

Among strained ring systems, bicyclo[1.1.0]butanes are of special significance due to their unique chemical bonding properties.1 Cleavage of the central bond in bicyclo[1.1.0]butane, formally a Csp3-Csp3 σ-bond, is facilitated by the release of ~65 kcal/mol in strain energy and represents the most common reaction pathway observed for these molecules. The similarity of the σ-bond to a Csp2=Csp2 π-bond has often been invoked to explain the chemical behavior of bicyclo[1.1.0]butanes,2 in particular, their participation in pericyclic, radical, and ionic reactions.1,3 As with other strained rings,4metal-catalyzedisomerizationreactionsofbicyclo[1.1.0]butanes have been studied extensively.5 Formation of 1,3-butadiene is a major reaction pathway, but the selectivity is strongly dependent on catalysts, reaction conditions, and substituents.5b For example, reactions catalyzed by Ag(I) are thought to proceed via an initial cleavage of the lateral C-C bond in bicyclo[1.1.0]butanes followed by skeletal rearrangement of a carbocation.6 In contrast, group 9 and 10 metals have been shown to operate through a mechanistically distinct pathway.7-9 Trapping experiments confirmed the formation of a carbene intermediate,8 but low yields as well as limited scope in these reactions prevented a further development of general synthetic methodologies.
We have recently shown that bicyclo[1.1.0]butylalkylamines undergo highly stereoselective intramolecular cycloadditions with alkenes and alkynes under mild reaction conditions.3 We further envisioned that a transient metal carbenoid species resulting from isomerization of the bicyclo[1.1.0]butane could be trapped intramolecularly by the N-allyl substituent before undergoing isomerization to a 1,3-butadiene. Mindful of literature precedence5 as well as the similarity of the carbene-forming step to the metal-catalyzed decomposition of diazocompounds,10 we selected Rh(I) precatalysts as potential promoters of the rearrangement reactions. Representative optimization studies for a rhodium-catalyzed cycloisomerization of bicyclobutanes are shown in Table 1.11a When 1 was heated with 5% [Rh(=)2Cl]2 in PhMe at 110 °C in the presence of 10% n-Bu3P, a 6:1 mixture of 2 and 3 was formed in good overall yield (entry 1). Addition of Ph3P (10%) improved the yield of pyrrolidine 2 to 87% with 8% of the azepine 3 (entry 2). The reaction was complete within 10 min and 2 was formed with excellent diastereoselectivity (>95:5).11b When additional Ph3P was added, or the amount of precatalyst was reduced (entries 3 and 4), the yield of 2 dropped. Phosphines with electron-donating or -deficient aromatic substituents had a detrimental effect (entries 5 and 6), and bulkier aliphatic ligands reduced the yield of 2 dramatically (entry 7). Precatalysts bearing large alkene ligands (entries 9 and 10) also led to diminished yields and chemoselectivity.
Table 1.
Precatalyst and Ligand Optimizations for the Cycloisomerization of 1
 | ||||
|---|---|---|---|---|
| entrya | pre-catalyst | ligand | yield [%]b |
|
| 2 | 3 | |||
| 1 | [Rh(=)2Cl]2 | n-Bu3P | 78 | 12 |
| 2 | “ | Ph3P | 87c | 8 |
| 3 | “ | Ph3P(20%) | 70 | 2 |
| 4 | “ (1 mol%) | Ph3P | 71 | 3 |
| 5 | “ | (4-MeOC6H4)3P | 43 | 4 |
| 6 | “ | (4-FC6H4)3P | 61 | 1 |
| 7 | “ | t-Bu3P | 22 | 5 |
| 8 | “ | Ph2MeP | 81 | 9 |
| 9 | [Rh(coe)2Cl]2 | Ph3P | 38 | 9 |
| 10 | [Rh(cod)2Cl]2 | Ph3P | 57 | 20 |
| 11 | RhCl(PPh3)3 (10 mol%) | - | 24 | 49 |
| 12 | [Rh(CO)2Cl]2 | dppe | <1 | 83d |
| 13 | “ | dppp | <1 | 29 |
| 14 | “ | dppb | <1 | 7 |
| 15e | “ | dppe | <1 | 63 |
| 16 | [Rh(=)2Cl]2 | - | 11 | 10 |
Reactions used 5% of precatalyst and 10% of phosphine ligand at 0.05 M concentration of 1 in PhMe at 110 °C.
NMR yields based on internal standard.
Isolated yield: 77%.
Isolated yield: 77%.
Concentration: 0.1 M.
