

Abstract
Sphingosine-1-phosphate is a ubiquitous, lipophilic cellular mediator that acts in part by activation of G-protein-coupled receptors. Modulation of S1P signaling is an emerging pharmacotherapeutic target for immunomodulatory drugs. Although multiple S1P receptor types exist in the CNS, little is known about their function. Here we report that S1P stimulated G- protein activity in the CNS, and results from [35S]GTPγS autoradiography using the S1P1-selective agonist SEW2871 and the S1P1/3-selective antagonist VPC44116 show that in several regions a majority of this activity is mediated by S1P1 receptors. S1P receptor activation inhibited glutamatergic neurotransmission as determined by electrophysiological recordings in cortical neurons in vitro, and this effect was mimicked by SEW2871 and inhibited by VPC44116. Moreover, central administration of S1P produced in vivo effects resembling the actions of cannabinoids, including thermal antinociception, hypothermia, catalepsy and hypolocomotion, but these actions were independent of CB1 receptors. At least one of the central effects of S1P, thermal antinociception, is also at least partly S1P1 receptor mediated because it was produced by SEW2871 and attenuated by VPC44116. These results indicate that CNS S1P receptors are part of a physiologically relevant and widespread neuromodulatory system, and that the S1P1 receptor contributes to S1P-mediated antinociception.
Keywords: S1P receptor, GPCR, Glutamate, Analgesia, Hypothermia, Autoradiography
INTRODUCTION
Cellular lysolipids are important in cell-cell communication and the field is rapidly growing. These important cellular mediators include sphingosine-1-phosphate (S1P), lysophosphatidic acid, arachidonylglycerol-based endocannabinoids, and related lysolipids. S1P, which is widely distributed throughout the body, has been implicated in diverse physiological functions, including immune modulation, vascular and nervous system development, regulation of smooth muscle and auditory and vestibular function (Spiegel and Milstien 2003; Brinkmann 2007; Choi et al. 2008). S1P produces most of its physiological effects by binding to five G-protein coupled receptors, termed S1P1-5 and formerly known as endothelial differential gene (EDG) receptors (Chun et al. 2002). At the cellular level, the balance between the actions of S1P and its precursors, ceramide and sphingosine, acts as a cellular rheostat that signals cell survival (S1P) or death (ceramide, sphingosine) (Spiegel and Milstien 2000). To date, the best defined therapeutic application of drugs targeting S1P receptors is the investigational immunomodulatory pro-drug FTY720 (fingolimod, 2-amino-2-(2-[4-octylphenyl]ethyl)-1,3-propanediol), a sphingosine analog that has shown promising results in clinical trials for treatment of multiple sclerosis (O'Connor et al. 2009).
S1P, its synthetic enzymes, sphingosine kinase 1 and 2, and S1P receptors are found in the CNS (Edsall and Spiegel 1999; Toman and Spiegel 2002; Blondeau et al. 2007; Bryan et al. 2008), where their role has been most clearly defined during neuronal development. In contrast, the putative function(s) of S1P in the adult CNS is only beginning to emerge. Recent studies suggest that S1P modulates neuronal excitability (Zhang et al. 2006; Kajimoto et al. 2007), but the mechanism underlying this action and its in vivo consequences are not well understood. S1P-stimulated G-protein activity has been reported, with activity widely distributed throughout the brain (Waeber and Chiu 1999). Localization of S1P receptor mRNA reveals that S1P1, S1P2 and S1P3 receptors are widely distributed in the brain, with region-specific distributions and differential localization on neurons versus glia (Beer et al. 2000). In contrast, S1P5 receptors are found primarily on oligodendrocytes and S1P4 receptors are absent from the CNS (Terai et al. 2003; Brinkmann 2007). Despite increasing interest in S1P-mediated therapeutics and the identification of S1P receptors in the CNS, little is known regarding their role in neural function in the adult. Elucidation of S1P receptor-mediated CNS effects might have important implications for the development of novel therapeutics and could indicate potential CNS side effects of these drugs. The present study assessed S1P receptor function in the rodent CNS at both the cellular and in vivo levels. Because S1P1 receptor-mediated G-protein activity appeared to predominate in several CNS regions, further studies concentrated on the role of S1P1 receptors in CNS function. The results indicate that S1P receptors are part of a novel neuromodulatory system, and the anatomical distribution of S1P receptor-mediated G-protein activity and in vivo profile of S1P activity suggest that these receptors contribute to diverse CNS functions.
METHODS
Materials
S1P was purchased from Biomol and SEW2871was purchased from Cayman Chemical. VPC44116 was synthesized as described in Foss et al. (2007). SR141716A was provided by the National Institute on Drug Abuse Drug Supply Program. [35S]GTPγS (1250 Ci/mmol) was purchased from Perkin Elmer. GDP was purchased from Sigma/RBI. All other reagent grade chemicals were obtained from Sigma or Fisher.
Agonist-stimulated [35S]GTPγS autoradiography
Assays were conducted as previously described with slight modification (Sim et al. 1995). Mice were sacrificed by decapitation and brains were frozen in isopentane at -30°C. Twenty-micron coronal sections were cut on a cryostat at -20°C, thaw-mounted onto gelatin-coated slides and stored desiccated at -80°C. For assay, slides were brought to room temperature, then equilibrated in 50 mM Tris-HCl, pH 7.4, with 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl (assay buffer) for 10 min at 25°C. Slides were transferred to assay buffer containing 0.5% bovine serum albumin (assay buffer + BSA) with 2 mM GDP and 9.5 mU/ml adenosine deaminase and incubated for 15 min at 25 °C. Slides were then incubated in assay buffer + BSA containing 2 mM GDP, 9.5 mU/ml adenosine deaminase and 0.04 nM [35S]GTPγS in the presence or absence (basal) of appropriate concentrations of drugs/vehicle for 2 hours at 25°C. Concentration-effect curves were generated using 3 to 60 μM S1P and 3 to 80 μM SEW2871. Maximally effective concentrations of S1P (60 μM) or SEW2871 (80 μM) were calculated and used in the assay. Antagonist studies were conducted using EC75 concentrations of S1P (10 μM) or SEW (20 μM) with and without 50 μM VPC44116. After incubation, slides were rinsed twice in 50 mM Tris buffer (pH 7.4) at 4°C, then in deionized water. Slides were exposed to Biomax MR film with [14C]microscales for 24 hours. Films were digitized and analyzed using NIH Image. Data are reported as mean values ± SEM of triplicate sections of brains from at least 7 mice per group. Net [35S]GTPγS binding is defined as (agonist-stimulated [35S]GTPγS binding - basal [35S]GTPγS binding). [14C] values were corrected for [35S] based upon incorporation of [35S] into sections of frozen brain paste. Statistical significance of the data was determined by ANOVA with appropriate post-hoc tests, as given in the figure legends.
