

Abstract
Recent studies indicate a role of chymase in the regulation of angiotensin II (AngII) formation in cardiovascular and renal tissues. We investigated a possible contribution of chymase to AngII formation and to renal fibrosis in unilateral ureteral obstruction (UUO). Eight-week-old Syrian hamsters were subjected to UUO and treated with vehicle, the specific chymase inhibitor (CI) 4-[1-(4-methyl-benzo[b]thiophen-3-ylmethyl)-1H-benzimidazol-2-ylsulfanyl]-butyric acid (50 mg/kg, twice a day, p.o.), or the selective AT1-receptor blocker olmesartan (10 mg/kg per day, p.o,) for 14 days. UUO-induced renal interstitial fibrosis was associated with increases in renal mRNA levels of α-smooth muscle actin (SMA), type I collagen, and transforming growth factor (TGF)-β. The UUO hamsters showed markedly higher AngII contents and increased AT1-receptor mRNA level in the obstructed kidney than sham-operated ones. In contrast, angiotensin-converting enzyme (ACE) protein expression was significantly lower in UUO hamsters. In UUO hamsters, treatment with CI or olmesartan significantly decreased AngII levels in renal tissue and mRNA levels of α-SMA, type I collagen, and TGF-β and ameliorated tubulointerstitial injury. On the other hand, neither CI nor olmesartan changed systolic blood pressure, renal ACE, and AT1-receptor protein levels. These data suggest that chymase-dependent intrarenal AngII formation contributes to the pathogenesis of interstitial fibrosis in obstructed kidneys of hamsters.
Keywords: chymase, angiotensin II, angiotensin-converting enzyme, unilateral ureteral obstruction
Introduction
Congenital obstructive nephropathy is a leading cause of chronic renal failure in children and adolescents, nearly 50% of whom progress to end-stage renal disease (1). Chronic ureteral obstruction also leads to tubulointerstitial fibrosis in adults (2). Although multiple mechanisms contribute to the progression of renal injury induced by chronic ureteral obstruction, a potential role of the renin-angiotensin system (RAS) has been suggested. El-Dahr et al. (3) showed that angiotensin II (AngII) contents in the kidney were significantly increased in rats with chronic unilateral ureteral obstruction (UUO). In these animals, gene expression of the AngII AT1 receptor and the binding affinity of AngII with AT1 receptor in the kidney were also increased (4). Treatment with AngII AT1–receptor blockers (5, 6) or angiotensin-converting enzyme (ACE) inhibitors (7, 8) is known to ameliorate UUO-induced renal injury in mice and rats. Other studies have shown that UUO-induced renal fibrosis is reduced in mice lacking angiotensinogen (9) or the AT1a receptor (10). However, the precise mechanisms responsible for the augmentation of intrarenal AngII are unclear.
A growing body of evidence indicates that the chymase-dependent (ACE-independent) pathway plays a critical role in forming AngII from AngI in cardiovascular and renal tissues (11, 12). Murakami et al. (13) showed that 20% of the AngII in the renal cortex was produced by chymase in dogs. Furthermore, it has been demonstrated that intraarterial infusion of the chymase inhibitor (CI) chemostatin significantly decreased intrarenal AngII contents in the clipped kidney of two-kidney, one-clip dogs (14). Importantly, human studies showed that an increase in chymase expression in mast cells is associated with the severity of interstitial fibrosis in kidneys (15). Collectively, these data show the potential contribution of chymase-dependent intrarenal AngII formation in the progression of renal injury.
In the present study, we investigated the possible role of chymase-dependent AngII formation in the pathogenesis of UUO-induced renal injury. Because the chymase-dependent AngII generating potency in humans is similar to that of hamster (12), we conducted studies in hamsters to examine the effects of a newly developed, orally active specific CI, 4-[1-(4-Methyl-benzo[b] thiophen-3-ylmethyl)-1H-benzimidazol-2-ylsulfanyl]-butyric acid, on intrarenal AngII levels and interstitial injury induced by UUO. We hereby demonstrated that treatment with CI prevented the development of ureteral obstruction–induced renal fibrosis with potent inhibition of renal AngII production.
Materials and Methods
Animal preparation
All experimental procedures were performed under the guidelines for the care and use of animals as established by the Kagawa University Medical School. Male Syrian hamsters (Japan SLC, Shizuoka), aged 6 – 7 weeks and weighing 95 – 110 g, underwent left proximal UUO or sham operations under sodium pentobarbital anesthesia (50 mg/kg, i.p.; Dainippon Pharmaceutical, Osaka). Hamsters were randomly treated for 2 weeks with one of the following combinations: sham-operation + vehicle (carboxymethyl cellulose sodium salt; Nacalai Tesque, Kyoto) (n = 7); UUO + vehicle (n = 10); UUO + CI (50 mg/kg, twice a day, p.o.; Teijin Pharma, Tokyo) (n=10); or UUO + an AT1-receptor blocker, olmesartan (10 mg/kg per day, p.o.; Daiichi-Sankyo, Tokyo) (n = 10). Preliminary pharmacokinetic data showed that twice-daily oral administration of CI at 50 mg/kg (100 mg/kg per day) was appropriate to obtain a sufficient plasma-unbound concentration (above 20 nM) in hamsters (K1 value for hamster chymase is 9.85nM, our unpublished data). Thus, the dose of CI used in the present study was sufficient to inhibit hamster chymase activity in vivo.
