

Abstract
Human polo-like kinase Plk1 localizes to the centrosomes, kinetochores and central spindle structures during mitosis. It plays an essential role in promoting mitosis and cytokinesis through phosphorylation of a number of different substrates. Kinase activity is regulated by a conserved C-terminal domain, termed the polo box domain (PBD), which acts both as an autoinhibitory domain and as a subcellular localization domain. We have determined the crystal structure of Plk1 PBD (residues 367–603) to 2.2 Å resolution and the structure of a phospho-peptide–PBD (residues 345–603) complex to 2.3 Å resolution. The two polo boxes of the PBD exhibit identical folds based on a six-stranded β-sheet and an α-helix, despite only 12% sequence identity. The phospho-peptide binds at a site between the two polo boxes. It makes a short antiparallel β-sheet connection and critical contacts to residues Trp414, Leu490, His538 and Lys540. Most of these residues had been shown to be important for biological activity through mutational studies. The results provide an explanation for phospho-peptide recognition and create the basis for new functional studies.
Keywords: cell cycle control/Plk1/polo box domain/phospho-peptide recognition/protein structure
Introduction
Polo-like kinases (Plks) are a family of serine/threonine kinases related to the polo gene product of Drosophila melanogaster, originally identified from a mitotic mutant that displayed abnormal spindle poles (Sunkel and Glover, 1988). Plks play essential roles in several stages of mitosis and cytokinesis with a dynamic pattern of localization to centrosomes, kinetochores and central spindle structures during the cell cycle (reviewed in Glover et al., 1998; Nigg, 1998; Donaldson et al., 2001). On entry into mitosis, Plks activate CDK1–cyclin B through phosphorylation of upstream regulators (Kumagai and Dunphy, 1996; Toyoshima-Morimoto et al., 2002; Nakajima et al., 2003) and cyclin B (Toyoshima-Morimoto et al., 2001; Yuan et al., 2002a; Jackman et al., 2003). At the same time, Plks also promote centrosome maturation (Casenghi et al., 2003), disassembly of the Golgi complex and dissociation of cohesin from the chromosomes (Sumara et al., 2002). Subsequently, Plks help initiate anaphase through regulation of the anaphase-promoting complex (APC) (see Glover et al., 1998; Nigg, 1998; Donaldson et al., 2001) and other proteins (Alexandru et al., 2001; May et al., 2002). Finally, Plks promote cytokinesis through roles in formation and positioning of the central spindle (Neef et al., 2003) and yeast septum (Song et al., 2000; Song and Lee, 2001). Although not all functions have been demonstrated in all species, it is clear that Plks have multiple substrates and multiple roles.
In mammals, four Plks have been identified (Plk1, Plk2/Snk, Plk3/Prk/Fnk and Sak), but in yeast there is a single Plk [Cdc5p (Saccharomyces cerevisiae) and Plo1p (Schizosaccharomyces pombe)]; there is also a single Plk in D.melanogaster (Polo), but Xenopus laevis expresses at least three Plks (Plx1, Plx2 and Plx3) (Duncan et al., 2001; and references therein). Some clues for the multiple roles of Plks are given by the organization of the protein. Plks contain an N-terminal kinase domain (residues 49–310 in the human Plk1 sequence) with a requirement for phosphorylation of a threonine in the activation segment of the kinase (Thr210 in human Plk1) by an upstream kinase (Lee and Erikson, 1997; Qian et al., 1999; Jang et al., 2002b; Kelm et al., 2002). The C-terminal region (residues 345–603) contains the polo box domain (PBD) so called because of conservation of two stretches of ∼80 amino acids among the polo kinases from different species. The PBD has at least two functions. It is an autoinhibitory domain (Lee and Erikson, 1997; Mundt et al., 1997); inhibition by the PBD can be relieved by phosphorylation of Thr210 or mutation of Thr210 to aspartate (Jang et al., 2002a). Secondly, the PBD plays a crucial role in subcellular localization (Song et al., 2000). Both polo boxes are required for localization, and overexpression of the PBD induces defective bipolar spindles (Seong et al., 2002; Reynolds and Ohkura, 2003).
The preferred substrate epitope for Plk phosphorylation has been identified as Glu/Asp-X-Ser/Thr-Φ, where X is any amino acid and Φ is usually hydrophobic, frequently leucine, from the sites phosphorylated with substrates such as Cdc25C (Toyoshima-Morimoto et al., 2002), SCC1 (Alexandru et al., 2001), BRAC2 (Lin et al., 2003), Myt1 (Nakajima et al., 2003), cyclin B (Jackman et al., 2003), NudC (Zhou et al., 2003) and MKlp2 (Neef et al., 2003). The epitope recognized by the PBD that results in localization of Plk1 to cellular substructures is less well understood. Major progress in understanding this process was achieved recently with a proteomic screen that identified an optimal phospho-peptide motif for binding to the PBD with the sequence Met.Gln.Ser.pThr.Pro.Leu (Elia et al., 2003). The phospho-peptide was able to disrupt Plk substrate binding and localization of the PBD to centrosomes. The work of Elia et al. (2003) provided the first clear example of the likely recognition sequence for Plk polo box targets. The work raised the question of how at the structural level the PBD interacts with phospho-peptides.
This question has been addressed in the present study through crystallization of the PBD with and without bound phospho-peptide. In the structural work reported here, we show that the two polo boxes of Plk1 PBD have identical folds based on a six-stranded antiparallel β-sheet and an α-helix. The phospho-peptide binds at a site between the two polo boxes. Contact residues include many residues that have been identified as important for function from mutational studies, and the structure also defines new residues involved in phospho-peptide recognition. Previously, the polo box structure for murine Sak, a Plk that exceptionally contains only one polo box, has been determined (Leung et al., 2002). The Plk1 PBD shows some similarities but also significant differences with the strand-exchanged dimer Sak structure.
Results
The structure of the PBD
The structure of the subtilisin-cleaved PBD (residues 367–603 with a subtilisin cleavage point between residues 494 and 495; see Materials and methods) was solved with SOLVE (Terwilliger and Berendzen, 1999) (Table I). The elongated molecule is composed of the two polo boxes [residues 411–492 for polo box 1 (PB1) and residues 511–592 for polo box 2 (PB2)] and an N-terminal extension (residues 372–410) that wraps around PB2 (Figure 1). Each polo box contains a continuous six-stranded antiparallel β-sheet and an α-helix (αB for PB1 and αD for PB2). There are several examples of non-polar clusters [e.g. tyrosines, 417 (β1), 421 (β2), 481 (αB) and 485 (αB)].
Table I. Summary of crystallographic data.