Cortical cell cultures
Primary cultures of neurons plus glia were prepared using a modified version of Huettner and Baughman (1986). Neocortices were isolated from 1 to 2 day old Sprague-Dawley rats and digested using a 10 U/mL papain solution. Following trituration, cells were plated at a density of approximately 100-200 cells/mm2 on a confluent layer of astrocytes in conditioned growth media consisting of MEM supplemented with 5% calf serum, 25 mM glucose, 100 U/mL penicillin, 100 ug/L streptomycin, and 500 nM glutamine. Cell cultures were incubated at 37°C an fed twice weekly with conditioned growth media. Cultures were utilized between 12-21 days in vitro (DIV).
Measurement of spontaneous excitatory postsynaptic currents (sEPSCs)
Using the whole-cell voltage clamp technique (Hamill et al. 1981), spontaneous excitatory postsynaptic currents were measured from individual cortical pyramidal neurons, identified by their characteristic morphology. Patch electrodes were made from borosilicate glass capillaries (WPI, Sarasota, FL) that were pulled to a tip resistance of 3-6 MΩ and filled with a solution containing (in mM): 135 K gluconate, 4 KCl, 2 NaCl, 10 EGTA, 0.2 CaCl2, 2 MgATP, 0.6 Na2GTP, 10 HEPES, pH 7.2. 285-295 mOsm The external recording solution contained (in mM): 130 NaCl, 4 KCl, 3 CaCl2, 2 MgCl2, 10 HEPES, 11 glucose, 0.01 glycine, and 0.05% BSA, pH 7.3, 295-305 mOsm. Solutions containing S1P, SEW2871, and VPC 44116 were made fresh daily from stock solutions of 10 mM drug in 5% 1N HCl:95% DMSO (vol:vol). All non-drug solutions contained 0.005% - 0.05% DMSO as a drug vehicle control. Drugs were applied using a SF-77 Perfusion Fast-Step system (Warner Ins. Co., Hamden, CT) as previously described (Goforth et al. 1999). Cells were continuously superfused with standard external solution at a rate of approximately 2 ml/min. Neurons used for analysis had stable series resistances (Rs) ≤30 MΩ and were compensated at ≥ 85%. All recordings were performed at room temperature.
Statistical analysis
sEPSC events were analyzed using Synaptosoft software (Decatur, GA). Individual events were detected using an amplitude threshold value of 4-6 pA and confirmed visually. Median sEPSC amplitude, 10-90% rise time, and half width were determined for each cell in the presence and absence of drug treatment and the mean median values of all cells were compared for each treatment. The total charge transfer resulting from sEPSC activity was calculated by integrating the current response over a 2 minute time period after baseline correction. Statistical analysis was performed using GraphPad Prism 2.0 (GraphPad Software Inc., San Diego, CA). Data are expressed as mean ± SEM. Data sets were tested for normality using the D'Agostino Pearson normality test. Significance was determined using ANOVA and Newman-Keuls multiple comparison post-hoc test (normal distributions) or Kruskal-Wallis and Dunn's post-hoc tests (non-parametric).
In vivo testing
Male ICR mice (Harlan Laboratories, Indianapolis, IN) weighing 25-30 g were housed in an animal facility maintained at 22 ± 2 °C on a 12-h light/dark cycle with ad libitum food and water. Mice were transported to the laboratory 6-8 h prior to testing to allow acclimation. All procedures were conducted in accordance with the regulations of the Institutional Animal Care and Use Committee at Virginia Commonwealth University and the Guide for the Care and Use of Laboratory Animals as promulgated by the National Institutes of Health. Six mice were tested per dose of drug or vehicle unless otherwise noted. S1P and SEW2871 were dissolved in acidified DMSO (5% 1N HCl:95% DMSO, vol:vol) and injected i.c.v. in a volume of 5μl/mouse (Pedigo et al. 1975). VPC44116 was prepared in acidified DMSO and administered i.c.v. 10 min prior to all drugs. SR141716A was dissolved in 1:1:18 (emulphor: ethanol: saline) and administered at a dose of 3 mg/kg, i.p. at 1 hr prior to i.c.v. administration of S1P, SEW2871 or vehicle. All drugs were freshly prepared before each use.
Antinociception
Baseline tail-flick latencies were determined prior to drug administration using the radiant heat tail-flick latency test (D'Amour and Smith 1941), and average values were between 2 and 4 sec. During testing, a cut-off time of 10 s was employed to prevent tail damage. Mice were not acclimated to the apparatus and were tested only twice - one baseline latency and one post-drug (test) latency. Antinociception was quantified using the percent maximal effect (%MPE) calculation (Harris and Pierson 1964): %MPE= [(test-baseline)/(maximum-baseline)] ×100 for each mouse. Mean % MPE ± SEM were recorded for each group of mice.
Rectal Temperature
Core temperature to the nearest 1°C was measured by inserting a rectal probe (2 cm into the rectum) connected to a Telethermometer. Baseline temperature was determined prior to drug administration. Following drug treatment, rectal temperature was measured at 1 min after test tail-flick latencies were determined, and values were expressed as the difference between control temperature (before injection) and temperatures following drug administration (°C).
Spontaneous Activity
After determination of tail-flick latency and rectal temperature, mice were placed in individual activity chambers, and spontaneous activity was measured for 10 min. Activity was measured as total number of interruptions of 16 photocell beams per chamber during the 10-min test and expressed as percentage inhibition of activity of the vehicle group.
Ring Immobility
Immediately after measurement of spontaneous activity, mice were placed on the ring immobility apparatus for 5 min (Pertwee 1972) and the total time (in seconds) that the mouse remained motionless was measured. The criterion for ring immobility was the absence of all voluntary movement, including snout and whisker movement. If a mouse fell or escaped from the ring apparatus during testing, it was immediately placed back on the ring. If it fell more than five times before 150 s had elapsed, data were omitted from analysis. The immobility is presented as the average of the total number of seconds that the mice remained immobile.