At the end of the 2-week treatment, blood and kidney samples were obtained in a chilled tube after decapitation. Half of the obstructed left kidney was collected for determination of AngII level and part of the left kidney was fixed in 10% formalin for histological examination. The AngII level in the kidney was measured by radio-immunoassay as described previously (16). The remaining tissues were prepared for protein or mRNA extraction (17). Blood urea nitrogen (BUN) was measured by using a commercially assay kit (BUN-test-Wako; Wako Pure Chemical, Osaka).
Systolic blood pressure and heart rate
Because it is impossible to perform tail-cuff blood pressure measurement in hamsters, conscious arterial blood pressure was measured directly from the femoral artery after the 2-week treatment period in a separate group of hamsters (n = 5 for each group). Under sodium pentobarbital anesthesia (50 mg/kg, i.p.), a polyethylene catheter (PF-10 connected to PE-50; Becton Dickinson, Franklin Lakes, NJ, USA) was placed in the femoral artery at a week before starting the treatment. The catheter was filled with heparinized saline (100 IU/mL), and exteriorized through a cutaneous tunnel at the back of the neck after confirmation of their tip locations by pressure tracings. The femoral arterial catheter was connected to a pressure transducer (model 361; NEC-San-ei, Tokyo). Heart rate was triggered by the blood pressure pulse wave form.
Histological examination
Kidneys were fixed with 10% formalin (pH 7.4), embedded in paraffin, and sectioned into 3-μm slices. The severity of tubulointerstitial fibrosis was assessed by automatic image analysis of the renal medulla occupied by interstitial tissue staining positively for collagen in Azan-stained sections (18, 19). Sections were also used for α-smooth muscle actin (SMA) immunohistochemistry with a commercially available antibody against α-SMA (Sigma, St. Louis, MO, USA) (19, 20). Enhanced expression of α-SMA is a marker for interstitial phenotypic changes in renal fibrosis, and the activated interstitial cells are known as myofibroblasts (2). Immunohistochemistry was performed using a robotic system (Dako Autostainer, Carpinteria, CA, USA) (19). For each microscopic field, the Azan staining- or α-SMA–positive area was automatically calculated by the software and the affected area was divided by the total area of the microscopic field, as previously described (19). Ten consecutive microscopic fields were examined for each hamster and the averaged percentages of positive lesions were obtained for each animal.
Western blotting
Protein levels of ACE and AT1 receptor in renal tissues were analyzed by western blotting. Briefly, protein samples were separated by 8% or 10% polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane. The membrane was incubated with a polyclonal anti-ACE antibody (1:400; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) or a polyclonal anti–AT1 receptor antibody (1:1,000; Santa Cruz Biotechnology, Inc.), followed by incubation with a horseradish peroxidase–conjugated secondary antibody (for ACE: donkey anti-goat IgG, 1:1,000; Santa Cruz Biotechnology Inc.; for AT1 receptor: goat anti-rabbit IgG, 1:1,000; Cell Signaling Technology, Inc., Beverly, MA, USA). Membranes were reprobed with an antibody against β-actin (Sigma). All values were normalized by arbitrarily setting the integrated densitometric values of sham + vehicle hamsters to 1.0 after normalization to β-actin.
Reverse transcription–PCR
The mRNA levels of 18S ribosomal RNA (18S), α-SMA, type I collagen, and transforming growth factor (TGF)-β were analyzed by Real-time PCR using a GeneAmp® 5700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA), Amplifications were earned out in a total volume, of 20 μL containing each primer at 0.1 μM, 10 μL SYBR® Green PCR Master Mix (Applied Biosystems), and 2 μL of diluted cDNA. The PCR conditions were: 2 min at 50°C and 10 min at 95 °C, followed by 40 cycles with denaturing for 15 s at 95°C and annealing and extension for 1 min at 60°C. The relative quantitative values of the transcripts were obtained using the comparative Ct method, as described elsewhere: Relative Quantitation of Gene Expression [User Bulletin No. 2], from Applied Biosystems. All data are expressed as the relative differences between sham operation + vehicle hamsters and UUO hamsters, UUO + CI or UUO + olmesartan after normalization to 18S expression. Table 1 presents the sequences of the primers used for the respective genes.
Table 1.
Primer sequences
| 18S | F: 5′-TAAGTCCCTGCCCTTTGTACACA-3′ |
| R: 5′-GATCCGAGGGCCTCACTAAAC-3′ | |
| Collagen I | F: 5′-CGAGGGCAACAGTCGCTTTA-3′ |
| R: 5′-CCCAACTTCCGGTGTGACTC-3′ | |
| α-SMA | F: 5′-CACCGACTACCTCATGAAGATCCT-3′ |
| R: 5′-CTCACGCTCAGCAGTAGTCACAA-3′ | |
| TGF-β | F: 5′-GCCCAGCATCTGCAAAGCT-3′ |
| R: 5′-GTCCTTGCGGAAGTCAATGTACAG-3′ | |
Detailed information of PCR conditions are described in the Methods section.
