

Abstract
Stimulation of human colonic epithelial T84 cells with the muscarinic receptor agonist carbachol, a stable analog of acetylcholine, induced Akt, p70S6K1 and ERK activation. Treatment of T84 cells with the selective inhibitor of EGF receptor (EGFR) tyrosine kinase AG1478 abrogated Akt phosphorylation on Ser473 induced by either carbachol or EGF, indicating that carbachol-induced Akt activation is mediated through EGFR transactivation. Surprisingly, AG1478 did not suppress p70S6K1 phosphorylation on Thr389 in response to carbachol, indicating the G protein-coupled receptor (GPCR) stimulation induces p70S6K1 activation, at least in part, via an Akt-independent pathway. In contrast, treatment with the selective MEK inhibitor U0126 (but not with the inactive analog U0124) inhibited carbachol-induced p70S6K1 activation, indicating that the MEK/ERK/RSK pathway plays a critical role in p70S6K1 activation in GPCR-stimulated T84 cells. These findings imply that GPCR activation induces p70S6K1 via ERK rather than through the canonical PI 3-kinase/Akt/TSC/mTORC1 pathway in T84 colon carcinoma cells.
Keywords: EGF, EGF receptor transactivation, tuberin, AG1487, protein kinase D, phorbol ester
INTRODUCTION
Hormones, neurotrasmitters and vasoactive peptides that signal through G protein-coupled receptors (GPCRs) act as potent cellular growth factors for a variety of cell types [1-4]. Mitogenic signaling emanating from GPCRs is also implicated in abnormal biological processes, including tumorigenesis [2-9]. In particular, GPCRs and their cognate agonists are expressed by a variety of human cancer cells, as well as by mesenchymal cells in the microenviroment of these tumors, leading to autocrine/paracrine loops that contribute to stimulation of cell proliferation, migration, invasiveness and angiogenesis [for reviews see [2-9]]. Consequently, the elucidation of the intracellular pathways that mediate the biological effects induced by GPCR agonists is of major importance because these studies may identify novel strategies for therapeutic interventions.
The mTOR (Target Of Rapamycin) pathway is the focus of interest because it integrates signals from growth factors, nutrients, cellular energy levels, GPCR agonists and stress conditions to stimulate protein synthesis and cell growth [10]. Two distinct mTOR complexes, mTORC1 (mTOR complex 1) and mTORC2 (mTOR complex 2), have been characterized [11]. A major downstream target of mTORC1 is p70S6K1, an enzyme implicated in the regulation of protein synthesis and cell proliferation [12]. The activity of p70S6K1 is controlled by multi-site phosphorylation [13], including phosphorylation at Thr389 by mTORC1 [14]. There have been important advances in understanding how growth factors stimulate mTORC1 signaling via tyrosine kinase receptors that stimulate the PI3kinase/Akt signaling pathway. Akt contains a PH domain and conserved residues (Thr308 and Ser473) which are critical for enzyme activation. Specifically, Akt, translocated to the plasma membrane in response to products of PI 3-kinase, is activated by phosphorylation at Thr308 in the activation loop and by phosphorylation within the carboxy-terminus at Ser473 [15]. Akt has been shown to phosphorylate the product of the tuberous sclerosis complex (TSC) 2 gene which forms a heterodimer with TSC1 and represses mTOR activity [16, 17]. Phosphorylation of TSC2 by Akt is thought to alleviate this inhibitory restraint on mTOR signaling, resulting in its activation. The prevalent model is that tyrosine kinase receptor agonists induce p70S6K1 activation via the canonical PI 3-kinase/Akt/TSC/mTOR pathway. However, an indispensable role for Akt in p70S6K has been disputed in other studies [4, 18-23], suggesting the operation of alternative pathways. Activation of GPCRs stimulates phosphorylation and activation of p70S6K1 in a variety of cell types [3, 4, 24-27] but the precise mechanism that mediates GPCR-induced mTOR/p70S6K activation has remained poorly defined.