Data Analysis
Analysis of variance (ANOVA) was used to determine significant differences between control and treatment groups followed by Dunnett's or Newman-Keuls post hoc analysis. Statistical analysis was performed using Stat View, version 5.0 (SAS Institute, Cary, NC). Significance is defined as a p < 0.05. ED50 values were calculated by least squares linear regression analysis using at least 3 doses of drug, followed by calculation of 95% confidence limits according to the method of Bliss (1967).
RESULTS
S1P and SEW2871 stimulate [35S]GTPγS binding in brain sections
The anatomical distribution of S1P-stimulated [35S]GTPγS autoradiography was determined and compared to that produced by the S1P1 receptor-selective agonist SEW2871. Visual inspection revealed that S1P and SEW2871 stimulated [35S]GTPγS binding throughout the brain, with levels of activity varying by region (Fig. 1a), as confirmed by quantitative densitometry (Fig. 1b). The highest levels of S1P-stimulated activity were seen throughout the cortex, molecular layer of the cerebellum (Cblm) and amygdala (Amyg) (Fig. 1a, b). SEW2871-stimulated activity was also highest in these regions and comprised approximately 65-75% of total S1P-stimulated activity (Fig 1c). Moderate levels of S1P-stimulated activity were found in the caudate-putamen (CPu), periaqueductal gray (PAG), hippocampus (Hip) and hypothalamus (Hyp) (Fig. 1a, b). SEW2871 also stimulated [35S]GTPγS binding in these regions and appeared to represent approximately 40% of total S1P-stimulated activity (Fig. 1c). Lower levels of S1P-stimulated [35S]GTPγS binding were detected in thalamus (Thal) and corpus callosum (cc) (Fig. 1a, b) and S1P1 receptors appeared to contribute only minimally to activity in these regions, as indicated by SEW2871 (Fig. 1b, c).
Figure 1.

Representative autoradiograms and densitometric analysis reveal region-specific S1P-and SEW2871-stimulated G-protein activity in mouse brain. a.) Autoradiograms show basal binding in the absence of agonist (left column) and stimulation of [35S]GTPγS binding by 60 μM S1P (center column) or 80 μM SEW2871 (S1P1-selective agonist, right column). b.) Densitometric analysis of S1P-and SEW2871-stimulated [35S]GTPγS binding in the brain reveal regional differences in the magnitude of G-protein activation. Data are expressed as net agonist-stimulated activity ± SEM. **, SEW2871 was p < 0.01 different from S1P by 2-way ANOVA (drug x region) with post-hoc Bonferroni test (n = 10-12). c.) SEW2871-stimulated [35S]GTPγS binding, expressed as a percent of S1P-stimulated activity in the brain, varies among brain regions, suggesting that S1P1 receptors contribute varying proportions of the total G-protein activation seen with S1P in different brain regions. Regions with different letter designations are p < 0.05 different from each other by ANOVA with post-hoc Newman-Keuls test (n = 10-12). Abbreviations: Amyg; amygdala, Cblm; cerebellum, cc; corpus callosum, CCtx; cingulated cortex, CPu; caudate-putamen, Hip; hippocampus, Hyp; hypothalamus, PAG; periaqueductal gray, Thal; thalamus.
S1P- and SEW2871-stimulated [35S]GTPγS binding are inhibited by the S1P1/3 receptor antagonist VPC44116
The contribution of S1P1 receptors to total S1P-stimulated [35S]GTPγS binding was further assessed by incubating the S1P1/3 antagonist VPC44116 with EC75 concentrations of S1P and SEW2871. VPC44116 is a competitive antagonist at S1P1 receptors with a Ki value of 25 nM, whereas it is approximately 10-fold less potent at S1P3 receptors (Foss et al. 2007). VPC44116 is also a partial agonist of S1P4 and S1P5 receptors, although it has very low (>6 μM) affinity for S1P4, and does not have significant affinity for S1P2 (Foss et al. 2007). As seen in cerebellum (Fig. 2a) and cingulate cortex (Fig. 2b), incubation with VPC44116 alone did not significantly alter [35S]GTPγS binding, but incubation of VPC44116 with either S1P or SEW2871 completely blocked agonist-stimulated G-protein activity. Similar results were obtained in most other regions examined (data not shown), including amygdala, PAG, and hypothalamus, where both S1P and SEW2871 produced significant stimulation that was completely blocked by incubation with VPC44116 (p < 0.001 by Newman-Keuls test). Interestingly, S1P at this lower concentration produced only minimal stimulation of [35S]GTPγS binding in regions including caudate-putamen, corpus callosum and thalamus, suggesting possible regional differences in the potency of S1P to activate G-proteins (data not shown).
Figure 2.

Net S1P- and SEW2871-stimulated G-protein activity in cerebellum and cingulate cortex is inhibited by the S1P1/3 antagonist VPC44116. Sections at the level of the a.) cerebellum or b.) cingulate cortex were incubated with vehicle (basal), S1P (10 μM) or SEW2871 (20 μM) in the presence and absence of the S1P1/3 antagonist VPC44116 (50 μM). VPC44116 blocked the stimulation of [35S]GTPγS binding by S1P or SEW2871, but did not affect [35S]GTPγS binding by itself. [35S]GTPγS binding is expressed as mean net stimulate [35S]GTPγS binding ± SEM. The p values shown were obtained by ANOVA with post-hoc Newman-Keuls test (n = 6-7).
S1P suppresses excitatory synaptic transmission
Exogenous S1P induces glutamate release in primary hippocampal neurons (Kajimoto et al. 2007). To determine whether the activation of S1P receptors and concomitant G-protein activity affects neurotransmission, we examined the effect of S1P receptor agonists on excitatory synaptic transmission between cortical neurons in vitro. Spontaneous excitatory postsynaptic currents (or sEPSCs) of individual cortical pyramidal neurons were isolated from GABAergic inhibitory postsynaptic currents (IPSCs), by voltage- clamping neurons to the reversal potential of the IPSCs (~ -65 mV). All remaining events were glutamatergic because they were blocked by the glutamate receptor antagonists CNQX (10 μM) and APV (20 μM; data not shown). Cultured cortical neurons exhibited spontaneous excitatory synaptic currents (Fig. 3) and 1 μM S1P reduced sEPSC frequency and amplitude. Thus, S1P significantly reduced mean sEPSC frequency by 27.3 ± 9.5% (n=11, p< 0.04, Fig. 3b) and mean median sEPSC amplitude by 12.7 ± 2.7% (n=10, p < 0.01, Fig. 3c). S1P had no significant effect on sEPSC rise time (10-90%) or half width (data not shown). The combined decrease in sEPSC frequency and amplitude resulted in significant reduction of the total charge transfer during excitatory synaptic transmission, as mean charge transfer in the presence of 1 μM S1P decreased by 42.0 ± 5.3% (n=10, p < 0.001, Fig. 3d). Thus, S1P appears to suppress overall excitatory input, with changes in sEPSC frequency most likely resulting from diminished presynaptic glutamate release.