Statistical analyses
Values are presented as means ± S.E.M. Statistical comparisons of differences were performed using one-way analysis of variance for repeated measures combined with the Newman-Keuls post hoc test. Values of P<0.05 were considered statistically significant.
Results
Systolic blood pressure and heart rate
Blood pressure and heart rate after the 2-week treatment were measured directly from the femoral artery (Table 2). There were no differences in systolic blood pressure, mean blood pressure, diastolic blood pressure, or heart rate between sham-operated and UUO hamsters. Treatment with CI or olmesartan for 2 weeks had no significant effects on systolic blood pressure or heart rate compared with vehicle-treated UUO hamsters.
Table 2.
Effects of CI (100 mg/kg per day) and olmesartan (10 mg/kg per day) on systolic blood pressure (SBP), diastolic blood pressure DBP), and heart rate (HR) in UUO hamsters
| Sham + Vehicle (n = 5) | UUO + Vehicle (n = 5) | UUO + CI (n = 5) | UUO + Olmesartan (n = 5) | |
|---|---|---|---|---|
| SBP (mmHg) | 114 ± 7 | 111 ± 7 | 118 ± 4 | 102 ± 7 |
| DBP (mmHg) | 77 ± 4 | 78 ± 5 | 77 ± 3 | 72 ± 6 |
| HR (beats/min) | 383 ± 14 | 369 ± 11 | 409 ± 17 | 372 ± 22 |
Data are means ± S.E.M.
Body and kidney weights and blood urea nitrogen
Body and kidney weights after the 2-week treatment are summarized in Table 3. All UUO groups had lower body weight compared with the sham group. Obstructed left kidney weight and left kidney/body weight ratio were lower in vehicle-treated UUO hamsters than sham-operated hamsters. In contrast, the contralateral right kidney weight and right kidney/body weight ratio were higher in vehicle-treated UUO hamsters than sham-operated hamsters, possibly for compensation of dysfunctional obstructed kidney. In UUO hamsters, treatment with CI or olmesartan tended to increase the left kidney weight and left kidney/body weight ratio; however, these changes were not statistically significant (Table 3). Neither treatment with CI nor olmesartan affected right kidney weight and right kidney/body weight ratio. UUO increased the BUN in either vehicle-treated (22.8 ± 1.0 mg/dL) or CI- or olmesartan-treated (CI: 22.5 ± 1.2 mg/dL, olmesartan: 21.9 ± 1.0 mg/dL) hamster compared with that in sham-operated hamsters (14.6 ± 0.7 mg/dL), and there was no difference in the BUN between the UUO hamsters.
Table 3.
Effects of CI (100 mg/kg per day) and olmesartan (10 mg/kg per day) on body weight (BW), left (obstructed) and right (contralateral) kidney weight (KW), and KW/BW ratio in UUO hamsters
| Sham + Vehicle (n=7) | UUO + Vehicle (n=10) | UUO + CI (n=10) | UUO + Olmesartan (n=10) | |
|---|---|---|---|---|
| BW (g) | 135 ±3 | 121 ± 3* | 119 ± 3* | 122 ± 2* |
| Left KW (g) | 0.48 ± 0.01 | 0.34 ± 0.02* | 0.36 ± 0.02* | 0.35 ± 0.05* |
| Left KW/BW Ratio (%) | 0.36 ± 001 | 0.28 ± 0.02* | 0.31 ±0.02* | 0.29 ± 0.04* |
| Right KW (g) | 0.46 ± 0.01 | 0.65 ± 0.03* | 0.66 ± 0.01* | 0.64 ± 0.02* |
| Right KW/BW Ratio (%) | 0.34 ± 0.01 | 0.54 ± 0.03* | 0.56 ± 0.02* | 0.52 ± 0.01* |
P<0.05: vs. sham + vehicle.
Histological findings
The interstitial histological findings with Azan staining of obstructed kidney are shown in Fig. 1A. UUO led to a markedly increased Azan staining-positive area in the medullary interstitium in comparison with sham operation. Treatment with CI or olmesartan significantly attenuated the UUO-induced increase in the Azan staining-positive area.
Fig. 1.