| Se-Met apo PBD | Native apo PBD | PBD co-crystallized with phospho-peptide | |
|---|---|---|---|
| Beamline | ESRF ID29 | Elettra | ESRF ID29 |
| Space group and unit cell (Å) | P1, a = 33.16, b = 42.72, c = 82.78 α = 102.19, β = 94.90, γ = 91.87 | P1, a = 32.74, b = 42.02, c = 80.73, α = 103.16 β = 93.88, γ = 91.32 | P1, a = 57.07, b = 56.89, c = 85.05, α = 91.45, β = 103.21 γ = 118.50 |
| Wavelength (Å) | 0.9792 | 0.998 | 0.9792 |
| Resolution (Å); | 25.6–3.0 | 20.0–2.2 | 24.7–2.3 |
| (highest range) | (3.16–3.0) | (2.26–2.2) | (2.42–2.3) |
| No. of reflections | 32 271 | 103 961 | 71 256 |
| No. of unique reflections | 8571 | 20 156 | 35 628 |
| Multiplicity | 3.1 | 2.2 | 1.9 |
| I/σI | 7.2 (3.4) | 8.4 (3.6) | 2.7 (2.1) |
| Completeness % | 97.5 (96.9) | 95.2 (92.0) | 95.7 (96.6) |
| Anomalous completeness | 92.4 | ||
| Rsym | 0.074 (0.151) | 0.050 (0.146) | 0.065 (0.258) |
| Wilson B-factor (Å2) | 39.46 | 31.25 | 49.67 |
| SAD phasing statistics | |||
| SOLVE Z-score | 45.8 | ||
| FOM (after density modification) | 0.42 (0.65) | ||
| Refinement statistics | |||
| Molecules per asymmetric unit | 2 | 3 | |
| Protein atoms | 3396 | 5520 | |
| Waters | 226 | 190 | |
| Rconv | 0.198 | 0.244 | |
| Rfree | 0.288 | 0.313 | |
| Mean main chain protein B-factor (Å2) | 31.3 | 46.4 | |
| R.m.s.d. bond lengths (Å) | 0.017 | 0.014 | |
| R.m.s.d. bond angles (°) | 1.72 | 1.67 |
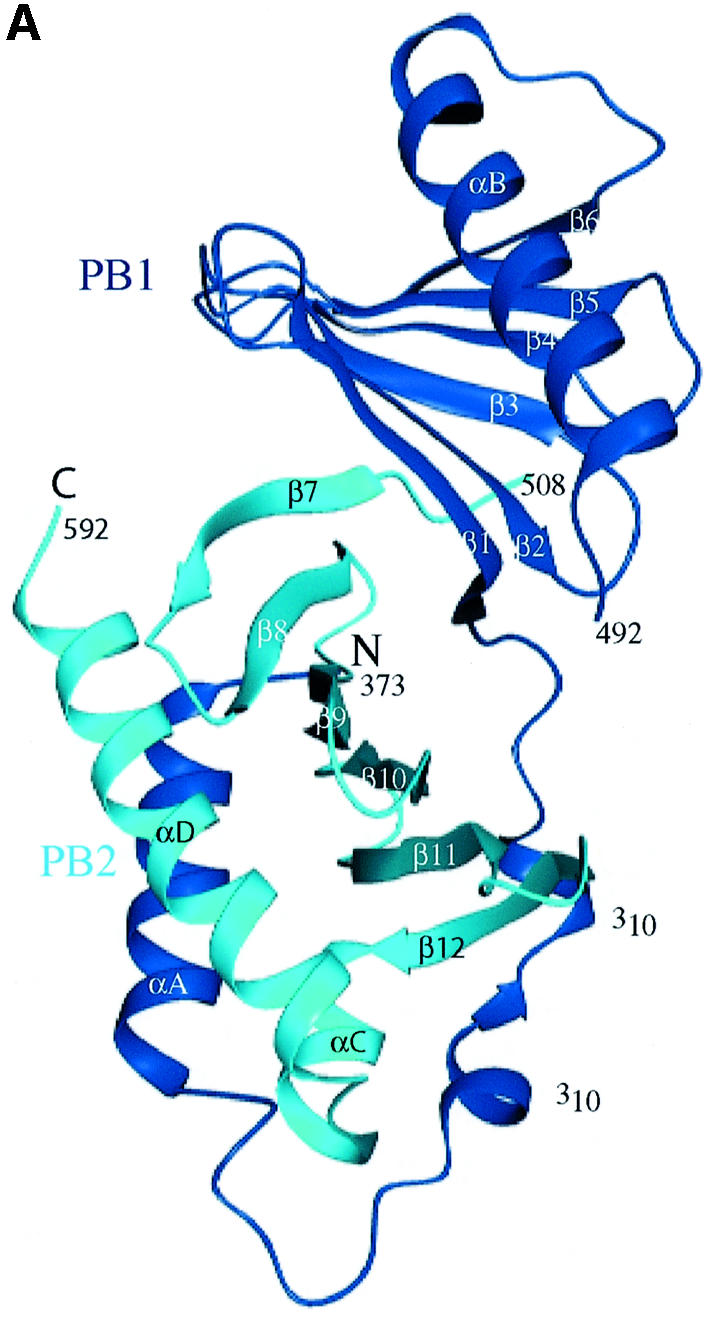

Fig. 1. Schematic diagram of the PBD of Plk1. (A) The N-terminal extension (residues 372–410) and polo box 1 (PB1: residues 411–492) are coloured blue, and polo box 2 (PB2: residues 508–592) cyan. Helices are shown as spirals and β-strands as arrows. There is a break in electron density for residues 493–507 that includes the subtilisin cleavage site. The view is down the approximate 2-fold symmetry axis that relates PB1 to PB2. The structure begins with a segment additional to the polo boxes, residues 372–410, which wraps around the second polo box PB2. This segment comprises a helix αA (residues 374–387), a bend, and then two turns of 310 helices that are separated by two residues in an extended conformation (residues 400–401). PB1 comprises a continuous region of six strands of antiparallel β-sheet (residues 411–466) followed by one helix αB (residues 469–489). The αB helix shields one side of the PB1 β-sheet. The second polo box PB2 begins at residue 511 and runs with a second continuous six-stranded antiparallel β-sheet from residues 511 to 563. The chain then wraps around the lower part of PB2 with two α helices (αC residues 564–570 and αD residues 574–592) that shield the PB2 β-sheet. The αD helix is antiparallel to αA. The main chain atoms of residues 400–401 from the N-terminal extension make two hydrogen bonds to main chain atoms 563 and 561 in the last β-strand, β12, of PB2. This figure and other graphics figures in this paper were prepared with Aesop (M.E.M. Noble, unpublished work). (B) The secondary structure assignment and amino acid sequence of the Plk1 PBD. Helices are indicated as rectangles and β-strands as arrows. The structure-based sequence alignment of Sak is shown below the PBD sequence. Residues coloured magenta are from the A subunit and residues coloured red from the B subunit. Sak residues in bold are structurally equivalent (r.m.s.d. in Cα position <2 Å) to residues in Plk1 PBD. Residues in contact with the phospho-peptide are marked with an asterisk.
The two polo boxes are related by an ∼2-fold symmetry axis (Figure 1A) and superimpose with an r.m.s.d. of 1.8 Å for 70 atoms with equivalent residues corresponding to 411–488 and 511–592 (Figure 2A). The sequence identity is 12% (Figure 2B). In a search for proteins with similar folds, several hits were obtained by Dali (Holm and Sander, 1995). The most interesting was the hit to arylsulfatase from Pseudomonas aeruginosa (Boltes et al., 2001). Fifty-one residues from the secondary structural elements of the PB2 sheet superimpose with four strands and a helix from the C-terminal domain of arylsulfatase with an r.m.s.d. of 1.8 Å and sequence identity of 16%. Since there is no functional similarity between the PBD and arylsulfatase, we assume that the superposition is an example of two different sequences adopting a common fold. The fold of the PBD is not a new fold.
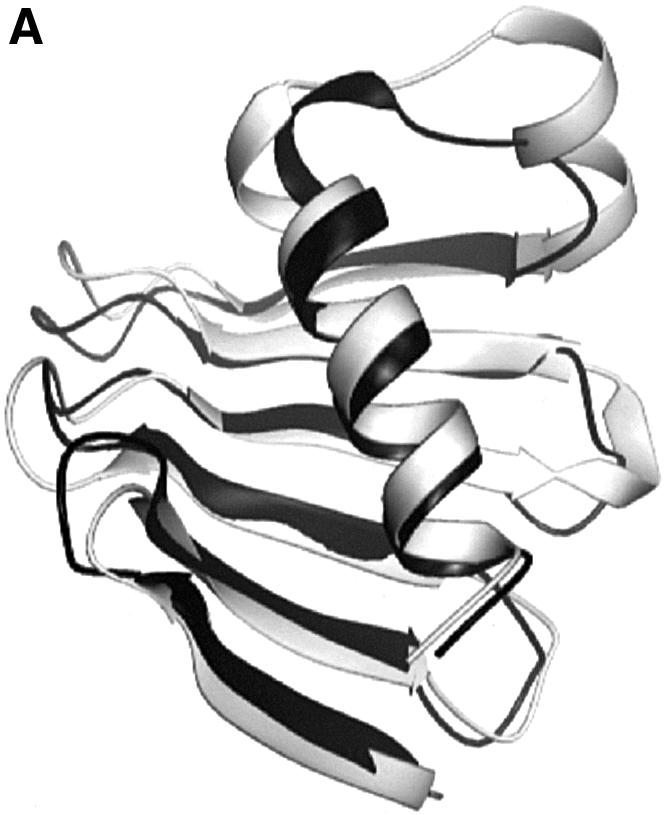

Fig. 2. Comparison of PB1 and PB2 and structure-based sequence alignment. (A) PB1 in black superimposed with PB2 in grey. The view is similar to that in Figure 1A for PB1. (B) Structure-based sequence alignment of PB1 and PB2. Structurally equivalent residues are shown in bold. Contact residues to the phospho-peptide are marked with an asterisk.