Figure 3.

S1P decreases sEPSC frequency and amplitude in cortical pyramidal neurons. a.) Whole-cell recording of sEPSCs (downward deflections) from an individual cortical pyramidal neuron. Application of 1 μM S1P (black bar) reduced sEPSC frequency and amplitude. b.) sEPSC parameters for each condition were normalized to the pre-drug value in each neuron. 1 μM S1P decreased mean sEPSC frequency by 27.3 ± 9.5% (n=11, p < 0.04 vs. control, ANOVA) and sEPSC frequency returned to 90.1 ± 9.5% of control frequency after removal of S1P. c.) 1 μM S1P decreased mean sEPSC amplitude by 12.7 ± 2.7% (n=10, p < 0.01 vs. control) and sEPSC amplitude returned to 94.7 ± 2.6% of control values after wash. d.) 1 μM S1P decreased mean sEPSC charge transfer by 42.0 ± 5.3% (n=10, p < 0.001 vs. control) and sEPSC charge transfer returned to 90.8 ± 10.8% of control values after wash. Statistical significance was determined by ANOVA with post-hoc Newman-Keuls test.
S1P-induced alterations of sEPSCs are mediated by S1P1 receptors
The S1P1/3 antagonist, VPC44116 was used to determine whether S1P modulation of sEPSCS required S1P1/3 receptor activation. VPC44116 partially reversed the effects of S1P on sEPSC amplitude, frequency, and charge transfer (Fig. 4). As previously demonstrated, 1 μM S1P significantly reduced mean sEPSC frequency by 57.1 ± 16.7% (n=5, p< 0.03 vs. control, Fig. 4a) while the subsequent co- application of 1 μM S1P + 5 μM VPC44116 returned sEPSC frequency to 69.0 ± 21.6% of control (n=5, p > 0.05 vs. control). Of note, sEPSC frequency varied between individual cells and cell preparations even without drug treatment, and the degree to which S1P reduced sEPSC frequency was also heterogeneous. This may reflect, in part, differing degrees of endogenous S1P activity, which we did not independently determine. Mean median sEPSC amplitude, while not significant in this data set, exhibited a similar trend and decreased to 75.0 ± 7.5% of control amplitude in the presence of 1 μM S1P only (n=5, p > 0.05 vs. control), but returned to 96.7 ± 15.8% of control amplitude following the addition of 5 μM VPC44116 (Fig. 4c). VPC44116 also reversed the reduction in overall charge transfer induced by S1P (Fig. 4d), whereby S1P decreased mean charge transfer by 54.4 ± 9.1% (n=5, p < 0.002 vs. control), and the subsequent addition of VPC44116 restored charge transfer to 87.3 ± 9.4% of control (n=5, p > 0.05 vs. control; p < 0.01 vs. 1 μM S1P only). The ability of VPC44116 to reverse S1P modulation of sEPSCs suggests that S1P modulation is mediated, at least in part, by the activation of S1P1 receptors. A similar suppression of sEPSC activity was observed using the selective S1P1 receptor agonist SEW2871 (Fig. 5). In 8/9 neurons, SEW2871 (20 μM) reduced sEPSC charge transfer by 43.8 ± 8.2% (n=8, p < 0.05), and the addition of VPC44116 in the continued presence of SEW2871 restored charge transfer to 85.5 ± 10.1% of control. The localization of S1P1 receptors in cortical cultures was confirmed by immunofluorescence with confocal microscopy (supplemental data). S1P1 receptor immunoreactivity was found in cortical pyramidal neurons, most of which appeared to be glutamatergic, as indicated by the presence of immunoreactivity for the vesicular glutamate transporter. These results extend those obtained using agonist-stimulated [35S]GTPγS autoradiography to show that S1P functionally modifies synaptic transmission in the cortex acutely.
Figure 4.

S1P-induced suppression of sEPSC activity is attenuated by the S1P1/3 receptor antagonist, VPC44116. a.) Whole-cell recording of sEPSCs from an individual cortical pyramidal neuron during the application of 1 μM S1P (black bar) and the subsequent addition of 5 μM VPC44116 (striped bar). The addition of VPC44116 partially attenuated the decrease in sEPSC frequency and amplitude induced by S1P. b.) sEPSC parameters for each condition were normalized to the pre-drug value in each neuron. 1 μM S1P decreased mean sEPSC frequency by 57.1± 16.7% (n=5, p < 0.03 vs. control) and the addition of 5 μM VPC44116 restored sEPSC frequency to 69.0 ± 21.6% of control frequency. c.) 1 μM S1P decreased mean median sEPSC amplitude by 25.0 ± 7.5% (n=5, p > 0.05 vs. control) and the addition of 5 μM VPC44116 restored sEPSC amplitude to 96.7 ± 15.8% of control amplitude. d.) 1 μM S1P decreased mean sEPSC charge transfer by 54.4 ± 9.1% (n=5, * p < 0.002 vs. control) and the addition of 5 μM VPC44116 restored sEPSC frequency to 87.3 ± 9.4% of control frequency (** p < 0.01 vs. S1P, p >0.05 vs. control). Statistical significance was determined by ANOVA with post-hoc Newman-Keuls test.
Figure 5.

The S1P1 receptor agonist, SEW2871, decreases sEPSC activity. a.) Whole-cell recording of sEPSCs from an individual cortical pyramidal neuron during the application of 20 μM SEW2871 (black bar) and the subsequent addition of 5 μM VPC44116 (striped bar).b.) 20 μM SEW2871 decreased mean sEPSC charge transfer by 43.8 ± 8.2 % (n=8, * p < 0.05 vs. control by) and the administration of 5 μM VPC44116 in the continued presence of SEW2871 restored sEPSC charge transfer to 85.5 ± 10.1% of control values (n=8, ** p<0.05 vs. SEW2871 only, p> 0.05 vs. control). Statistical significance was determined by ANOVA with post-hoc Newman-Keuls test.