Here, we examined the relationship of Akt activation to mTORC1 activation, as revealed by phosphorylation of p70S6K1 on Thr389, a site phosphorylated by mTOR in the context of mTORC1. We report that stimulation of human colonic epithelial T84 cells with the muscarinic GPCR agonist carbamylcholine (carbachol), a stable analog of acetylcholine, induces p70S6K1 phosphorylation on Thr389 via an ERK-dependent but Akt-independent pathway in T84 cells. The results dissociate Akt from p70S6K1 activation in T84 colon cancer cells and imply that GPCR activation induces p70S6K1 via ERK rather than through the canonical PI 3-kinase/Akt/TSC/mTORC1 pathway in these cells.
EXPERIMENTAL PROCEDURES
Cell culture
The human colon carcinoma cell line T84 was purchased from American Type Culture Collection. Stock cultures of T84 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F12 media (1:1) supplemented with 10% fetal bovine serum. Cells were cultured in an atmosphere of 5% CO2 at 37°C with medium changes every 3-4 days.
Cell stimulation and cell lysates
Cultures of T84 cells were washed twice with serum-free DMEM/Ham’s F12, equilibrated in the same medium at 37°C for 3 h, and then treated with agonists and/or inhibitors as described in the individual experiments. The stimulation was terminated by aspirating the medium and lysing the cells in 200μl of 2x SDS-polyacrylamide gel electrophoresis sample buffer (20 mM Tris/HCl, pH 6.8, 6% SDS, 2 mM EDTA, 4% 2-mercaptoethanol, 10% glycerol).
Western Blotting
After SDS-PAGE, proteins were transferred to immobilon membranes. After transfer, membranes were blocked using 5% nonfat dried milk in PBS, pH 7.2 and incubated overnight at 4°C with the anti-phospho Akt Ser473 Ab (0.1 μg/ml), anti-phospho p70S6K1 Thr389 (0.1 μg/ml), anti-Akt Ab (0.1 μg/ml), anti-phospho-p44/42 MAP kinase(0.1 μg/ml) or anti-ERK2 (0.1 μg/ml) as indicated. The membranes were washed three times with PBS-0.1% Tween 20 and then incubated with secondary antibodies (horseradish peroxidase-conjugated donkey antibodies to rabbit, NA 934V or Mouse, NA931V) (1:5000) for 1 h at 22°C. After washing three times with PBS-0.1% Tween 20, the immunoreactive bands were visualized using enhanced chemilumiescence (ECL) detection reagents.
Materials
Carbachol, U-0126, and U-0124 were purchased from Calbiochem (San Diego, CA). EGF was obtained from Sigma (St. Louis, Mo). Horseradish peroxidase-conjugated donkey antibodies to rabbit (NA 934V) or mouse (NA931V) and ECL reagents were from GE Healthcare (Piscataway, NJ). Anti-ERK2 polyclonal antibody was from Santa Cruz Biotechnology Inc (Santa Cruz, Ca). The phosphospecific polyclonal Abs to Akt Ser473, p70S6K1 Thr389 and anti-phospho-ERK1/2 MAb were obtained from Cell Signaling Technology (Beverly, MA). All other reagents used were of the purest grade available.
RESULTS and DISCUSSION
Carbachol induces Akt and tuberin phosphorylation in human colon carcinoma T84 cells
In order to determine whether GPCR agonists regulate Akt phosphorylation in human colon carcinoma T84 cells, cultures of these cells were stimulated with the muscarinic agonist carbachol (100 μM), which acts in these cells via a Gq-coupled M3 receptor subtype [28, 29]. Akt activation was scored by Western blot analysis using an antibody that detects the phosphorylated state of Akt on Ser473. As shown in Fig. 1 A, carbachol induced robust Akt phosphorylation on Ser473 in a time-dependent fashion. A small increase was observed after 5 min of exposure to carbachol with maximal stimulation of Akt phosphorylation at Ser473 evident after 15 min of incubation. In addition, carbachol induced a marked increase in Akt phosphorylation on Thr308 (results not shown).