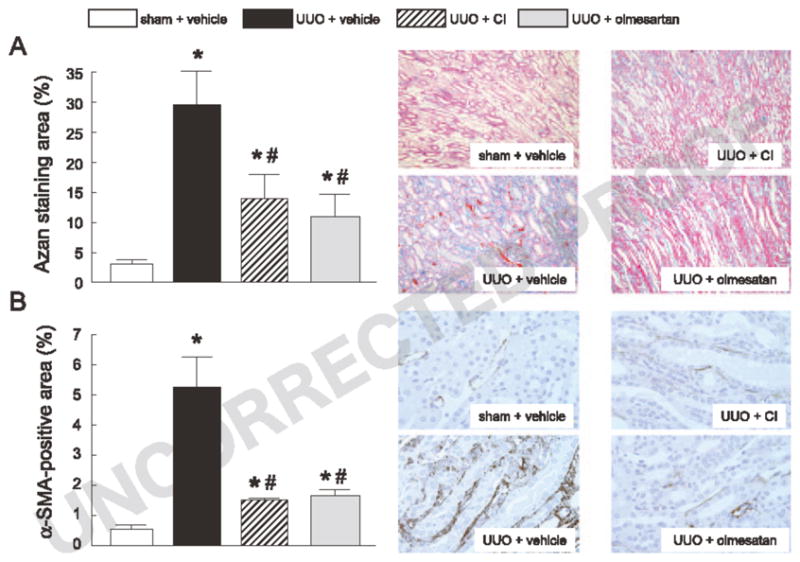
Azan staining (A) and α-SMA immunohistochemistry (B) in the different groups (original magnification: ×100 and ×200, respectively). The Azan staining- or α-SMA–positive area was calculated for each microscopic field, and the affected area was divided by the total area of the microscopic field. Ten consecutive microscopic fields were examined for each hamster, and the averaged percentages for the respective positive lesions were obtained for each animal. *P<0.05: vs. sham + vehicle and *P<0.05: vs. UUO + vehicle.
Immunohistochemistry with an α-SMA antibody of the obstructed kidney is shown in Fig. IB. In sham-operated hamsters, α-SMA was observed only on the blood vessel walls. However, the vehicle-treated UUO group had markedly increased α-SMA staining in the medullary interstitium. Treatment with CI or olmesartan attenuated, to a similar level, the UUO-induced increase in the α-SMA-positive area.
Ang II contents and protein expression of ACE and AT1 receptor
Vehicle-treated UUO hamsters showed markedly increased AngII content in the obstructed kidney (168 ± 7 vs. 1156 ± 154 fmol/g, Fig. 2A) compared with the sham-operated hamsters. In contrast, ACE protein expression was significantly lower in the obstructed kidney of vehicle-treated UUO hamsters (51 ± 9% of sham, Fig. 2B). UUO induced a 2.5 ± 0.1–fold higher renal AT1-receptor protein expression compared with sham operation (Fig. 2C). Treatment with CI or olmesartan attenuated the UUO-induced increases in renal AngII levels (530 ± 100 and 488 ± 90 fmol/g, respectively). However, neither CI nor olmesartan altered ACE or AT1 receptor protein levels in the kidney of UUO hamsters.
Fig. 2.
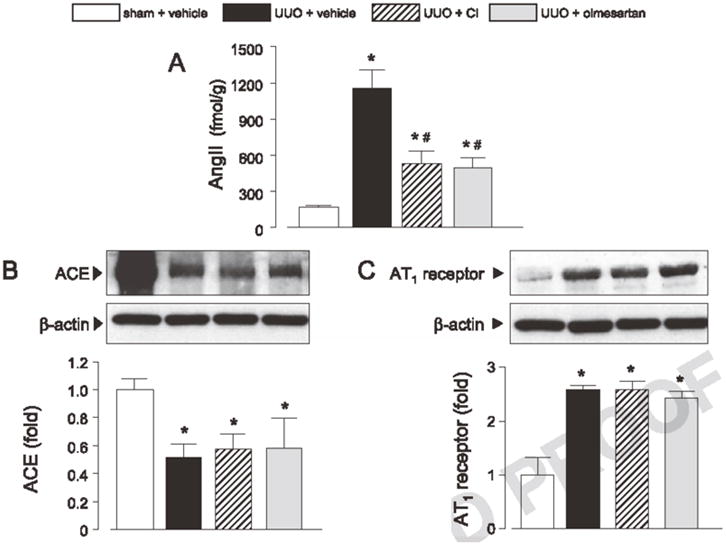
AngII contents (A) and protein levels of ACE (B) and AT1 receptor (C) are shown. All values for ACE and AT1 receptor protein levels were normalized by arbitrarily setting the integrated densitometric values of sham + vehicle to 1.0 after normalization to β-actin. *P<0.05: vs. sham + vehicle and *P<0.05: vs. UUO + vehicle.
Gene expression of α-SMA, TGF-β and type I collagen in renal tissues
The gene expression of α-SMA, TGF-β and type I collagen in the obstructed kidney are represented in Fig. 3. Compared with the sham-operated hamsters, UUO increased the mRNA levels of α-SMA, type I collagen, and TGF-β by 9.6 ± 0.2–, 25.8 ± 4.1–, and 3.2 ± 0.6–fold, respectively. Treatment with CI or olmesartan significantly decreased the mRNA levels of α-SMA (−47 ± 5% and −48 ± 4%, respectively), type I collagen (−39 ± 3% and −38 ± 4%, respectively), and TGF-β (−34 ± 5% and −31 ± 5%, respectively).
Fig. 3.