S1P produces antinociception to thermal stimulus via S1P1 receptors
Because S1P produced changes in both receptor-mediated G-protein activity and functional synaptic transmission, it was next determined whether S1P-mediated changes could produce in vivo effects. The effect of S1P on thermal nociception in mice using the tail withdrawal was selected because it is a reflex and would not be confounded by possible effects on motor activity or core body temperature (Lichtman et al. 1993). Dose-response curves were generated for S1P-mediated antinociception using the tail flick assay after i.c.v. injection of S1P (Fig. 6a). Time-course studies determined that the peak time of antinociceptive activity was 15-20 minutes and the offset of antinociceptive activity occurred within 30 minutes (data not shown). S1P elicited dose-dependent antinociception, with an ED50 value of 19.1 μg [17.6-20.8]. Due to solubility limitations, doses higher than 25 μg/mouse i.c.v. were not tested. Thermal antinociception was also observed with the S1P1-selective agonist, SEW2871. The peak effect in the tail flick test was 45.7 ± 21.6 %MPE, observed 10 minutes after the i.c.v. administration of 50 μg SEW2871. However, solubility of the drug was poor at concentrations needed to test higher doses, so no ED50 value could be calculated. In all studies, the vehicle control (acidified DMSO) was administered i.c.v. and produced 18 ± 5 %MPE.
Figure 6.

S1P produces antinociception to thermal stimulus of the tail in mice via S1P1 receptors. Mice were injected i.c.v with S1P with or without VPC44116. a. Dose-response curve for S1P-mediated antinociception in the tail-flick assay. S1P was injected i.c.v. and tail-flick latencies were obtained 20 min after injection. S1P produced dose-dependent antinociception. Data are mean %MPE values ± SEM. b. Antinociception expressed as mean %MPE ± SEM, determined following i.c.v. injection of S1P (25 μg) or the S1P1-selective agonist SEW2871 (SEW, 50 μg) with or without prior injection (i.c.v.) of vehicle or the S1P1/3 antagonist VPC44116 (25 μg). VPC44116 blocked the antinociceptive action of S1P or SEW2871, but administration of VPC44116 alone had no effect. Statistical significance of the data was determined by ANOVA with post-hoc Newman-Keuls test (n = 6).
To further examine the contribution of specific S1P receptor types to in vivo activity, the effect of the S1P1/3-selective antagonist VPC44116 was also assessed in the tail-flick assay (Fig. 6b). Administration of vehicle or VPC44116 alone (25 μg i.c.v.) had no significant effect on tail withdrawal latency (10 ± 4 %MPE, 24 ± 5 %MPE, respectively). S1P (25 μg i.c.v.) and SEW2871 (50 μg i.c.v.) were administered and the tail-flick assay was conducted 10 (SEW2871) or 15 (S1P) minutes following injection. Under these conditions, S1P produced 87 ± 7 %MPE whereas SEW2871 produced 40 ± 28 %MPE. For antagonist studies, VPC44116 (25 μg i.c.v.) was administered 10 (S1P) or 15 (SEW) minutes prior to agonist and mice were tested at 25 minutes post-VPC44116 injection. In both cases, pretreatment with VPC44116 significantly inhibited antinociception produced by the agonist (p <0.01) [12.6 ± 2 %MPE, 4 ± 2 %MPE, respectively, for S1P and SEW2871]. These results provide further evidence that S1P produces thermal antinociception via an S1P1 receptor-mediated mechanism.
S1P produces hypoactivity, catalepsy, hypothermia and antinociception
Studies were next conducted to determine whether there were additional in vivo effects of centrally administered S1P. Because of the homology between S1P and cannabinoid receptors (Toman and Spiegel 2002), their similar anatomical distributions in the CNS (Herkenham et al. 1991; present study) and the ability of both cannabinoids and S1P to mediate thermal antinociception, a tetrad of in vivo measures that are characteristic of the CNS-mediated effects of cannabinoids was assessed (Compton et al. 1993). Mice were injected with S1P (25 μg i.c.v.) and tested as described in Methods. Antinociception was determined at 15 minutes post-administration of S1P followed immediately by the determination of rectal temperature, spontaneous activity (assessed by beam breaks) and catalepsy (ring immobility). As expected, a significant antinociceptive effect of S1P was again observed (Fig. 7a). Interestingly, mice that received S1P also exhibited significant hypothermia (Fig. 7b), catalepsy (Fig. 7c) and inhibition of locomotor activity (Fig. 7d).
Figure 7.

S1P administered i.c.v. produces antinociception (a), hypothermia (b), catalepsy (c) and locomotor inhibition (d) in mice. Mice were injected (i.c.v.) with either vehicle or VPC44116 (25 μg) prior to injection (i.c.v.) with vehicle or S1P (25 μg), and tested following the final injection as described in Methods. S1P alone produced significant effects in all four tests. *, **, p < 0.05, 0.01 different from the vehicle/vehicle-treated group by ANOVA with post-hoc Dunnett's test (n = 6). VPC44116 significantly inhibited S1P-mediated antinociception (a.) as determined by ANOVA with post-hoc Newman-Keuls test (n = 6). VPC44116 alone produced catalepsy (d.) and partially inhibited S1P-induced locomotor inhibition (c.).
To determine whether these effects of S1P were S1P1/3 receptor mediated, the S1P1/3 antagonist, VPC44116, or vehicle were administered 10 minutes prior to administration of S1P or vehicle, and mice were then tested as described above. Surprisingly, the administration of VPC44116 alone produced significant hypothermia, catalepsy and decreased spontaneous activity when compared to vehicle administration (Fig. 7, b-d). In contrast, VPC44116 alone did not produce antinociception, but significantly blocked the antinociceptive effects of S1P (Fig. 7a), similar to the results presented above. VPC44116 did not enhance the hypothermia, catalepsy or hypoactivity produced by S1P, and in fact appeared to produce partial reversal of S1P-induced catalepsy (Fig. 7d).