Figure 1. Carbachol induces Akt phosphorylation on Ser473 and p70S6K phosphorylation on Ser389in a time-dependent fashion in T84 cells.
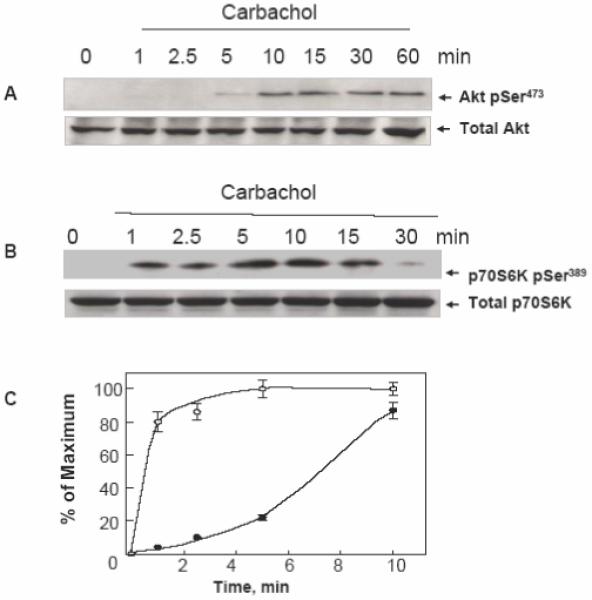
Cultures of T84 cells grown on 35 mm dishes were stimulated with 100 μM carbachol for the indicated times. All cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by SDS-PAGE and immunoblotting with antibodies that detect either Akt phosphorylated on Ser473 (panel A) or p70S6K phosphorylated on Ser389 (panel B). Antibodies that detect total Akt or p70S6K were used to verify equivalent loading of the gel. Autoluminograms were quantified by densitometric scanning. The results shown are the mean ± S.E.M. n=3 and are expressed as percentage of the maximum increase induced by treatment with carbachol for either AKT pSer473 closed symbols or p70S6K pSer389 open symbols (panel C).
p70S6K1, which is implicated in cell growth and G1 cell cycle progression, is considered a major downstream target of the canonical PI 3-kinase/Akt/TSC/mTORC1 pathway. To determine whether carbachol activates p70S6K1 in T84 cells, lysates of these cells stimulated with carbachol for various times (1-30 min) were subjected to Western blot analyses using an antibody that detects p70S6K1 phosphorylated at Thr389, a site targeted by mTORC1 [13]. As shown in Fig. 1B, carbachol induced a rapid and striking increase in p70S6K1 phosphorylation in T84 cells. It is noteworthy that an increase in p70S6K1 phosphorylation at Thr389 was evident within 1 min of carbachol stimulation, clearly preceding Akt phosphorylation (see Fig. 1 C). These results showing that carbachol induces p70S6K1 activation more rapidly than Akt in colonic T84 cells are inconsistent with the operation of a canonical PI 3-kinase/Akt/TSC/mTORC1 pathway leading to p70S6K1 in these cells.
Role of EGFR in carbachol- induced Akt and p70S6K1 phosphorylation in T84 cells
GPCRs stimulate EGFR tyrosine kinase in a variety of cell types, a process termed EGFR transactivation [4]. Previous reports demonstrated that carbachol induces EGFR transactivation in human colonic T84 epithelial cells [28, 30, 31]. Here, we determined whether EGFR transactivation mediates Akt and p70S6K1 activation induced by carbachol in these cells. Specifically, T84 cells were treated with or without the specific EGFR tyrosine kinase inhibitor AG1478 (1 μM) and subsequently challenged with either carbachol or EGF (as a control). The increase of Akt phosphorylation at Ser473 induced by carbachol was comparable to that induced by EGF in parallel cultures. As shown in Fig. 2A, treatment with AG1478 completely prevented Akt phosphorylation on Ser473 induced by either carbachol or EGF in T84 cells. These results indicate that stimulation with carbachol strikingly stimulates Akt activation through EGFR tyrosine kinase in T84 cells.