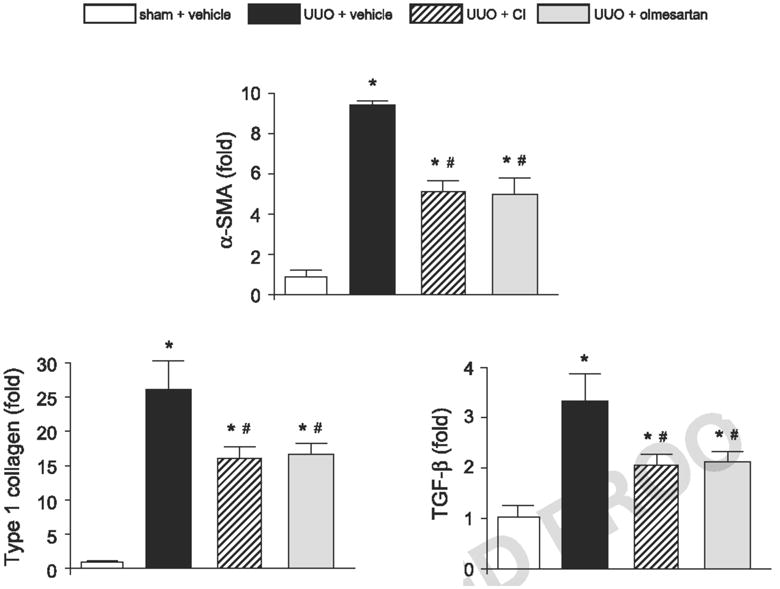
α-SMA, type I collagen, and TGF-β mRNA levels in renal tissues. Data are expressed as the relative differences in UUO, UUO + CI, or UUO + olmesartan compared with sham + vehicle after normalization to the expression of 18S. *P<0.05: vs. sham + vehicle and #P<0.05: vs. UUO + vehicle.
Discussion
Chronic UUO is a well established model of experimental renal injury characterized by interstitial fibrosis (21), and renal RAS has been implicated in the development of renal interstitial fibrosis (22, 23). In fact, the present study showed that UUO-induced interstitial fibrosis is associated with an increase in intrarenal AngII level and AT1-receptor expression in hamsters, which is consistent with a previous finding that UUO for 5 weeks increases kidney AngII contents in young rats (3). Furthermore, in the present study, the AT1-receptor blocker significantly alleviated the UUO-induced renal injury. Collectively, our data using hamsters support the hypothesis proposed in earlier studies in mice and rats (3 – 10) that RAS activation plays a critical role in the pathogenesis of UUO-induced renal injury. However, despite considerable evidence for the involvement of renal RAS in UUO-induced injury, the effect of ACE on UUO-induced injury is still controversial. The improvement of renal fibrosis was obtained by 5- or 10-day treatment with an ACE inhibitor (enalapril) in UUO animals (7, 8). However, Turan et al. (24) reported that treatment with enalapril for 14 days has no effect on the renal injury induced by UUO. This discrepancy may depend on the experimental conditions, such as duration of treatment and/or doses of ACE inhibitors. However, it is also possible that an alternative ACE-independent RAS activating pathway contributes to the pathogenesis of UUO-induced renal injury. Chymase, a chymotrypsin-like serine protease, is synthesized in mast cells, endothelial cells, and mesenchymal cells; it is secreted directly into the interstitium and contributes for up to 60% – 80% of AngII synthesis in the human heart (25, 26). Chymase activity is inactivated in blood immediately after being released, indicating that chymase has enzymatic activity only in tissues (11, 17). The high AngII-generating potency of chymase is well-documented in humans, monkeys, dogs, and hamsters, but is much higher than that in mice and rats (12,17, 27). In the present study, we observed a significant augmentation of renal AngII level and a reduction of renal ACE protein expression in the obstructed kidney of hamsters. Furthermore, treatment with a specific CI significantly decreased the kidney AngII contents and ameliorated the interstitial fibrosis, but had no effects on arterial blood pressure in hamsters subjected to chronic UUO. These data suggest that chymase-dependent intrarenal AngII formation rather than ACE-dependent AngII formation contributes to the pathogenesis of UUO-induced renal fibrosis in hamsters.
We observed similar beneficial effect against UUO-induced renal injury and increase in renal AngII content either by CI or olmesartan. However, the mechanism by which each drug suppressed the UUO-induced renal AngII content would be different. We and another group have previously shown that AngII that is filtered from glomeruli is internalized into tubular cells, especially proximal tubular cells, via the AT1 receptor-mediated endocytosis (28 – 30). The accumulation of internalized AngII stimulates the transcription of angiotensinogen that can induce further increase in AngII content in the kidney (28–30). Thus, it could be speculated that the decrease in renal AngII level by olmesartan in the present study results from the prevention of AT1 receptor-mediated AngII internalization and subsequent accumulation of AngII in the kidney.
El-Dahr et al. (3) showed that intrarenal ACE activity is increased by chronic UUO in young rats. However, we observed a significantly reduced expression of renal ACE protein in UUO hamsters. The reasons for these discrepancies are not clear; however, there may be differences between species and in the enzymes responsible for AngII generation in the kidney. It has been suggested that the AngII-generating potency of chymase in rats is much lower than that in either hamsters or humans (11, 12, 26). Because techniques for measuring the chymase activity in the kidney of hamsters are limited, direct evidence of renal chymase activity is not shown in the present study. However, treatment with a specific CI and an AT1-receptor blocker showed similar protective effects against UUO-induced renal injury in hamsters, effects that were associated with decreased kidney AngII levels. Thus, it seems likely that the chymase-dependent pathway contributes substantially to AngII production and renal injury in hamsters.