Comparison of the polo boxes of Plk1 and Sak
Attempts to solve the Plk1 PBD by molecular replacement using the Sak PBD as search object were not successful. A Dali search with Plk1 PBD failed to identify Sak. The Sak polo box has an unusual topology arising from the strand exchange between the two subunits of the dimer in which four strands come from one subunit and two strands from the other (Figure 3A). The Plk1 PBD has two continuous six-stranded sheets with no strand exchange between the two polo boxes. The Sak polo box structure was aligned with Plk1 in O using 13 residues (PBD 432–444, the β3–β4 turn), which Leung et al. (2002) had identified as homologous between the two proteins. The alignment was then elaborated using the complete structures. [A similar alignment was achieved with LSQMAN (Kleywegt and Jones, 1997) for a single chain of Sak followed by improvement with O in order to detect the strand exchange components.] Part of the Sak dimer polo box structure superimposed on the Plk1 PB1 with an r.m.s.d. of 1.5 Å for 55 Cα atoms (Figures 3A and 1B). A similar alignment was obtained for the alignment of the Sak PBD on PB2 (Figure 1B) where the superposition gave an r.m.s.d. of 2.0 Å for 57 atoms. In this alignment, the last β-strand from Sak (subunit A) superimposed with β7, the first strand of the PB2. The structure-based alignment shown in Figure 1B gives a sequence identity of 22% for 67 residues of PB1 and 8% for 73 residues of PB2. The ion pair contact observed in the Sak polo box between Asp838 and Lys906 is conserved in Plk1 PB1 and PB2 (Asp438:Lys475 and Asp537:Arg579). Despite the close superposition of the β-sheet and part of the αB helix, the two complete structures show quite different arrangements (Figure 3A and B). The mechanisms for shielding the non-polar core of the β-sheets are different. In Sak PBD, the two β-sheets pack so as to orient the non-polar groups between the sheets and to place the polar groups outside (Figure 3A). In Plk1 PBD, the sheets are protected mostly by contacts to the αB helix (PB1) and αD helix (PB2) (Figure 3B). The Plk1 PBD phospho-peptide-binding site (described below) is different from that predicted as a possible binding site from the Sak PBD structure. Further studies are required to establish the binding site in Sak PBD.

Fig. 3. Comparison of the Plk1 PBD and the Sak PBD. (A) The crystal structure of the Sak PBD (Leung et al., 2002) is a dimer comprised of two strand-exchanged subunits A (coloured magenta) and B (coloured red). The secondary structural elements are labelled, with labels of subunit B followed by ′. (B) The structurally conserved regions between the Plk PBD and Sak PBD. Plk1 PB1 and PB2 are in blue and cyan as in Figure 1A. The orientations and scale are identical to (A) and the orientation also corresponds to Figure 1A. Part of the Sak PBD subunit A (magenta; elements β1, β2 and β3) superimposes on strands (blue) β2, β3 and β4 of Plk PB1 (not labelled) and part of subunit B (red; elements β4′, β5′ and αA′) superimposes on strands β5, β6 and part of helix αB of Plk1 PB1 (not labelled). A similar superposition is observed for Sak PBD and Plk1 PB2 (not shown). The arrangement of the Sak dimer (A) is quite different from that of Plk1 PB1 and PB2 (in B and Figure 1A).
The polo box domain–phospho-peptide structure
The intact polo box (residues 345–603) was co-crystallized with the phospho-peptide Met.Gln.Ser.pThr.Pro.Leu, the peptide identified by Elia et al. (2003). The three molecules of the asymmetric unit are nearly identical, with small differences that are noted below (Table I). The chain can be traced continuously from residues 373 to 593, but there is no electron density for the first 27 residues from the N-terminal end and the last 11 residues at the C-terminus. The peptide is well supported by electron density in all three subunits (Figure 4A). The linker region between PB1 and PB2 (residues 493–507), previously absent in the subtilisin-cleaved apo PBD structure, is also well supported by density in the A and B subunits, but there is a break between residues 495 and 501 in the C subunit. The C subunit is slightly less well ordered than the A and B subunits.



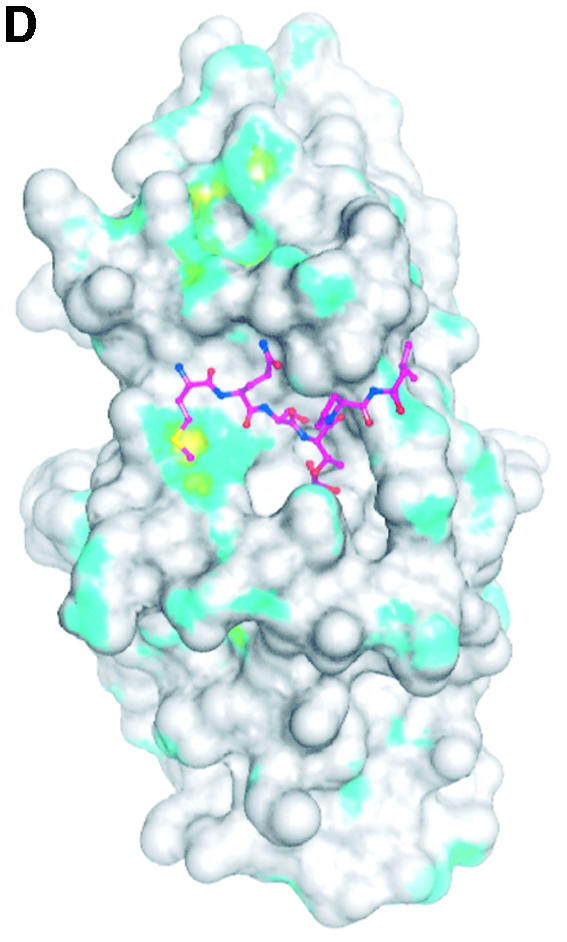
Fig. 4. The structure of the phospho-peptide–PBD complex. (A) Stereo diagram of the final 2Fo – Fc σA-weighted electron density map in the region of the phospho-peptide contoured at 1.1σ. The kink in the chain between the phospho-Thr and the proline is apparent. (B) Schematic diagram showing the Plk1 PBD and the bound phospho-peptide (magenta). The view is ∼90° to the view in Figure 1A. The N-terminal region and PB1 are in blue, and PB2 is in cyan. The linker region is in pink. The phospho-peptide (in magenta) binds between PB1 and PB2 making a short antiparallel β-sheet with β1 from PB1. Secondary structural elements that contribute contact residues are labelled. This figure is depth-cued so that parts further from the viewer appear in fainter colours. (C) Stereo diagram of the interactions between the phospho-peptide Met.Gln.Ser.pThr.Pro.Leu and the B subunit of Plk1 PBD. The view is similar to Figure 1A. Hydrogen bonds are shown as black dotted lines. The colour code is similar to that in (B) except that those residues from PB1 are in pale blue. (D) The van der Waals surface of the Plk1 PBD domain with the hydrophobic potential calculated by GRID (Goodford, 1985) displayed. The deepest hydrophobic potential is yellow, the medium cyan, and the neutral surface is grey. The atoms of the phospho-peptide are superimposed. The view is similar to Figure 1A. The threonine phosphate is partly obscured by the van der Waals surface of His538.
