google Scholar]
- 109.Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol. 2005;7:742–749. doi: 10.1038/ncb0805-742. [DOI] [PubMed] [Google Scholar]
- 110.Raasi S, Wolf DH. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin Cell Dev Biol. 2007;18:780–791. doi: 10.1016/j.semcdb.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 111.Jentsch S, Rumpf S. Cdc48 (p97): a ‘molecular gearbox’ in the ubiquitin pathway? Trends Biochem Sci. 2007;32:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 112.Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 115.Medicherla B, Kostova Z, Schaefer A, Wolf DH. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 2004;5:692–697. doi: 10.1038/sj.embor.7400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rumpf S, Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol Cell. 2006;21:261–269. doi: 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 117.Kim I, Ahn J, Liu C, Tanabe K, Apodaca J, Suzuki T, Rao H. The Png1-Rad23 complex regulates glycoprotein turnover. J Cell Biol. 2006;172:211–219. doi: 10.1083/jcb.200507149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim I, Mi K, Rao H. Multiple interactions of rad23 suggest a mechanism for ubiquitylated substrate delivery important in proteolysis. Mol Biol Cell. 2004;15:3357–3365. doi: 10.1091/mbc.E03-11-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carlson EJ, Pitonzo D, Skach WR. p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 2006;25:4557–4566. doi: 10.1038/sj.emboj.7601307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kothe M, Ye Y, Wagner JS, De Luca HE, Kern E, Rapoport TA, Lencer WI. Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J Biol Chem. 2005;280:28127–28132. doi: 10.1074/jbc.M503138200. [DOI] [PubMed] [Google Scholar]
- 121.Lipson C, Alalouf G, Bajorek M, Rabinovich E, Atir-Lande A, Glickman M, Bar-Nun S. A proteasomal ATPase contributes to dislocation of ERAD substrates. J Biol Chem. 2008;283:7166–7175. doi: 10.1074/jbc.M705893200. [DOI] [PubMed] [Google Scholar]
- 122.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Haynes CM, Caldwell S, Cooper AA. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J Cell Biol. 2002;158:91–101. doi: 10.1083/jcb.200201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Spear ED, Ng DT. Stress tolerance of misfolded carboxypeptidase Y requires maintenance of protein trafficking and degradative pathways. Mol Biol Cell. 2003;14:2756–2767. doi: 10.1091/mbc.E02-11-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kelly SM, Vanslyke JK, Musil LS. Regulation of ubiquitin-proteasome system mediated degradation by cytosolic stress. Mol Biol Cell. 2007;18:4279–4291. doi: 10.1091/mbc.E07-05-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 127.Akiyama Y, Ito K. Reconstitution of membrane proteolysis by FtsH. J Biol Chem. 2003;278:18146–18153. doi: 10.1074/jbc.M302152200. [DOI] [PubMed] [Google Scholar]
- 128.Liu CW, Corboy MJ, DeMartino GN, Thomas PJ. Endoproteolytic activity of the proteasome. Science. 2003;299:408–411. doi: 10.1126/science.1079293. [DOI] [PMC free article] [PubMed] [Google Scholar]