Because the tetrad of effects described above is reliably produced by cannabinoids, these tests were repeated with S1P in the presence and absence of the CB1-selective antagonist rimonabant (SR141716A). Moreover, because certain sphingosine derivatives can act as CB1 antagonists (Paugh et al. 2006), it was important to determine if there is any involvement of the cannabinoid system in mediation of these effects. As observed above, S1P produced antinociception (Fig. 8a) and hypothermia (Fig. 8b). Administration of the CB1 antagonist rimonabant (3mg/kg) did not significantly block these S1P-mediated effects, nor did the cannabinoid antagonist have significant intrinsic effects in any of the tests at the doses administered (Fig. 8a, b). Similar results were obtained with hypomotility and catalepsy (data not shown). These results demonstrate that while S1P produced cannabinoid-like in vivo effects, these actions were independent of CB1 receptors. However, due to the apparent intrinsic effects of VPC44116 on body temperature and motor activity, only antinociception could be definitively attributed to the activation of S1P1/3 receptors.
Figure 8.
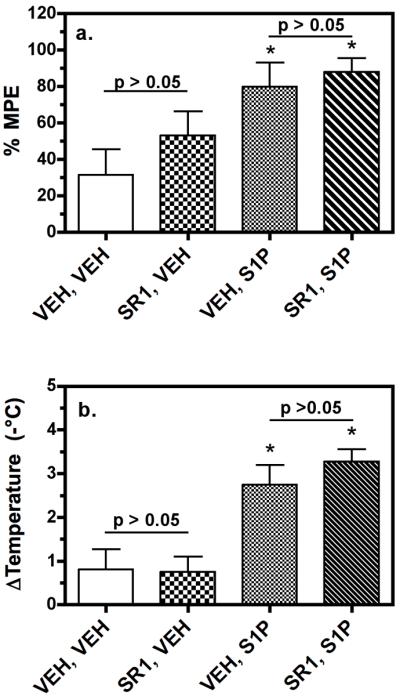
S1P-mediated antinociception and hypothermia does not require cannabinoid CB1 receptor activation. Mice were injected (i.p.) with 3mg/kg of the CB1-selective antagonist rimonabant (SR141716A) prior to injection (i.c.v.) of S1P (25 μg), and tested following the final injection as described in Methods. S1P produced significant antinociception (a.) and hypothermia (b.). *, **, p < 0.05, 0.01 different from the vehicle/vehicle-treated group by ANOVA with post-hoc Dunnett's test (n = 6). Rimonabant did not inhibit S1P-induced antinociception (a.) or hypothermia (b.) and rimonabant alone had no significant effects on either measure (a., b.), as determined by ANOVA with post-hoc Newman-Keuls test (n = 6).
DISCUSSION
The current study revealed that S1P activated G-proteins in anatomically distinct brain regions and reduced excitatory neuronal activity in cortical neurons, presumably by reducing synaptically released glutamate, consistent with the hypothesis that S1P is a functionally active neuromodulator. The effect of S1P on agonist-stimulated [35S]GTPγS binding appeared to result largely from activity at S1P1 receptors in certain regions of the brain, including cortex, cerebellum and amygdala. Additionally, in vivo administration of S1P produced antinociception, hypoactivity, catalepsy and hypothermia, although only antinociception could be definitively attributed to S1P1 receptors, as indicated by the antinociceptive action of the S1P1-selective agonist SEW2871 and reversibility by the S1P1/3-selective antagonist VPC44116. To our knowledge, this is the first study to elucidate the role of a specific S1P receptor type in CNS-mediated effects in adult animals, and the results suggest that S1P1 receptors play an important functional role in the adult brain. This finding is particularly significant because the immunomodulatory agent FTY720 produces reversible lymphopenia via activity at S1P1 receptors (Mandala et al. 2002) and shows promise for treatment of multiple sclerosis (O'Connor et al. 2009) and possibly other autoimmune disorders (Brinkmann 2007). Thus, S1P1 receptors are an emerging therapeutic target and the results of this study suggest additional applications, for example as analgesics or modulators of glutamatergic neurotransmission. Additionally, the possibility that drugs acting at S1P receptors will produce CNS side effects, such as motor impairment or hypothermia, should be considered. Alternatively, S1P might also produce intracellular effects independently of its action at S1P receptors in the CNS, as previously observed in some non-neural cell types (Spiegel and Milstien 2000).
The current studies utilized S1P1-selective ligands to assess the role of this receptor in CNS function. In contrast, compounds selective for S1P2, S1P3 and S1P5 receptors have proven to be more challenging to develop (Lynch and Macdonald 2008). Although S1P1 receptors are clearly involved in CNS function, the current results suggest that other S1P receptor types might also be involved. Most notably, measures of hypomotility, hypothermia and catalepsy induced by S1P were confounded by the intrinsic in vivo effects of VPC44116. Moreover, S1P1 receptor-mediated G-protein activity stimulated by S1P was not fully mimicked by SEW2871. In fact, the contribution of S1P1 receptors to total G-protein activity based on a comparison between S1P and SEW2871 varied from approximately 10% in thalamus and corpus callosum to approximately 75% in cortex and cerebellum. It is also possible that the relative contribution of S1P1 receptors attributed to SEW2871 could have been underestimated if this ligand acted as a partial agonist relative to S1P. However, SEW2871 appeared to be a full agonist for stimulation of [35S]GTPγS binding in CHO cells stably expressing S1P1 receptors (Sanna et al. 2004). Nonetheless, it should be noted that the relative efficacy of GPCR partial agonists could appear to be greater in transfected cell preparations than brain membranes due to differences in receptor reserve (Selley et al. 1998). This concept might also explain why Sanna et al. observed a >15-fold greater EC50 value of SEW2871 relative to S1P, whereas preliminary concentration-effect curves for stimulation of [35S]GTPγS binding to cerebellar sections indicated only 1.5-fold difference in the potency of these two drugs (data not shown).
Because selective ligands are lacking for most of the S1P receptor types, information on the localization of specific S1P receptors has been inferred from in situ hybridization studies (Beer et al. 2000). S1P1 receptor mRNA was highest in cerebellum, and S1P3 receptor mRNA, while only weakly expressed, was highest in the cortex. S1P2 receptor mRNA was most abundant among hippocampal neurons. In contrast, S1P5 receptor mRNA is reportedly confined to oligodendrocytes (Terai et al. 2003). The present findings, combined with the CNS distribution of mRNA for other S1P receptor types, suggest that S1P3 and possibly S1P2 receptors might also contribute to neural function. Similarly, S1P receptor types exhibit distinct signaling profiles (Takabe et al. 2008). All of the S1P receptors activate Gi/o and all except S1P1 receptors couple to G12/13. In addition, S1P2 and S1P3 receptors couple to Gq/11, suggesting that activation of these receptors might produce different intracellular effects compared to S1P1 receptors.