Vuruskan et al. (31) reported that patients with ureteral stones exhibited increased plasma TGF-β levels. Consistent with previous animal studies performed in mice and rats (5, 10, 32), our data showed that UUO-induced renal fibrosis was associated with increased expression of α-SMA, type I collagen, and TGF-β in hamsters. Shin et al. (33) reported that the suppression of angiotensinogen expression by using an antisense method significantly inhibited the augmentation of TGF-β expression level in the kidney of UUO rats. We also observed that treatment with a specific CI or an AngII-receptor blocker similarly attenuated UUO-induced renal fibrosis and increases in renal expression of α-SMA, type I collagen, and TGF-β Because the effects of a CI on expression of these genes were accompanied by reductions in intrarenal AngII levels, chymase-dependent AngII formation may contribute to the stimulation of these fibrotic pathways during the progression of UUO-induced renal injury.
Mice that have systemic human renin overexpression with kidney-specific human angiotensinogen overexpression developed hypertension (34), indicating that enhancement of local (renal) AngII production induced significant blood pressure elevation. The evidence could be inconsistent with our present result showing no blood pressure change with the increase in renal AngII level in UUO hamsters. However, there are reports showing that the increase in AngII level in the kidney is not correlated with blood pressure changes, for example, in streptozotocin-induced diabetic nephropathy mice (35) and in IgA nephropathy model mice (36). Thus, evidence shows the increase in AngII level in the kidney is associated with renal injury, but is not correlated with blood pressure elevation. Physiologically, AngII induced constriction of renal vasculature and enhanced sodium reabsorption (37). In contrast, AngII stimulated nitric oxide production, a molecule that inhibits sodium reabsorption (38), in medullary thick ascending limb (37). Because blood flow and tubules tightly regulate sodium balance of each other (39), it seems likely that AngII in the kidney regulates blood pressure by highly complicated mechanisms, an acceleration of sodium reabsorption in vessels, and an inhibition of sodium reabsorption in tubules. Thus, we can speculate about whether the effect of AngII on blood pressure is determined by the part of the kidney responsible for increasing the AngII level. Because it is, so far, technically difficult to analyze the location of AngII accumulation in vivo, future studies need to characterize the role of renal AngII in each pathological condition.
UUO elevated the blood urea nitrogen level, suggesting aggravated glomerular filtration. Although the protective effects of the drugs were strong in (the renal medulla, like those shown in Fig. 1, treatments with CI or olmesartan were ineffective on the elevation of BUN. RAS may not be responsible for the reduction of glomerular function in the UUO hamster model.
To the best of our knowledge, the present study provides the first evidence that shows the contribution of chymase-dependent AngII formation on the progression of UUO-induced renal injury. Among different species, the AngII-generating potency of chymase is highest in humans (11, 12, 31). Therefore, CIs might help prevent the progression of renal fibrosis in humans. Further studies are needed to elucidate the specific role of chymase in the pathogenesis of renal injury.
Acknowledgments
We are grateful to Teijin Pharma Ltd. for providing CI. This work was supported by grants from the Salt Science Research Foundation (07C2) and Kagawa University Research Project 2007 and 2008 (to Akira Nishiyama).
References
- 1.Warady BA, Hébert D, Sullivan EK, Alexander SR, Tejani A. Renal transplantation, chronic dialysis, and chronic renal insufficiency in children and adolescents. The 1995 Annual report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Nephrol. 1997;11:49–64. doi: 10.1007/s004670050232. [DOI] [PubMed] [Google Scholar]
- 2.Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol. 2006;17:17–25. doi: 10.1681/ASN.2005070757. [DOI] [PubMed] [Google Scholar]
- 3.el-Dahr SS, Gee J, Dipp S, Hanss BG, Vari RC, Chao J. Upregulation of renin-angiotensin system and downregulation of kallikrein in obstructive nephropathy. Am J Physiol. 1993;264:F874–F881. doi: 10.1152/ajprenal.1993.264.5.F874. [DOI] [PubMed] [Google Scholar]
- 4.Yoo KH, Norwood VF, el-Dahr SS, Yosipiv I, Chevalier RL. Regulation of angiotensin II AT1 and AT2 receptors in neonatal ureteral obstruction. Am J Physiol. 1997;273:R503–R509. doi: 10.1152/ajpregu.1997.273.2.R503. [DOI] [PubMed] [Google Scholar]