Figure 2. Carbachol induced Akt phosphorylation on Ser473 is EGFR-dependent but p70S6K phosphorylation on Ser389 is EGFR-independent.
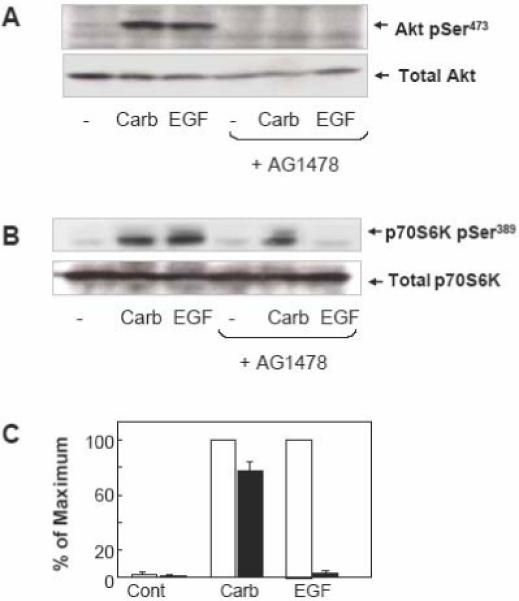
A and B. T84 cells were incubated in the absence or presence of the EGFR inhibitor AG1478 (1 μM) for 1 h prior to stimulation of the cells with 100 μM carbachol (carb) or 50 ng/ml EGF for 10 min. The cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by SDS-PAGE and immunoblotting with phospho Akt Ser473 and total Akt in panel A and phospho p70S6K Ser389 and total p70S6K to verify equal loading in panel B. Shown here are representative autoluminograms; similar results were obtained in 3 independent experiments. Autoluminograms were quantified by densitometric scanning. The results shown are the mean ± S.E.M. n=3 and are expressed as percentage of the maximum increase induced by treatment with carbachol, in cells preincubated in the absence (open bars) or the presence (closed bars) of AG1478 (panel C).
In striking contrast to the results seen with Akt, treatment with AG1478 resulted in only a modest attenuation of p70S6K1 phosphorylation on Thr389 induced by carbachol (Fig. 2B and C). We verified that AG1478, under our experimental conditions, completely prevented p70S6K1 phosphorylation on Thr389 induced by exogenous EGF, demonstrating the effectiveness of the inhibitor at the concentration used in T84 cells. The results shown in Fig. 2 indicate that carbachol induces Akt activation via EGFR transactivation but, in striking contrast, p70S6K1 phosphorylation in response to this agonist appears to be mediated by a different pathway in human colonic T84 cells. This conclusion is in agreement with the kinetics depicted in Fig. 1.
Role of ERK in carbachol-induced p70S6K1 phosphorylation in T84 cells
Recently, a novel mechanism of mTORC1 activation involving the ERK pathway but separate from Akt has been elucidated in response to phorbol esters [32] and expression of constitutively active MEK1 [22, 32]. To elucidate the mechanism(s) by which carbachol induces p70SK1 activation independently from Akt, we examined whether the ERKs link the M3 GPCR to p70S6K1 in carbachol-stimulated T84 cells. Initially, we examined the kinetics of carbachol-induced ERK activation in T84 cells. As shown in Fig 3A, ERK activation in T84 cells was detectable within 1 min of stimulation and reached a maximum within 5 min of carbachol stimulation. These results indicated that the kinetics of ERK activation is comparable to that of p70S6K1 phosphorylation at Thr389 and clearly faster than Akt phosphorylation on Ser473, implying that ERKs could lead to p70S6K1 activation in carbachol-stimulated T84 cells.
Figure 3. Carbachol induced p70S6K phosphorylation through an ERK-dependent pathway.