There are no conformational changes in the PBD on binding peptide (the r.m.s.d. in the Cα atom positions is 0.65 Å for 205 atoms excluding the linker region). The phospho-peptide binds in a groove between PB1 and PB2, contacting residues in PB1 from strand β1 and helix αB and residues in PB2 from the end of strand β7, and the loop between β9 and β10 (Figure 4B). There is a short stretch of β-sheet in which the peptide interacts with the protein via three hydrogen bonds from the main chain N and O of Met1 to the main chain O and N of Asp416, respectively, and from the main chain N of Ser3 to the main chain O of Trp414. These interactions with β1 effectively extend the six-stranded sheet of PB1 to seven strands (Figure 4B). There is a pronounced kink in the chain between the phospho-threonine and proline residues (Figure 4A).
The peptide makes a number of specific polar and non-polar contacts (Figure 4C and summarized in Table II). The side chain of Met1 makes non-polar contacts to the PB1 residue Trp414 (β1) and to the PB2 side chains of Arg516 (β7) and Phe535 (β9). The side chain of Gln2 hydrogen-bonds via the NE2 atom to the side chain OH of Tyr485 (αB), one of the tyrosines of the tyrosine cluster (see above), and to a water molecule. The side chain hydroxyl of Ser3 is directed into the groove. In the A and C subunits, the side chain adopts a gauche conformation and contacts one water molecule that links to the main chain oxygen of pThr4. In the B subunit (illustrated in Figure 4C), the side chain has a trans conformation and contacts two waters and the main chain nitrogen of pThr4. One of the waters contacts the phosphate of the phospho-threonine. The phospho-threonine is also directed into the groove at a site that contains two basic residues and several water molecules. One phosphate oxygen contacts ND2 of His538 (β9–β10 loop) and another phosphate oxygen contacts NZ of Lys540 (β10). In addition, the phosphate contacts between two and five water molecules (Table II) (differences correspond to the three subunits with rather more waters located in the B subunit than in the other two subunits). The waters form an extensive hydrogen-bonded network. The main chain O of pThr4 hydrogen-bonds to the main chain N of Leu491. Pro5, in the trans conformation, is partially shielded by the side chain of Gln2 and by van der Waals interactions with Leu490. Finally, Leu6 makes non-polar interactions with His489, Leu490 and Leu491. Arg557 is in the vicinity of the pThr side chain but it is well ordered and >6 Å from the phosphate group. Lys492 as also in the vicinity of the phospho-threonine site, but its side chain is directed away.
Table II. Summary of contacts between the phospho-peptide and PBD.
| Polar contactsa | ||||
|---|---|---|---|---|
| Peptide | PBD residue | Distance (Å) |
||
| A | B | C | ||
| Met1 | N Asp416 O | 3.6 | 3.3 | 3.2 |
| O Asp416 N | 2.8 | 2.9 | 2.7 | |
| Gln2 | NE2 Tyr48 OH | 2.4 | 2.5 | 2.4 |
| NE2 Watb (70, 113, 194) | 3.6 | 2.6 | 3.1 | |
| Ser3 | N Trp414 O | 2.9 | 3.1 | 2.9 |
| OG pThr4 O2P | – | 3.4 | – | |
| OG Wat (24, 8, 135) | 2.7 | 2.5 | 2.8 | |
| OG Wat (–, 83, –) | – | 2.8 | – | |
| OG N pThr4 | – | 3.1 | – | |
| pThr4 | O Leu491 N | 3.0 | 2.9 | 2.9 |
| O Wat (24, 8, 135) | 2.6 | 2.6 | 2.8 | |
| O1P Lys540 NZ | 2.8 | 2.4 | 2.5 | |
| O1P Wat (117, 202, 2) | 2.4 | 2.8 | 2.4 | |
| O2P Wat (165, 83, 92) | 2.7 | 3.4 | 2.8 | |
| O2P Wat (–, 62, –) | – | 2.6 | – | |
| O3P His538 ND1 | 2.8 | 2.6 | 2.9 | |
| O3P Wat (–, 100, –) | – | 2.9 | – | |
| O3P Wat (–, 33, –) | – | 2.7 | – | |
| Pro5 | – | – | – | |
| Leu6 |
N His489O |
3.0 |
3.0 |
3.0 |
| Non-polar contactsc | ||||
| Peptide | PBD residue | No. of contacts |
||
| |
|
A |
B |
C |
| Met1 | Trp414 | 4 | 5 | 3 |
| Asp416 | 1 | 1 | 1 | |
| Arg516 | 0 | 4 | 6 | |
| Phe535 | 6 | 5 | 0 | |
| Gln2 | Trp414 | 8 | 7 | 7 |
| Tyr485 | 0 | 2 | 0 | |
| Leu490 | 1 | 2 | 2 | |
| Ser3 | Lys413 | 1 | 1 | 0 |
| Trp414 | 8 | 10 | 9 | |
| Leu490 | 2 | 1 | 0 | |
| pThr4 | Leu490 | 1 | 1 | 0 |
| His538 | 1 | 1 | 2 | |
| Pro5 | Leu490 | 2 | 3 | 2 |
| Leu491 | 0 | 1 | 0 | |
| Leu6 | Glu488 | 1 | 1 | 1 |
| His489 | 4 | 3 | 3 | |
| Leu490 | 4 | 3 | 3 | |
| Leu491 | 1 | 3 | 1 | |
| Total | 45 | 55 | 36 | |
aPolar contacts are listed if the contact in at least one subunit is <3.3 Å.
bWater numbers are given in parentheses for the A, B and C subunits.
cNon-polar contacts are defined as contacts <4.3 Å between non-polar groups.
The total binding energy is the sum of favourable contributions to the enthalpy from the polar and non-polar interactions and to the entropy from shielding of non-polar groups and displacement of water molecules, combined with unfavourable contributions to the enthalpy from breaking of hydrogen bonds to water molecules and to the entropy arising from localization of the phospho-peptide. The hydrogen bonds, especially those that align the peptide to the PBD through the antiparallel β-sheet arrangement and those to the pThr, appear important for specificity. For the non-polar contacts, Trp414 makes a dominant contribution. Trp414 contributes ∼44% of the total non-polar contacts between the phospho-peptide and the PBD (Table II). Figure 4D illustrates the van der Waals representation of the surface of the uncomplexed PBD with hydrophobic potential (Goodford, 1985) superimposed. The phospho-peptide position is shown. Met1 docks into a significant hydrophobic pocket (yellow), which includes contributions from Trp414, Arg516, Phe535 and Ile521. This last residue is at the back of the site and ∼6 Å from Met1. Leu6 also docks against a hydrophobic surface (cyan). The molecular surface area buried on binding the phospho-peptide to PBD corresponds to 434 Å2 (phospho-peptide) and 486 Å2 (PBD), indicating complementarity of the two surfaces. The phospho-peptide is 63% buried, a fairly typical percentage burial for peptides binding to a surface site (e.g. Lowe et al., 2002).
The peptide binds between PB1 and PB2. The two polo boxes are related by a pseudo 2-fold axis (Figure 1A). Operation of the pseudo 2-fold symmetry generates an equivalent binding site. Structural comparison shows that although the pseudo-symmetry-related site is reasonably well conserved in structural terms, there are significant changes in contact residues (Figure 2B). Trp414 is conserved (Trp514). The position of Lys540 is taken by Arg441 and, although this residue could contribute to the phosphate recognition site, its side chain position would be >6 Å away from the phosphate group. A further major difference concerns the position of His538, which is taken by Ser439, and the shortened loop (residues 438–440 compared with 536–540) places the serine so that it partly blocks the phosphate site. Other residues at this site are also changed in character compared with the phospho-peptide-binding site. The positions of Arg516 and Phe535 are taken by Asp416 and Asn437, respectively, and these residues cannot contribute to the hydrophobic stabilization of peptide binding. The differences in the ends of the αB and αD helices result in no equivalent residues to His489, Leu490 and Leu491, so there are no contributions to the proline- and leucine-binding sites for the phospho-peptide. Overall, the cumulative effect of the changes results in a disrupted site for the phospho-group and, despite conservation of the tryptophan, many of the other interactions that are made at the recognition site with the peptide are lost at the pseudo-symmetry-related site.