- 5.Ishidoya S, Morrissey J, Mccracken R, Reyes A, Klahr S. Angiotensin II receptor antagonist ameliorates renal tubulo-interstitial fibrosis caused by unilateral, ureteral obstruction. Kidney Int. 1995;47:1285–1294. doi: 10.1038/ki.1995.183. [DOI] [PubMed] [Google Scholar]
- 6.Sugiyama H, Kobayashi M, Wang DH, Sunami R, Maeshima Y, Yamasaki Y, et al. Telmisartan inhibits both oxidative stress and renal fibrosis after unilateral ureteral obstruction in acatalasemic mice. Nephrol Dial Transplant. 2005;20:2670–2680. doi: 10.1093/ndt/gfi045. [DOI] [PubMed] [Google Scholar]
- 7.El Chaar M, Chen J, Seshan SV, Jha S, Richardson I, Ledbetter SR, et al. Effect of combination therapy with enalapril and the TGF-beta antagonist 1D11 in unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2007;292:F1291–F1301. doi: 10.1152/ajprenal.00327.2005. [DOI] [PubMed] [Google Scholar]
- 8.Guo G, Morrissey J, Mccracken R, Tolley T, Klahr S. Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am J Physiol. 1999;277:F766–F772. doi: 10.1152/ajprenal.1999.277.5.F766. [DOI] [PubMed] [Google Scholar]
- 9.Fern RJ, Yesko CM, Thornhill BA, Kim HS, Smithies O, Chevalier RL. Reduced angiotensinogen expression attenuates renal interstitial fibrosis in obstructive nephropathy in mice. J Clin Invest. 1999;103:39–46. doi: 10.1172/JCI4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satoh M, Kashihara N, Yamasaki Y, Maruyama K, Okamoto K, Maeshima Y, et al. Renal interstitial fibrosis is reduced in angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol. 2001;12:317–325. doi: 10.1681/ASN.V122317. [DOI] [PubMed] [Google Scholar]
- 11.Urata H, Boehm KD, Philip A, Kinoshita A, Gabrovsek J, Bumpus FM, et al. Cellular localization and regional distribution of an angiotensin II-forming chymase in the heart. J Clin Invest. 1993;91:1269–1281. doi: 10.1172/JCI116325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyazaki M, Takai S. Tissue angiotensin II generating system by angiotensin-converting enzyme and chymase. J Pharmacol Sci. 2006;100:391–397. doi: 10.1254/jphs.cpj06008x. [DOI] [PubMed] [Google Scholar]
- 13.Murakami M, Matsuda H, Kubota E, Wakino S, Honda M, Hayashi K, et al. Role of angiotensin II generated by angiotensin converting enzyme-independent pathways in canine kidney. Kidney Int Suppl. 1997;63:S132–S135. [PubMed] [Google Scholar]
- 14.Tokuyama H, Hayashi K, Matsuda H, Kubota E, Honda M, Okubo K, et al. Differential regulation of elevated renal angiotensin II in chronic renal ischemia. Hypertension. 2002;40:34–40. doi: 10.1161/01.hyp.0000022060.13995.ed. [DOI] [PubMed] [Google Scholar]
- 15.Yamada M, Ueda M, Naruko T, Tanabe S, Han YS, Ikura Y, et al. Mast cell chymase expression and mast cell phenotypes in human rejected kidneys. Kidney Int. 2001;59:1374–1381. doi: 10.1046/j.1523-1755.2001.0590041374.x. [DOI] [PubMed] [Google Scholar]
- 16.Nishiyama A, Seth DM, Navar LG. Renal interstitial fluid concentrations of angiotensins I and II in anesthetized rats. Hypertension. 2002;39:129–134. doi: 10.1161/hy0102.100536. [DOI] [PubMed] [Google Scholar]
- 17.Zhang GX, Ohmori K, Nagai Y, Fujisawa Y, Nishiyama A, Abe Y, et al. Role of AT1 receptor in isoproterenol-induced cardiac hypertrophy and oxidative stress in mice. J Mol Cell Cardiol. 2007;42:804–811. doi: 10.1016/j.yjmcc.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Nagai Y, Yao L, Kobori H, Miyata K, Ozawa Y, Miyatake A, et al. Temporary angiotensin II blockade at the prediabetic stage attenuates the development of renal injury in type 2 diabetic rats. J Am Soc Nephrol. 2005;16:703–711. doi: 10.1681/ASN.2004080649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobori H, Ozawa Y, Suzaki Y, Nishiyama A. Enhanced intrarenal angiotensinogen contributes to early renal injury in spontaneously hypertensive rats. J Am Soc Nephrol. 2005;16:2073–2080. doi: 10.1681/ASN.2004080676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun GP, Kohno M, Guo P, Nagai Y, Miyata K, Fan YY, et al. Involvements of Rho-kinase and TGF-beta pathways in aldosterone-induced renal injury. J, Am Soc Nephrol. 2006;17:2193–2201. doi: 10.1681/ASN.2005121375. [DOI] [PubMed] [Google Scholar]
- 21.Klahr S, Morrissey L. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol. 2002;283:F861–F875. doi: 10.1152/ajprenal.00362.2001. [DOI] [PubMed] [Google Scholar]
- 22.de Borst MH, van Timmeren MM, Vaidya VS, de Boer RA, van Dalen MB, Kramer AB, et al. Induction of kidney injury molecule-l in homozygous Ren2 rats is attenuated by blockade of the renin-angiotensin system or p38 MAP kinase. Am J Physiol Renal Physiol. 2007;292:F313–F320. doi: 10.1152/ajprenal.00180.2006. [DOI] [PubMed] [Google Scholar]