The generation of mice lacking specific S1P receptors has provided further insights into the role of the different S1P receptor types, but few studies have directly examined effects in the CNS (Choi et al. 2008). S1P1 receptor deletion is lethal due to inadequate vascular maturation and resulting embryonic hemorrhage (Liu et al. 2000). S1P3 receptor null mice have significantly reduced litters, suggesting reproductive/developmental deficits, and alterations in receptor signaling (Ishii et al. 2001), but do not exhibit a marked phenotype. S1P2 receptor deficient mice have auditory and vestibular deficits due to cochlear hair cell loss and vascular disturbances (Brinkmann 2007; Choi et al. 2008). Deletion of S1P2 receptors also increased seizure activity due to hyperexcitability of cortical pyramidal neurons (MacLennan et al. 2001), but not all S1P2 null lines exhibit this phenotype.
The results observed in cortical neurons are consistent with the possibility that certain S1P receptors negatively regulate neuronal activity. In the present study, S1P1 receptors were found to inhibit synaptic glutamate release in cortical neurons, as indicated by the effectiveness of the S1P1 agonist SEW2871 and reversal by the S1P1/3 antagonist VPC44116. While these data appear to contradict a recent report of S1P-induced stimulation of glutamate secretion in hippocampal neurons (Kajimoto et al. 2007), the measurement of EPSCs provides a more direct assay of synaptic glutamatergic function and raises the possibility that the previously observed increase in overall extracellular glutamate might not be localized synaptically (Kajimoto et al. 2007). It is also possible that S1P-mediated effects on neuronal activity are region and receptor dependent. Thus, S1P-stimulated glutamate release in primary cultures of hippocampal neurons was attributed to both S1P1 and S1P3 receptors, based on PCR analysis and inhibition of the effects by siRNA knockdown (Kajimoto et al. 2007). S1P was also reported to increase excitability of small diameter sensory neurons isolated from dorsal root ganglia via GPCR-mediated effects on Na+ and K+ channels (Zhang et al. 2006). These authors suggested that S1P might therefore contribute to the hypersensitivity observed during inflammation. Indeed, FTY720 was recently reported to reduce antinociception in the formalin assay, a model of inflammation (Coste et al. 2008), but interpretation is complicated by the fact that FTY720 can inhibit cPLA2 (Payne et al. 2007) and thus prostaglandins, whereas FTY720-P is an agonist at four of the S1P receptors. Nevertheless, considered in light of the present finding that S1P produces antinociception to thermal stimuli, the S1P system is emerging as a novel analgesic target.
In summary, this report demonstrates that S1P receptors are part of a novel neuromodulatory system in the CNS and that administration of S1P agonist regulates nociception, body temperature and motor activity. Moreover, the application of S1P1 selective ligands in both cellular and in vitro assays revealed that S1P1 receptors produce high levels of G-protein activity throughout multiple brain regions and regulate neuronal excitability via the inhibition of glutamatergic transmission in the cortex. Additionally, S1P1 receptors mediate antinociception to thermal stimuli. The data further reveal that activation of other types of S1P receptors might produce G-protein activity and might contribute to S1P-mediated modulation of hypothermia, motor function and catalepsy, although these behavioral effects could be mediated in part via non-receptor effects of S1P. These studies support the concept that S1P and S1P receptors in the brain are important regulators of CNS function and should be considered both for novel therapeutic targets and potential side effects.
Supplementary Material
Acknowledgements
These studies were supported by USPHS grants R01 DA014277 (LJS), R01 DA10770 (DES), R03 DA022581 (SPW), R01 GM043880 (SS), R01 NS049519 (LSS), R01 GM067958 (KRL & TLM), the Thomas F. and Kate Miller Jeffress Memorial Trust (LJS, SPW) and the NIMH Intramural Research Program (SM). The authors thank Dr. Katherine Falenski, James Burston, Peter Nguyen and David Stevens for assistance.
REFERENCES
- Beer MS, Stanton JA, Salim K, Rigby M, Heavens RP, Smith D, McAllister G. EDG receptors as a therapeutic target in the nervous system. Ann. N. Y. Acad. Sci. 2000;905:118–131. doi: 10.1111/j.1749-6632.2000.tb06544.x. [DOI] [PubMed] [Google Scholar]
- Bliss CI. Statistics in Biology. McGraw-Hill; New York: 1967. [Google Scholar]
- Blondeau N, Lai Y, Tyndall S, Popolo M, Topalkara K, Pru JK, Zhang L, Kim H, Liao JK, Ding K, Waeber C. Distribution of sphingosine kinase activity and mRNA in rodent brain. J. Neurochem. 2007;103:509–517. doi: 10.1111/j.1471-4159.2007.04755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: Mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007;115:84–105. doi: 10.1016/j.pharmthera.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Bryan L, Kordula T, Spiegel S, Milstien S. Regulation and functions of sphingosine kinases in the brain. Biochim. Biophys. Acta. 2008 doi: 10.1016/j.bbalip.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Lee CW, Chun J. Biological roles of lysophospholipid receptors revealed by genetic null mice: An update. Biochim. Biophys. Acta. 2008 doi: 10.1016/j.bbalip.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Goetzl EJ, Hla T, Igarashi Y, Lynch KR, Moolenaar W, Pyne S, Tigyi G. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 2002;54:265–269. doi: 10.1124/pr.54.2.265. [DOI] [PubMed] [Google Scholar]
- Compton DR, Rice KC, DeCosta BR, Razdan RK, Melvin LS, Johnson MR, Martin BR. Cannabinoid structure-activity relationships: Correlation of receptor binding and in vivo activities. J. Pharmacol. Exp. Ther. 1993;265:218–226. [PubMed] [Google Scholar]