The linker region between PB1 and PB2
The linker region between PB1 and PB2, residues 493–507, is present in the intact PBD in complex with the phospho-peptide. It forms an exposed loop (Figure 4B). The linker regions for the A and B subunits have essentially identical conformations, but the C subunit is slightly different due to lattice contacts. The B-factors of the atoms in this region are ∼10 Å2 higher than the average B-factor for the whole chain. From residues 492 to 495 and 494 to 497, there are two turns of the 310 helix. Thereafter the linker contains no secondary structural elements until residue 511, where the chain enters β7 of PB2. The linker region makes no direct contacts to the phospho-peptide, but residues prior to the start (Leu490 and Leu491) make important contacts and the linker residues 493–495 are shielded by the phospho-threonine. These interactions appear to provide sufficient stabilization of the linker to allow crystals for the intact PBD–phospho-peptide complex to form. In the native subtilisin-cleaved PBD crystals, the region of the missing linker is partly occupied by a lattice-related molecule.
Discussion
The crystal structure of the PBD shows that the two polo boxes (residues 411–489 and 511–592) have identical folds. These comprise a six-stranded antiparallel β-sheet that is shielded by one α-helix. Additional elements provided by the N-terminal region, residues 373–410, also appear important for stabilizing the fold. Despite the structural similarity (r.m.s.d. of 1.8 Å for 70 residues), the two polo boxes exhibit only 12% sequence identity.
The binding site for a cognate phospho-peptide is located at the interface of the two polo boxes. This result is consistent with studies with Plk1 in U2 OS cells (Seong et al., 2002) and fission yeast Plo1 (Reynolds and Ohkura, 2003) that have shown that both polo boxes are essential for cellular function and cell cycle-regulated localization. We observe a 1:1 complex between Plk1 PBD and the phospho-peptide, consistent with the results of Elia et al. (2003). The pseudo 2-fold symmetry between the PB1 and PB2 domains generates an equivalent site related by the pseudo 2-fold axis, but sequence differences between PB1 and PB2 result in disruption of phospho-peptide recognition at the second site. The PBD domain behaves as a rigid body and there are no conformational changes on binding peptide.
The construct used for the co-crystallization of the PBD with the phospho-peptide comprised residues 345–603. Elia et al. (2003) observed that the construct of 326–603 bound the phospho-peptide but that the construct 377–603 did not. There are no data on intermediate length constructs. The present work shows that the construct 345–603 is sufficient for binding. The PBD N-terminal region (first located residue 371) is on the opposite side of the molecule from the phospho-peptide-binding site (Figure 4B), and hence the N-terminal region is unlikely to affect phospho-peptide binding directly. Conformational instability may provide a possible explanation as to why the shorter construct 377–603 did not bind the phospho-peptide. The N- and C-terminal regions of the PBD are close together, as is commonly found for protein interaction domains (Pawson and Nash, 2003). The antiparallel helices αA and αD make several non-polar contacts (e.g. Met377 packs against Val587, and Val374 against Val591). It is possible that loss of the first turn of helix αA could weaken the packing of the two helices leading to exposure of a short non-polar stretch at the end of the αD helix and conformational instability.
The structure of the PBD–phospho-peptide (Met. Gln.Ser.pThr.Pro.Leu) complex indicates that the conformation of the PBD is crucially important for binding in order to allow the main chain interactions that characterize the short antiparallel β-sheet between peptide and PBD β1 strand. Four residues dominate the remaining interactions: Trp414, Leu490, His538 and Lys540. Trp414 is shielded by the three N-terminal residues of the peptide (Met1.Gln2.Ser3) and makes a hydrogen bond via its NE1 group through a water molecule to the threonine phosphate. Leu490 provides non-polar interactions to the C-terminal Pro5.Leu6 of the phospho-peptide. His538 and Lys540 interact with the threonine phosphate group. These four residues, Trp414, Leu490, His538 and Lys540, are conserved in the PBDs of yeast, Drosophila, Xenopus, Caenorhabditis elegans and mammalian Plk1s (see alignment in Leung et al., 2002). Exceptionally, these residues are not conserved in Trypansoma brucei Plk where the following differences are noted: Trp414Phe, Leu490Arg, His538Lys and Lys540Glu. These differences suggest that the PBD of T.brucei might recognize different biological epitopes from those recognized in mammals, and the PBD may therefore provide a selective target for drugs against the causative agent of sleeping sickness. Peptide binding to cognate domains can exhibit degenerate specificity. Moreover, biological target recognition epitopes may not be identical to those defined by in vitro binding studies that are designed to establish optimal binding sequences. Hence, while the results obtained with the single phospho-peptide provide definitive evidence on contact residues, further studies with other biological target peptides are necessary to see if the result is general.
In mammals, in addition to Plk1 and Sak, there are two other Plks, Plk2 (previously called Snk) and Plk3 (previously called Prk or Fnk). Plk2 and Plk3 were originally identified as immediate-early response genes in mitogenically stimulated cultured cells (reviewed in Glover et al., 1998), suggesting a role in the regulation from a quiescent state to a proliferative state that is different from that of Plk1. However, Plk3 activity levels increase as cells move from G1 to M phase, and Plk3 may play a role during later stages of the cell cycle (Ouyang et al., 1997, 1999). More recently, there is evidence that Plk3 plays a role as a stress response protein that becomes phosphorylated following DNA damage and associates with and phosphorylates Chk2 and p53 (el Bahassi el et al., 2002). The structural results show that in the core of the PBD, Plk1, Plk2 and Plk3 are very similar (39 and 36% sequence identity, respectively, to Plk1 over the PBD residues 410–593), but there is little similarity in the linker region comprising residues 367–410. Examination of the phospho-peptide-binding region in models of Plk2 and Plk3 shows identity of 75% of the 12 residues involved in binding (including the four key residues noted above) and conservative changes for most of the other residues. Where there are apparent significant differences in sequence (e.g. Asp416 to glycine in Plk3), we note that only the main chain atoms of this residue are involved in contacts to the phospho-peptide. Thus the structural studies suggest that all three Plks can bind the phospho-peptide. Their differing cellular functions might be explained by differences in expression, by differences in the linker region between the kinase and PBD, by differences in kinase specificity for substrates, or by other targeting regions.
The structure results are consistent with a wealth of mutational and functional data that exist in the literature and provide an explanation for many previous observations. Mutational studies have identified a number of polo box amino acids whose mutation results in impaired function. Trp414 was shown to be critical for function. A Trp414Phe mutant disrupted the capacity of Plk1 to complement the defect associated with a cdc5-1 temperature-sensitive mutation in budding yeast apparently by preventing Plk localization at the spindle poles and cytokinetic neck (Lee et al., 1998). Curiously, the equivalent mutant in budding yeast cdc5-1 still possessed some capacity to complement the temperature-sensitive mutant. However, the triple mutant with residues equivalent to Trp414Phe, Val415Ala and Leu427Ala was not able to restore growth to a detectable level (Song et al., 2000). Further studies with Plk1 in U-2 OS cells showed that the triple mutation impaired the ability of the PBD to localize at the centrosomes and mid-body (Seong et al., 2002). In fission yeast Plo1, a number of mutations have been shown to abolish mitotic arrest seen on overexpression of wild-type protein Plo1 but without preventing the untimely septation (Reynolds and Ohkura, 2003). The results suggest that some functions of Plo1 can be separated. Mutations that abolished mitotic arrest include residues equivalent in Plk1 to Trp414Phe, Leu490Ala, and a triple mutation: Asp537Ala, His538Ala, Thr539Ala. These results are consistent with the phospho-peptide binding studies. Trp414 is a key residue in recognition of the phospho-peptide, contributing to peptide localization, non-polar interactions and a contact through water to the phosphate. Val415 and Leu427 are not directly involved in peptide interactions. The role of these non-polar side chains appears to be in maintenance of structural stability. The Val415 side chain is in van der Waals contacts with Tyr417, Phe482 and Tyr485. Leu427, at the centre of PB1, is surrounded by Phe409, Val411, Tyr425, Cys428, Ile443 and Leu508. Leu490 plays a role in non-polar contacts to Pro5 and Leu6 of the peptide, while His538, in the triple mutant of Reynolds and Ohkura (2003), is one of the two basic groups that contacts the phosphate of the pThr in the peptide.