- 23.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol. 2007;292:F330–F339. doi: 10.1152/ajprenal.00059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turan T, van Harten JG, de Water R, Tuncay OL, Kok DJ. Is enalapril adequate for the prevention of renal tissue damage caused by unilateral ureteral obstruction and/or hyperoxaluria? Urol Res. 2003;31:212–217. doi: 10.1007/s00240-003-0320-7. [DOI] [PubMed] [Google Scholar]
- 25.Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem. 1990;265:22348–22357. [PubMed] [Google Scholar]
- 26.Petrie MC, Padmanabhan N, Mcdonald JE, Hillier C, Connell JM, Mcmurray JJ. Angiotensin converting enzyme (ACE) and non-ACE dependent angiotensin II generation in resistance arteries from patients with heart failure and coronary heart disease. J Am Coll Cardiol. 2001;37:1056–1061. doi: 10.1016/s0735-1097(01)01111-1. [DOI] [PubMed] [Google Scholar]
- 27.Miyazaki M, Takai S. Local angiotensin II-generating system in vascular tissues: the roles of chymase. Hypertens Res. 2001;24:189–193. doi: 10.1291/hypres.24.189. [DOI] [PubMed] [Google Scholar]
- 28.Kobori H, Prieto-Carrasquero MC, Ozawa Y, Navar LG. AT1 receptor mediated augmentation of intrarenal angiotensinogen in angiotensin II-dependent hypertension. Hypertension. 2004;43:1126–1132. doi: 10.1161/01.HYP.0000122875.91100.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishiyama A, Yoshizumi M, Rahman M, Kobori H, Seth DM, Miyatake A, et al. Effects of AT1 receptor blockade on renal injury and mitogen-activated protein activity in Dahl salt- sensitive rats. Kidney Int. 2004;65:972–981. doi: 10.1111/j.1523-1755.2004.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Kats JP, de Lannoy LM, Jan Danser AH, van Meegen JR, Verdouw PD, Schalekamp MA. Angiotensin II type 1 (AT1) receptor-mediated accumulation of angiotensin II in tissues and its intracellular half-life in vivo. Hypertension. 1997;30:42–49. doi: 10.1161/01.hyp.30.1.42. [DOI] [PubMed] [Google Scholar]
- 31.Vuruskan H, Caliskan Z, Kordan Y, Ozakin C, Yavascaoglu I, Oktay B. Elevated plasma concentrations of transforming growth factor-beta 1 in patients with unilateral ureteral obstruction. Urol Res. 2005;33:465–469. doi: 10.1007/s00240-005-0509-z. [DOI] [PubMed] [Google Scholar]
- 32.Trachtman H, Weiser AC, Valderrama E, Morgado M, Palmer LS. Prevention of renal fibrosis by spironolactone in mice with complete unilateral ureteral obstruction. J Urol. 2004;172:1590–1594. doi: 10.1097/01.ju.0000140445.82949.54. [DOI] [PubMed] [Google Scholar]
- 33.Shin GT, Kim WH, Yim H, Kim MS, Kim H. Effects of suppressing intrarenal angiotensinogen on renal transforming growth factor-betal expression in acute ureteral obstruction. Kidney Int. 2005;67:897–908. doi: 10.1111/j.1523-1755.2005.00154.x. [DOI] [PubMed] [Google Scholar]
- 34.Kobori H, Ozawa Y, Satou R, Katsurada A, Miyata K, Ohashi N, et al. Kidney-specific enhancement of ANG II stimulates endogenous intrarenal angiotensinogen in gene-targeted mice. Am J Physiol Renal Physiol. 2007;293:F938–F945. doi: 10.1152/ajprenal.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ichihara A, Hayashi M, Kaneshiro Y, Suzuki F, Nakagawa T, Tada Y, et al. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest. 2004;114:1128–1135. doi: 10.1172/JCI21398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohashi N, Katsurada A, Miyata K, Satou R, Saito T, Urushihara M, et al. The role of activated intrarenal reactive oxygen species and renin-angiotensin system in IgA nephropathy model mice. Clin Exp Pharmacol Physiol. 2009 doi: 10.1111/j.1440-1681.2009.05172.x. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mori T, Cowley AW, Jr, Ito S. Molecular mechanisms and therapeutic strategies of chronic renal injury: physiological role of angiotensin II-induced oxidative stress in renal medulla. J Pharmacol Sci. 2006;100:2–8. doi: 10.1254/jphs.fmj05003x2. [DOI] [PubMed] [Google Scholar]
- 38.Ohkita M, Takaoka M, Matsumura Y. Drug discovery for overcoming chronic kidney disease (CKD): the endothelin ETB receptor/nitric oxide system functions as a protective factor in CKD. J Pharmacol Sci. 2009;109:7–13. doi: 10.1254/jphs.08r10fm. [DOI] [PubMed] [Google Scholar]
- 39.Nakano D, Pollock DM. Contribution of endothelin A receptors in endothelin 1-dependent natriuresis in female rats. Hypertension. 2009;53:324–330. doi: 10.1161/HYPERTENSIONAHA.108.123687. [DOI] [PMC free article] [PubMed] [Google Scholar]