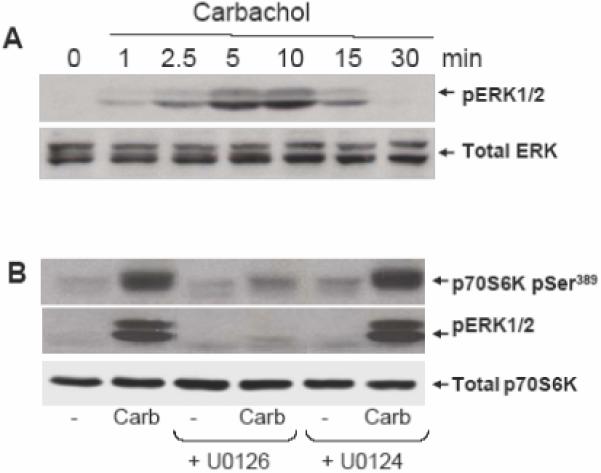
A. Cultures of T84 cells were stimulated with 100 μM carbachol for the indicated times, lysed with 2×SDS-PAGE sample buffer and analyzed by SDS-PAGE and immunoblotting with phospho-ERK antibody (pERK1/2). Shown here are representative autoluminograms; similar results were obtained in 3 independent experiments. B, T84 cells were incubated in the absence or presence of the MEK1/2 inhibitor U0126 (10 μM) or the inactive analogue U0124 for 1 h prior to stimulation of the cells with 100 μM carbachol (carb) for 10 min. The cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by SDS-PAGE and immunoblotting with the following antibodies: phospho ERK1/2, p70S6K Ser389 and total p70S6K. Shown here are representative autoluminograms; similar results were obtained in 3 independent experiments.
In order to determine whether ERK activation mediates p70S6K1 phosphorylation at Thr389 in response to carbachol in T84 cells, cultures of these cells were incubated for 1 h in the absence or presence of U0126 and subsequently stimulated with carbachol. As shown in Fig. 3B, treatment with U0126 dramatically inhibited P70S6K1 phosphorylation at Thr389 induced by carbachol. To substantiate the specificity of the inhibitory effects of U0126, we also examined the effect of the structurally related but inactive compound U0124, at an identical concentration. Treatment with U0124 did not produce any inhibitory effect on carbachol-iduced p70S6K1 phosphorylation on Thr389. We verified that U0126 (but not U0124), at the concentration used (10 μM), completely blocked carbachol-induced ERK signaling (Fig. 3 B). These results imply that carbachol induces p70S6K1 activation via ERK rather than through Akt.
Conclusions and Implications
While the role and mechanism of activation of the canonical PI 3-kinase/Akt/mTOR/p70S6K1 pathway in signal transduction linked to cell division, survival, and tumorigenesis by tyrosine kinase receptors are well recognized in many cell types, the operation of this pathway in the action of GPCR agonists in epithelial intestinal cells has been much less explored. The experiments presented here were designed to determine pathways leading to Akt and p70S6K1 activation in human colonic epithelial T84 cells stimulated via an endogenously expressed Gq-coupled receptor, the muscarinic subtype 3 receptor (M3R). Our results produced several lines of evidence indicating that carbachol induced p70S6K1 activation independently of Akt and EGFR transactivation: 1) Kinetic experiments showed that p70S6K1 phosphorylation on Thr389 could be detected earlier than Akt phosphorylation on Ser473. Indeed, at early time-points (1-2.5 min) p70S6K1 phosphorylation occurred in the absence of any detectable Akt activation. 2) Cell treatment with the specific EGFR tyrosine kinase inhibitor AG1478 completely prevented Akt activation in response to carbachol. In contrast, p70S6K1 activation was still evident in cells with suppressed EGFR tyrosine kinase activity. Thus, these kinetic and pharmacological results dissociate Akt from p70S6K1 activation in T84 colon cancer cells. 3) Further results show that carbachol induces p70S6K1 in these cells via MEK/ERK rather than through the canonical PI 3-kinase/Akt/TSC/mTOR pathway.
Studies on mechanisms of M3R-induced signaling in colon cancer cells assume an added relevance in view of recent reports showing that muscarinic receptor activation stimulates colon cancer cell proliferation in vitro and in vivo and by the fact that a substantial number of human colon cancers display increased expression of muscarinic receptors [29, 33, 34]. Our results showing the operation of an ERK/p70S6K1 pathway independently of Akt in the context of GPCR signaling in colon cancer cells may have important implications for identifying combinations of inhibitors of signal transduction pathways that provide novel strategies for therapeutic interventions.