Membrane-permeable polo box peptides have been reported to inhibit cancer cell proliferation for MCF-7, Saos-2 and HeLa S3 cells (Yuan et al., 2002b). The peptide comprising residues 410–429 of the Plk1 PBD fused to an Antennapedia peptide was internalized into cells and found to inhibit the proliferation of tumour cell lines by inducing apoptosis. The triply mutated peptide with Trp414Phe, Val415Ala and Leu427Ala was much less effective. It is curious that the wild-type peptide is able to be effective. Residues 410–429 comprise the first two strands β1 and β2 of the PB1. They contain the critical residue Trp414 and the residues that contribute to the short stretch of antiparallel β-sheet of the peptide to the PBD, but they do not contain the phosphate recognition residues nor other residues that contribute to phospho-peptide binding (Table II). It could be that the fragment is able to fold so as to resemble part of the recognition site and that this is sufficient at high concentrations to provide a dominant-negative effect.
Elia et al. (2003) defined the phospho-peptide motif for binding to the Plk1 PBD from a proteomic screen based on a peptide library of phospho-peptides containing the sequence pThr.Pro. The optimal peptide (Kd = 280 nM) was shown to disrupt the binding of the Plk1 PBD to centrosomes, while the non-phospho-peptide showed no disruption. A direct interaction between the Plk1 PBD and Cdc25C, phosphorylated at Thr130 in the sequence Ser.Thr.Pro, was demonstrated. These results indicate that the phospho-peptide is present in known Plk1 substrates and is relevant for biological function of Plk1. A strong selection for serine (Ser3) at the –1 position (relative to the pThr position) was observed. In the structure of the phospho-peptide complex, the Ser3 side chain hydroxyl makes either a single polar contact to a water or contacts to two waters and the pThr4 residue (Table II). The serine site is restricted, suggesting that larger non-polar amino acids such as valine would be excluded because no polar contacts could be made. Alanine could be tolerated but would be unable to make the polar contacts. The results of Elia et al. (2003) demonstrated a clear preference for serine relative to threonine at this site. Model building, in which the serine is substituted by threonine with its side chain in the trans conformation, shows that a threonine residue would result in some close packing but appears not to be totally excluded. The preference for a phospho-group (pThr4) at site 0 is explained by the interactions made between the phosphate and the two basic groups, His538 and Lys540, and the water molecules. However, the model suggests no strong preference for a phospho-threonine compared with a phospho-serine, while the results of Elia et al. (2003) indicated a 7-fold preference for phospho-threonine. The WD40 domain of Cdc4 also shows a 6-fold preference for pThr over pSer, but the basis for this selection was not obvious from the structure (Orlicky et al., 2003). The methyl group of the threonine side chain imposes some conformational restriction on torsion angles that could help direct the phospho group to its cognate conformation. The preference for proline in the +1 site is explained by the non-polar contacts made by this side chain and the kink that it induces to facilitate these contacts. The results of Elia et al. (2003) showed that a proline was not absolutely required. The structure suggests that other residues could be accommodated here and there are no constraints from the contacts to the Plk1 PBD for a proline residue at this site. This is in contrast to the conformation of CDK2 where a clear geometrical requirement for proline in the substrate peptide was recognized from the structure of pCDK2–cyclin A in complex with a cognate peptide (Brown et al., 1999). As demonstrated in Figure 4D, Met1 in the –3 position and Leu6 at the +2 position contact hydrophobic pockets. The screening results of Elia et al. (2003) showed some preference for aliphatic and aromatic residues at these sites. The structural results provide an explanation for the specificity determinants of the phospho-peptide, which is derived from a natural target Cdc25C. There is need for further work to define the binding sites for the other interacting partners of Plk1 PBD in order to determine if they also contain similar specificity determinants.
The PBD appears to be a domain that is unique for Plks. A BLAST (Altschul et al., 1990) search revealed that the motif did not occur among other proteins. The PBD, therefore, is not a modular domain present in other proteins, and its function appears to be localization and intramolecular regulation of Plk kinase activity. This is in contrast to other phospho-peptide-binding domains such as SH2, 14-3-3, WW and WD40 that are ubiquitous and appear to be used by nature in a cassette-like fashion in which they are fused to other signalling regulatory molecules (Pawson and Nash, 2003). Phosphoserine/threonine-binding domains have been reviewed (Yaffe and Elia, 2001). The structures of these domains show a variety of motifs and ways in which phospho-peptides contact these motifs. These include an eight bladed β-propeller (WD40 from Cdc4; (Orlicky et al., 2003), a three-stranded antiparallel β-sheet (WW from Pin1; Verdecia et al., 2000), a nine α-helical assembly (14-3-3 ζ; Yaffe et al., 1997) and an 11-stranded antiparallel β-sheet (FHA1 from Rad53p; Durocher et al., 2000). The closest in fold to Plk1 PBD is the FHA1 domain, but the topology of the β-strands and the ways in which the phospho-peptide contacts the domain are quite different. In FHA1, the peptide is above the sheet and parallel to the plane of the β-sheets. In the Plk1 PBD, the peptide is antiparallel to an edge strand and makes main chain interactions with the strand. The five phosphoserine/threonine-binding domains exhibit phospho recognition sites that vary from apparently tight with at least three basic groups (WD40 and 14-3-3 domains) to intermediate with two or one basic group (PBD, WW and FHA1 domains). Specificity seems to be dominated by both the phospho group recognition and an additional site. In FHA1, there is high specificity for an aspartate in +3 position, while the Plk1 PBD has specificity for serine at –1 and a deep hydrophobic site at the –3 position. All domains, except FHA1, contain distinct hydrophobic sites for residues following the phospho group, with WD40 and WW domains providing tight interactions for proline immediately following the phospho residue, while 14-3-3 provides a recognition site for a cis proline in the +2 position from the phospho-serine. The proline +1 recognition site in PBD makes significant non-polar contacts, but these are not as dominant as those observed in the other domains. The two (or at least two) pronged recognition can provide specificity, but also allows plasticity for selection of different residues at other sites among different targets. All of the domains, with the exception of FHA1, utilize tryptophan at the binding site, but the roles of the tryptophan in each of the phospho-peptide recognitions are different.
The structure of the PBD opens up the possibility of targeted mutations to probe cellular function and the design of peptido-mimics that might inhibit the function of Plk in tumour cells. It is of interest that microinjection of anti-Plk1 antibodies severely impaired the ability of cells to divide (Lane and Nigg, 1996). While immortalized HeLa cells responded to Plk1 inhibition by mitotic catastrophe, non-immortalized Hs68 cells arrested primarily as single mono-nucleated cells in G2, presumably reflecting a more stringent requirement for Plk1 function at the G2/M transition. This differential response of tumour and normal cells to Plk1 inhibition indicates promise for selective targeting.
Materials and methods
Expression, purification and crystallization
Full-length Plk1 was expressed in Sf9 insect cells using standard baculovirus expression protocols and purified using a two-stage Ni-NTA affinity/gel filtration procedure. Subsequent analysis by gel electrophoresis after 2 weeks storage indicated proteolysis. A number of distinct species were characterized by N-terminal sequencing and approximate molecular weight. One such intermediate (∼30 kDa) showed an N-terminal sequence (KGLENPLPER) indicative of proteolytic cleavage at residue 345 between the Plk1 kinase and the PBD. The PBD of Plk1 encoding residues 345–603 was cloned into the pGEX-6P1 vector (Pharmacia) and expressed as a GST fusion protein in B834 (DE3) pLysS cells (Novagen). An overnight culture (5 ml) was inoculated into 500 ml of LB medium and grown for 2.5 h at 37°C until OD ∼0.6. Expression of GST–PBD was induced by 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were harvested after overnight incubation at 20°C. Seleno-methionine PBD was expressed by inoculating an overnight culture as above into 500 ml of SelenoMet medium base plus nutrient mix containing 40 µg/ml seleno-methionine (Molecular Dimensions Ltd).