- Coste O, Pierre S, Marian C, Brenneis C, Angioni C, Schmidt H, Popp L, Geisslinger G, Scholich K. Antinociceptive activity of the S1P-receptor agonist FTY720. J Cell Mol Med. 2008;12:995–1004. doi: 10.1111/j.1582-4934.2008.00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amour FE, Smith DC. A method for determining the loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
- Edsall LC, Spiegel S. Enzymatic measurement of sphingosine 1-phosphate. Anal. Biochem. 1999;272:80–86. doi: 10.1006/abio.1999.4157. [DOI] [PubMed] [Google Scholar]
- Foss FW, Jr., Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, Macdonald TL. Synthesis and biological evaluation of gamma-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg. Med. Chem. 2007;15:663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goforth PB, Ellis EF, Satin LS. Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J. Neurosci. 1999;19:7367–7374. doi: 10.1523/JNEUROSCI.19-17-07367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Harris LS, Pierson AK. Some Narcotic Antagonists In The Benzomorphan Series. J. Pharmacol. Exp. Ther. 1964;143:141–148. [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW. Primary culture of identified neurons from the visual cortex of postnatal rats. J. Neurosci. 1986;6:3044–3060. doi: 10.1523/JNEUROSCI.06-10-03044.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii I, Friedman B, Ye X, Kawamura S, McGiffert C, Contos JJ, Kingsbury MA, Zhang G, Brown JH, Chun J. Selective loss of sphingosine 1-phosphate signaling with no obvious phenotypic abnormality in mice lacking its G protein-coupled receptor, LP(B3)/EDG-3. J. Biol. Chem. 2001;276:33697–33704. doi: 10.1074/jbc.M104441200. [DOI] [PubMed] [Google Scholar]
- Kajimoto T, Okada T, Yu H, Goparaju SK, Jahangeer S, Nakamura S. Involvement of sphingosine-1-phosphate in glutamate secretion in hippocampal neurons. Mol. Cell. Biol. 2007;27:3429–3440. doi: 10.1128/MCB.01465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Smith FL, Martin BR. Evidence that the antinociceptive tail-flick response is produced independently from changes in either tail-skin temperature or core temperature. Pain. 1993;55:283–295. doi: 10.1016/0304-3959(93)90003-8. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Invest. 2000;106:951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KR, Macdonald TL. Sphingosine 1-phosphate chemical biology. Biochim. Biophys. Acta. 2008;1781:508–512. doi: 10.1016/j.bbalip.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan AJ, Carney PR, Zhu WJ, Chaves AH, Garcia J, Grimes JR, Anderson KJ, Roper SN, Lee N. An essential role for the H218/AGR16/Edg-5/LP(B2) sphingosine 1-phosphate receptor in neuronal excitability. Eur. J. Neurosci. 2001;14:203–209. doi: 10.1046/j.0953-816x.2001.01634.x. [DOI] [PubMed] [Google Scholar]
- Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- O'Connor P, Comi G, Montalban X, Antel J, Radue EW, de Vera A, Pohlmann H, Kappos L. Oral fingolimod (FTY720) in multiple sclerosis: two-year results of a phase II extension study. Neurology. 2009;72:73–79. doi: 10.1212/01.wnl.0000338569.32367.3d. [DOI] [PubMed] [Google Scholar]
- Paugh SW, Cassidy MP, He H, Milstien S, Sim-Selley LJ, Spiegel S, Selley DE. Sphingosine and its analog, the immunosuppressant 2-amino-2-(2-[4- octylphenyl]ethyl)-1,3-propanediol, interact with the CB1 cannabinoid receptor. Mol. Pharmacol. 2006;70:41–50. doi: 10.1124/mol.105.020552. [DOI] [PubMed] [Google Scholar]
- Payne SG, Oskeritzian CA, Griffiths R, Subramanian P, Barbour SE, Chalfant CE, Milstien S, Spiegel S. The immunosuppressant drug FTY720 inhibits cytosolic phospholipase A2 independently of sphingosine-1-phosphate receptors. Blood. 2007;109:1077–1085. doi: 10.1182/blood-2006-03-011437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedigo NW, Dewey WL, Harris LS. Determination and characterization of the antinociceptive activity of intraventricularly administered acetylcholine in mice. J. Pharmacol. Exp. Ther. 1975;193:845–852. [PubMed] [Google Scholar]
- Pertwee RG. The ring test: a quantitative method for assessing the `cataleptic' effect of cannabis in mice. Br. J. Pharmacol. 1972;46:753–763. doi: 10.1111/j.1476-5381.1972.tb06900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna MG, Liao J, Jo E, Alfonso C, Ahn MY, Peterson MS, Webb B, Lefebvre S, Chun J, Gray N, Rosen H. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- Selley DE, Liu Q, Childers SR. Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTP gamma S binding in mMOR-CHO cells and rat thalamus. J. Pharmacol. Exp. Ther. 1998;285:496–505. [PubMed] [Google Scholar]
- Sim LJ, Selley DE, Childers SR. In vitro autoradiography of receptor-activated G-proteins in rat brain by agonist-stimulated guanylyl 5'-[γ-[35S]thio]-triphosphate binding. Proceedings of the National Academy of Sciences USA; 1995. pp. 7242–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: signaling inside and out. FEBS Lett. 2000;476:55–57. doi: 10.1016/s0014-5793(00)01670-7. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol. Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, Soga T, Takahashi M, Kamohara M, Ohno K, Yatsugi S, Okada M, Yamaguchi T. Edg-8 receptors are preferentially expressed in oligodendrocyte lineage cells of the rat CNS. Neuroscience. 2003;116:1053–1062. doi: 10.1016/s0306-4522(02)00791-1. [DOI] [PubMed] [Google Scholar]
- Toman RE, Spiegel S. Lysophospholipid receptors in the nervous system. Neurochem. Res. 2002;27:619–627. doi: 10.1023/a:1020219915922. [DOI] [PubMed] [Google Scholar]
- Waeber C, Chiu ML. In vitro autoradiographic visualization of guanosine-5'-O-(3-[35S]thio)triphosphate binding stimulated by sphingosine 1-phosphate and lysophosphatidic acid. J. Neurochem. 1999;73:1212–1221. doi: 10.1046/j.1471-4159.1999.0731212.x. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Fehrenbacher JC, Vasko MR, Nicol GD. Sphingosine-1-phosphate via excitation of a G-protein-coupled receptor(s) enhances the excitability of rat sensory neurons. J. Neurophysiol. 2006;96:1042–1052. doi: 10.1152/jn.00120.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.