Acknowledgments
This work was supported by NIH Grants R21CA137292, RO1DK56930, RO1DK55003 and P30DK41301 (to ER).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Rozengurt E. Early signals in the mitogenic response. Science. 1986;234:161–166. doi: 10.1126/science.3018928. [DOI] [PubMed] [Google Scholar]
- [2].Rozengurt E. Neuropeptides as growth factors for normal and cancer cells. Trends Endocrinol Metabol. 2002;13:128–134. doi: 10.1016/s1043-2760(01)00544-6. [DOI] [PubMed] [Google Scholar]
- [3].Rozengurt E, Walsh JH. Gastrin, CCK, signaling, and cancer. Annu. Rev. Physiol. 2001;63:49–76. doi: 10.1146/annurev.physiol.63.1.49. [DOI] [PubMed] [Google Scholar]
- [4].Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- [5].Rozengurt E. Growth factors and cell proliferation. Curr. Opin. Cell Biol. 1992;4:161–165. doi: 10.1016/0955-0674(92)90027-a. [DOI] [PubMed] [Google Scholar]
- [6].Rozengurt E. Autocrine loops, signal transduction, and cell cycle abnormalities in the molecular biology of lung cancer. Curr. Opin. Oncol. 1999;11:116–122. doi: 10.1097/00001622-199903000-00007. [DOI] [PubMed] [Google Scholar]
- [7].Heasley LE. Autocrine and paracrine signaling through neuropeptide receptors in human cancer. Oncogene. 2001;20:1563–1569. doi: 10.1038/sj.onc.1204183. [DOI] [PubMed] [Google Scholar]
- [8].Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- [9].Spiegelberg BD, Hamm HE. Roles of G-protein-coupled receptor signaling in cancer biology and gene transcription. Curr. Opin. Genet. Development. 2007;17:40–44. doi: 10.1016/j.gde.2006.12.002. [DOI] [PubMed] [Google Scholar]
- [10].Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- [11].Bhaskar PT, Hay N. The Two TORCs and Akt. Developmental Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- [12].Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- [13].Long X, Muller F, Avruch J. TOR action in mammalian cells and in Caenorhabditis elegans. Curr Top Microbiol Immunol. 2004;279:115–138. doi: 10.1007/978-3-642-18930-2_8. [DOI] [PubMed] [Google Scholar]
- [14].Ali SM, Sabatini DM. Structure of S6 Kinase 1 Determines whether Raptor-mTOR or Rictor-mTOR Phosphorylates Its Hydrophobic Motif Site. J. Biol. Chem. 2005;280:19445–19448. doi: 10.1074/jbc.C500125200. [DOI] [PubMed] [Google Scholar]
- [15].Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- [16].Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- [18].Radimerski T, Montagne J, Rintelen F, Stocker H, van der Kaay J, Downes CP, Hafen E, Thomas G. dS6K-regulated cell growth is dPKB/dPI(3)K-independent, but requires dPDK1. Nat Cell Biol. 2002;4:251. doi: 10.1038/ncb763. [DOI] [PubMed] [Google Scholar]
- [19].Tang X, Wang L, Proud CG, Downes CP. Muscarinic receptor-mediated activation of p70 S6 kinase 1 (S6K1) in 1321N1 astrocytoma cells: permissive role of phosphoinositide 3-kinase. Biochem. J. 2003;374:137–143. doi: 10.1042/BJ20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fan Q-W, Cheng C, Knight ZA, Haas-Kogan D, Stokoe D, James CD, McCormick F, Shokat KM, Weiss WA. EGFR Signals to mTOR Through PKC and Independently of Akt in Glioma. Sci. Signal. 2009;2:ra4. doi: 10.1126/scisignal.2000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sawhney RS, Cookson MM, Sharma B, Hauser J, Brattain MG. Autocrine Transforming Growth Factor {alpha} Regulates Cell Adhesion by Multiple Signaling via Specific Phosphorylation Sites of p70S6 Kinase in Colon Cancer Cells. J. Biol. Chem. 2004;279:47379–47390. doi: 10.1074/jbc.M402031200. [DOI] [PubMed] [Google Scholar]