GST fusion proteins were purified using glutathione–Sepharose beads (Pharmacia). The beads were washed using HBS buffer [10 mM HEPES pH 7.5, 200 mM NaCl, 3.4 mM EDTA and 0.01% (v/v) monothioglycerol] and the GST–PBD eluted with 20 mM glutathione (Sigma) in HBS buffer. The GST tag was cleaved by digestion with 1/50 (w/w) GST-3C precision protease. The mixture of GST and PBD was filtered through a Superdex SD200 gel filtration column (Pharmacia) and then passed through a second glutathione–Sepharose column. Typical yields were 25 mg of pure PBD and 8 mg of seleno-methionine PBD per 1 l of bacterial culture.
Crystallization using the sitting drop vapour diffusion method at 4°C was achieved from pure PBD (1 µl at 10 mg/ml) mixed with 1 µl of mother liquor [5–10% (v/v) PEG 4000, 0.1 M sodium citrate pH 6.0 and 0.1 M ammonium acetate]. Crystals formed under this condition were needle like and did not diffract. Limited proteolysis was performed in order to delete possible flexible surface-exposed loops that could inhibit crystal growth. Pure PBD was mixed with subtilisin [1/200 (w/w)] and incubated for 1 h at room temperature. The reaction was stopped with 1/1000 phenylmethylsulfonyl fluoride (PMSF). PBD (30 kDa) was digested to give two fragments of ∼10 and 12 kDa as indicated by SDS–PAGE. The resulting mixtures were filtered through a Superdex SD 200 gel filtration column. The two fragments co-migrated as a single complex. N-terminal amino acid sequencing (Tony Willis, MRC Immunochemistry Unit, Oxford, UK) identified the N-terminal sequences of the two fragments as 367GEVVDCHLSD and 495ANITPREGDE. The complex of the two fragments was crystallized under the same conditions as the uncut PBD domain. X-ray quality crystals were obtained after 1 week. The Se-Met PBD was treated with a similar protocol for subtilisin digestion and crystallization.
The peptide Met.Gln.Ser.pThr.Pro.Leu was synthesized by Dr Graham Bloomberg (University of Bristol, UK). To obtain crystals of PBD–peptide complex, undigested PBD (residues 345–603) was mixed with phospho-peptide in a molar ratio of 1:5. The intact PBD (10 mg/ml)–phospho-peptide complex was mixed with an equal volume of mother liquor (1–10% PEG 20000, 0.1 M MES pH 6.5) and crystallized using the sitting drop vapour diffusion method at room temperature with a 24-well plate. Crystals appeared after 2–3 days.
Data collection and structure solution
Data were collected from crystals of subtilisin-treated Se-Met PBD to a resolution of 3 Å at ESRF beamline ID29 and processed using MOSFLM (Leslie, 1992) (Table I). The structure was solved with single wavelength anomalous dispersion (SAD) phasing using SOLVE (Terwilliger and Berendzen, 1999). An initial model was built into the SOLVE/RESOLVE map using O (Jones et al., 1991). The model obtained from the SAD-phased map was then refined against the native data set collected at Elettra to 2.2 Å resolution using REFMAC (Murshudov et al., 1997) with iterative rebuilding in O and addition of water molecules with ARP/wARP (Lamzin and Wilson, 1993). The final structure comprises residues 372–593 with a break in electron density for residues 493–507 that includes the subtilisin cleavage site at residue 495.
Data from intact PBD co-crystallized with phospho-peptide were collected at ESRF beamline ID29 to a resolution of 2.3 Å. The structure was solved by molecular replacement using the refined model of subtilisin-treated apo-PBD as a search model with the program MOLREP (Vagin and Teplyakov, 2000). A solution was found containing three molecules within the asymmetric unit. The resulting electron density difference map showed clear density for both the bound phospho-peptide and the linker region, previously absent in the subtilisin-cleaved structure. The structure was refined as above. Analysis of both the apo native PBD and PBD–phospho-peptide complex structures with PROCHECK showed 89% of residues in the most favoured main chain conformation, 10% in the additionally allowed region and 1% in the generously allowed region. Coordinates have been deposited in the Protein Data Bank ID numbers 1Q4O (apo native PBD) and 1Q4K (PBD/phospho-peptide complex).
Acknowledgments
Acknowledgements
We wish to thank the beamline scientists at Elettra, Trieste and ESRF, Grenoble for their excellent support. This work has been supported by the MRC and the Max-Planck Society. K.-Y.C. is supported by the Croucher Foundation.
References
- Alexandru G., Uhlmann,F., Mechtler,K., Poupart,M.-A. and Nasmyth,K. (2001) Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast. Cell, 105, 459–472. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Boltes I., Czapinska,H., Kahnert,A., von Bulow,R., Dierks,T., Schmidt,B., von Figura,K., Kertesz,M.A. and Uson,I. (2001) 1.3 Å structure of arylsulfatase from Pseudomonas aeruginosa establishes the catalytic mechanism of sulfate ester cleavage in the sulfatase family. Structure (Camb.), 9, 483–491. [DOI] [PubMed] [Google Scholar]
- Brown N.R., Noble,M.E.M., Endicott,J.A. and Johnson,L.N. (1999) The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nature Cell Biol., 1, 438–443. [DOI] [PubMed] [Google Scholar]
- Casenghi M., Meraldi,P., Weinhart,U., Duncan,P.I., Korner,R. and Nigg,E.A. (2003) Polo-like kinase 1 regulates Nlp, a centrosome protein involved in microtubule nucleation. Dev. Cell, 5, 113–125. [DOI] [PubMed] [Google Scholar]
- Donaldson M.M., Tavares,A.A., Hagan,I.M., Nigg,E.A. and Glover,D.M. (2001) The mitotic roles of Polo-like kinase. J. Cell Sci., 114, 2357–2358. [DOI] [PubMed] [Google Scholar]
- Duncan P.I., Pollet,N., Niehrs,C. and Nigg,E.A. (2001) Cloning and characterization of Plx2 and Plx3, two additional Polo-like kinases from Xenopus laevis. Exp. Cell Res., 270, 78–87. [DOI] [PubMed] [Google Scholar]
- Durocher D., Taylor,I.A., Sarbassova,D., Haire,L.F., Westcott,S.L., Jackson,S.P., Smerdon,S.J. and Yaffe,M.B. (2000) The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell, 6, 1169–1182. [DOI] [PubMed] [Google Scholar]
- el Bahassi M., Conn,C.W., Myer,D.L., Hennigan,R.F., McGowan,C.H., Sanchez,Y. and Stambrook,P.J. (2002) Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene, 21, 6633–6640. [DOI] [PubMed] [Google Scholar]
- Elia A.E., Cantley,L.C. and Yaffe,M.B. (2003) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science, 299, 1228–1231. [DOI] [PubMed] [Google Scholar]
- Glover D.M., Hagan,I.M. and Tavares,A.A.M. (1998) Polo-like kinases: a team that plays throughout mitosis. Genes Dev., 12, 3777–3787. [DOI] [PubMed] [Google Scholar]
- Goodford P.J. (1985) A computational procedure for determing energetically favourable binding sites on biologically important macromolecules. J. Med. Chem., 28, 849–857. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1995) Dali: a network tool for protein structure comparison. Trends Biochem. Sci., 20, 478–480. [DOI] [PubMed] [Google Scholar]
- Jackman M., Lindon,C., Nigg,E.A. and Pines,J. (2003) Active cyclin B1–Cdk1 first appears on centrosomes in prophase. Nature Cell Biol., 5, 143–148. [DOI] [PubMed] [Google Scholar]
- Jang Y.J., Lin,C.Y., Ma,S. and Erikson,R.L. (2002a) Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc. Natl Acad. Sci. USA, 99, 1984–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y.J., Ma,S., Terada,Y. and Erikson,R.L. (2002b) Phosphorylation of threonine 210 and the role of serine 137 in the regulation of mammalian polo-like kinase. J. Biol. Chem., 277, 44115–44120. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved method for building models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kelm O., Wind,M., Lehmann,W.D. and Nigg,E.A. (2002) Cell cycle-regulated phosphorylation of the Xenopus polo-like kinase Plx1. J. Biol. Chem., 277, 25247–25256. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J. and Jones,T.A. (1997) Detecting folding motifs and similarities in protein structures. Methods Enzymol., 277, 525–545. [DOI] [PubMed] [Google Scholar]
- Kumagai A. and Dunphy,W.G. (1996) Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopus egg extracts. Science, 273, 1377–1380. [DOI] [PubMed] [Google Scholar]
- Lamzin V.S. and Wilson,K.S. (1993) Automated refinement of protein models. Acta Crystallogr. D, 49, 129–147. [DOI] [PubMed] [Google Scholar]
- Lane H.A. and Nigg,E.A. (1996) Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J. Cell Biol., 135, 1701–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.S. and Erikson,R.L. (1997) Plk is a functional homolog of Saccharomyces cerevisiae Cdc5 and elevated Plk activity induces multiple septation structures. Mol. Cell. Biol., 17, 3408–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.S., Grenfell,T.Z., Yarm,F.R. and Erikson,R.L. (1998) Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc. Natl Acad. Sci. USA, 95, 9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A.G.W. (1992) MOSFLM. In Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography. Daresbury Laboratory, Warrington, Vol. 26. [Google Scholar]
- Leung G.C., Hudson,J.W., Kozarova,A., Davidson,A., Dennis,J.W. and Sicheri,F. (2002) The Sak polo-box comprises a structural domain sufficient for mitotic subcellular localization. Nature Struct. Biol., 9, 719–724. [DOI] [PubMed] [Google Scholar]
- Lin H.R., Ting,N.S., Qin,J. and Lee,W.H. (2003) M-phase specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with dissociation of p300/CBP-associated factor, P/CAF. J. Biol. Chem., 278, 35979–35987. [DOI] [PubMed] [Google Scholar]
- Lowe E.D., Tews,I., Cheng,K.-Y., Brown,N.R., Gul,S., Noble,M.E.M., Gamblin,S.J. and Johnson,L.N. (2002) Specificity determinants of recruitment peptides bound to phospho-CDK2/cyclin A. Biochemistry, 41, 15625–15634. [DOI] [PubMed] [Google Scholar]
- May K.M., Reynolds,N., Cullen,C.F., Yanagida,M. and Ohkura,H. (2002) Polo boxes and Cut23 (Apc8) mediate an interaction between polo kinase and the anaphase-promoting complex for fission yeast mitosis. J. Cell Biol., 156, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundt K.E., Golsteyn,R.M., Lane,H.A. and Nigg,E.A. (1997) On the regulation and function of human Polo-like kinase 1 (Plk1): effects of overexpression on cell cycle progression. Biochem. Biophys. Res. Commun., 239, 377–385. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagen,A.A. and Dodson,E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nakajima H., Toyoshima-Morimoto,F., Taniguchi,E. and Nishida,E. (2003) Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem., 278, 25277–25280. [DOI] [PubMed] [Google Scholar]
- Neef R., Preisinger,C., Sutcliffe,J., Kopajitch,R., Nigg,E.A., Mayer,T.U. and Barr,F.A. (2003) Phosphorylation of MKlp2 by Plk1 is required for cytokinesis. J. Cell Biol., 162, 863–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg E. (1998) Polo-like kinases: positive regulators of cell division from start to finish. Curr. Opin. Cell Biol., 10, 776–783. [DOI] [PubMed] [Google Scholar]
- Orlicky S., Tang,X., Willems,A., Tyers,M. and Sicheri,F. (2003) Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell, 112, 243–256. [DOI] [PubMed] [Google Scholar]
- Ouyang B., Pan,H., Lu,L., Li,J., Stambrook,P., Li,B. and Dai,W. (1997) Human Prk is a conserved protein serine/threonine kinase involved in regulating M phase functions. J. Biol. Chem., 272, 28646–28651. [DOI] [PubMed] [Google Scholar]
- Ouyang B., Wang,Y. and Wei,D. (1999) Caenorhabditis elegans contains structural homologs of human prk and plk. DNA Seq., 10, 109–113. [DOI] [PubMed] [Google Scholar]
- Pawson T. and Nash,P. (2003) Assembly of cell regulatory systems through protein interaction domains. Science, 300, 445–452. [DOI] [PubMed] [Google Scholar]
- Qian Y.-W., Erikson,E. and Maller,J.L. (1999) Mitotic events of a constitutively active mutant of the Xenopus Polo-like kinase Plx1. Mol. Cell. Biol., 19, 8625–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds N. and Ohkura,H. (2003) Polo boxes form a single functional domain that mediates interactions with multiple proteins in fission yeast polo kinase. J. Cell Sci., 116, 1377–1387. [DOI] [PubMed] [Google Scholar]
- Seong Y.S., Kamijo,K., Lee,J.S., Fernandez,E., Kuriyama,R., Miki,T. and Lee,K.S. (2002) A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J. Biol. Chem., 277, 32282–32293. [DOI] [PubMed] [Google Scholar]
- Song S. and Lee,K.S. (2001) A novel function of Saccharomyces cerevisiae CDC5 in cytokinesis. J. Cell Biol., 152, 451–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Grenfell,T.Z., Garfield,S., Eroikson,R.L. and Lee,K.S. (2000) Essential function of the Polo box of Cdc5 in subcellular localization and induction of cytokinetic structures. Mol. Cell. Biol., 20, 286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara I., Vorlaufer,E., Stukenberg,P.T., Kelm,O., Redemann,N., Nigg,E.A. and Peters,J.M. (2002) The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell, 9, 515–525. [DOI] [PubMed] [Google Scholar]
- Sunkel C.E. and Glover,D.M. (1988) polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell Sci., 89, 25–38. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima-Morimoto F., Tanguchi,E., Shinya,N., Iwanatsu,A. and Nishida,E. (2001) Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature, 410, 215–220. [DOI] [PubMed] [Google Scholar]
- Toyoshima-Morimoto F., Taniguchi,E. and Nishida,E. (2002) Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep., 3, 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov,A. (2000) An approach to multi-copy search in molecular replacement. Acta Crystallogr. D, 56, 1622–1624. [DOI] [PubMed] [Google Scholar]
- Verdecia M.A., Bowman,M.E., Lu,K.P., Hunter,T. and Noel,J.P. (2000) Structural basis for phosphoserine–proline recognition by group IV WW domains. Nature Struct. Biol., 7, 639–643. [DOI] [PubMed] [Google Scholar]
- Yaffe M.B. and Elia,A.E. (2001) Phosphoserine/threonine-binding domains. Curr. Opin. Cell Biol., 13, 131–138. [DOI] [PubMed] [Google Scholar]
- Yaffe M.B., Rittinger,K., Volinia,S., Caron,P.R., Aitken,A., Leffers,H., Gamblin,S.J., Smerdon,S.J. and Cantley,L.C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell, 91, 961–971. [DOI] [PubMed] [Google Scholar]
- Yuan J., Eckerdt,F., Bereiter-Hahn,J., Kurunci-Csacsko,E., Kaufmann,M. and Strebhardt,K. (2002a) Cooperative phosphorylation including the activity of polo-like kinase 1 regulates the subcellular localization of cyclin B1. Oncogene, 21, 8282–8292. [DOI] [PubMed] [Google Scholar]
- Yuan J., Kramer,A., Eckerdt,F., Kaufmann,M. and Strebhardt,K. (2002b) Efficient internalization of the polo-box of polo-like kinase 1 fused to an Antennapedia peptide results in inhibition of cancer cell proliferation. Cancer Res., 62, 4186–4190. [PubMed] [Google Scholar]
- Zhou T., Aumais,J.P., Liu,X., Yu-Lee,L.Y. and Erikson,R.L. (2003) A role for Plk1 phosphorylation of NudC in cytokinesis. Dev. Cell, 5, 127–138. [DOI] [PubMed] [Google Scholar]