- [22].Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- [23].Rolfe M, McLeod LE, Pratt PF, Proud CG. Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2) Biochem J. 2005;388:973–984. doi: 10.1042/BJ20041888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Withers DJ, Seufferlein T, Mann D, Garcia B, Jones N, Rozengurt E. Rapamycin dissociates p70(S6K) activation from DNA synthesis stimulated by bombesin and insulin in Swiss 3T3 cells. J. Biol. Chem. 1997;272:2509–2514. doi: 10.1074/jbc.272.4.2509. [DOI] [PubMed] [Google Scholar]
- [25].Billington CK, Kong KC, Bhattacharyya R, Wedegaertner PB, Panettieri RA, Chan TO, Penn RB. Cooperative Regulation of p70S6 Kinase by Receptor Tyrosine Kinases and G Protein-Coupled Receptors Augments Airway Smooth Muscle Growth. Biochemistry. 2005;44:14595–14605. doi: 10.1021/bi0510734. [DOI] [PubMed] [Google Scholar]
- [26].Williams JA. Intracellular signaling mechanisms activated by cholecystokinin-regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu Rev Physiol. 2001;63:77–97. doi: 10.1146/annurev.physiol.63.1.77. [DOI] [PubMed] [Google Scholar]
- [27].Chiu T, Santiskulvong C, Rozengurt E. EGF receptor transactivation mediates ANG II-stimulated mitogenesis in intestinal epithelial cells through the PI3-kinase/Akt/mTOR/p70S6K1 signaling pathway. Am J Physiol Gastrointest Liver Physiol. 2005;288:G182–194. doi: 10.1152/ajpgi.00200.2004. [DOI] [PubMed] [Google Scholar]
- [28].Keely SJ, Uribe JM, Barrett KE. Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells. Implications for carbachol-stimulated chloride secretion. J. Biol. Chem. 1998;273:27111–27117. doi: 10.1074/jbc.273.42.27111. [DOI] [PubMed] [Google Scholar]
- [29].Frucht H, Jensen RT, Dexter D, Yang W-L, Xiao Y. Human Colon Cancer Cell Proliferation Mediated by the M3 Muscarinic Cholinergic Receptor. Clin Cancer Res. 1999;5:2532–2539. [PubMed] [Google Scholar]
- [30].Keely SJ, Calandrella SO, Barrett KE. Carbachol-stimulated Transactivation of Epidermal Growth Factor Receptor and Mitogen-activated Protein Kinase in T84 Cells Is Mediated by Intracellular Ca2+, PYK-2, and p60src. J. Biol. Chem. 2000;275:12619–12625. doi: 10.1074/jbc.275.17.12619. [DOI] [PubMed] [Google Scholar]
- [31].Santiskulvong C, Rozengurt E. Protein kinase Calpha mediates feedback inhibition of EGF receptor transactivation induced by G(q)-coupled receptor agonists. Cell Signal. 2007;19:1348–1357. doi: 10.1016/j.cellsig.2007.01.006. [DOI] [PubMed] [Google Scholar]
- [32].Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004;101:13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Raufman J-P, Samimi R, Shah N, Khurana S, Shant J, Drachenberg C, Xie G, Wess J, Cheng K. Genetic Ablation of M3 Muscarinic Receptors Attenuates Murine Colon Epithelial Cell Proliferation and Neoplasia. Cancer Res. 2008;68:3573–3578. doi: 10.1158/0008-5472.CAN-07-6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shah N, Khurana S, Cheng K, Raufman J-P. Muscarinic receptors and ligands in cancer. Am J Physiol Cell Physiol. 2009;296:C221–232. doi: 10.1152/ajpcell.00514.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]