Wilkinson’s catalyst led to a 1:2 ratio of 2 to 3, but a more striking reversal of selectivity was observed when bidentate phosphines were used in combination with [Rh(CO)2Cl]2 (entries 11-15). We found that addition of 10% dppe provided the desired azepine 3 in high yield and excellent diastereoselectivity.11b The combination of [Rh(=)2Cl]2 and dppe provided the azepine in 77% yield. Other bidentate phosphines furnished 3 in lower yields and selectivities, but formation of pyrrolidine 2 was completely suppressed in all cases. In the absence of ligand, both 2 and 3 were formed in low yield (entry 16). Solvents such as EtOAc, THF, CH2ClCH2Cl, and t-BuOMe did not provide improvements. In addition, we found that the best yields were obtained when the reactions were carried out at low concentration (0.05 M) (entry 15). Finally, under the optimized conditions for the formation of 2 or 3, isomerization of 1 to the butadiene did not exceed 1%.11a
The systematic variation of precatalysts and ligands for the cycloisomerization of model system 1 provided selective access to synthetically useful pyrrolidines and azepines. The scope of this methodology was further explored using various aromatic, heteroaromatic, and functionalized aliphatic substrates (Table 2). In addition to excellent scaffold chemoselectivity (pyrrolidine 5 vs azepine 6), all reactions proceeded in high diastereoselectivity. The degree of substitution around the bicyclo[1.1.0]butane ring appears to play a dominant role in controlling the reactivity of the transient rhodium carbene species. For example, large R groups on amide 4 considerably reduced the reaction rate, and chain-substituted allylic as well as homoallylic amides failed to give pyrrolidines or azepines in satisfactory yields.
Table 2.
Rh(I)-Catalyzed Isomerizations of Bicyclo[1.1.0]butanes 4
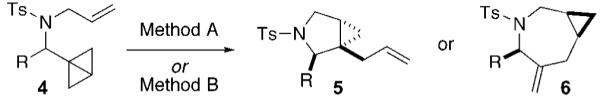 | |||
|---|---|---|---|
| entry | substrate | method Aa | method Bb |
| 1 | 4a, R = 3,5-(MeO)2C6H3 | 5a, 67% | 6a, 87% |
| 2 | 4b, R = 4-ClC6H4 | 5b, 55% | 6b, 80% |
| 3 | 4c, R = 2-furyl | 5c, 75% | 6c, 58% |
| 4 | 4d, R = BnOCH2 | 5d, 63% | 6d, 68% |
| 5 | 4e, R = cyclohexyl | 5e, 65% | 6e, 75% |
Method A: [Rh(=)2Cl]2 (5 mol %), Ph3P (10 mol %), PhMe (0.05 M), 110 °C.
Method B: [Rh(CO)2Cl]2 (5 mol %), dppe (10 mol %), PhMe (0.05 M), 110 °C.
We also briefly explored the cycloisomerization of allylic ethers (Scheme 1). With [Rh(=)2Cl]2 precatalyst, an NMR based yield of 66% of a 2.3:1 ratio of furanyl diene 8 and oxepane 9 was obtained. Other catalysts led to a diminished yield.
Scheme 1.
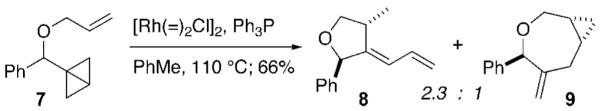
Furan and Oxepane from Allylic Ether
We propose that the isomerization reactions of bicyclo[1.1.0]butanes proceed via the mechanism depicted in Scheme 2. Oxidative addition of Rh(I) across the central σ-bond in bicyclo[1.1.0]butane 10 results in the formation of intermediate 11, which subsequently undergoes rearrangement to carbenes 12 and 13. Formation of the tricyclic intermediate 11 has been proposed for reactions of electron-deficient bicyclo[1.1.0]butanes, but it is worth noting that 11 may also exist in equilibrium with its isomers formed by insertion of the metal into the lateral bonds of bicyclo[1.1.0]butane 10.12 The selectivity of the rearrangement of 11 is controlled by the steric bias exerted by the phosphine ligands. Monodentate phosphines allow for the formation of internal carbene 12, a process most likely favored by the proximity of the allyl group.13 Alternatively, in the presence of bidentate ligands, the saturated complex 11 rearranges to carbene 13. Although no specific mechanistic data are available, formation of carbenes 12 and 13 could also be a reversible process.14 Subsequent cyclopropanation of the allyl group proceeds via transition states in which the α-substituent adopts a pseudoaxial orientation leading to 14 and 15. In cases where the allyl group is not accessible for a facile reaction, carbenes 12 and 13 undergo hydride migration to afford 1,3-butadienes.15
Scheme 2.
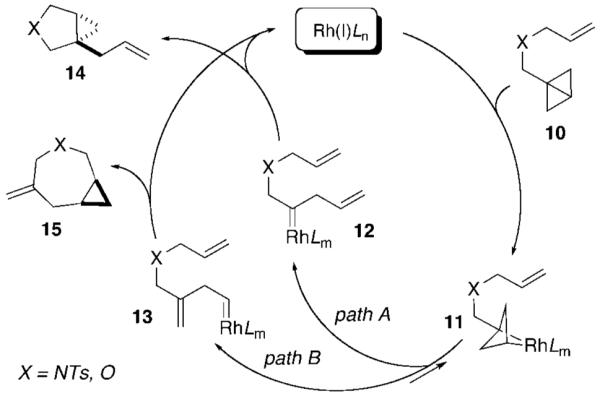
Proposed Mechanism of Cycloisomerization Reactions
In order to further extend the utility of the Rh-catalyzed isomerizations of bicyclo[1.1.0]butanes, sulfonamides 16a-c were subjected to reaction conditions that promoted formation of pyrrolidines (Scheme 3). Upon completion of the rearrangement, Ru metathesis catalyst16 was added and the novel tricyclic pyrrolidines 17a-c were formed in good yields. In combination with the ligand-controlled isomerization pathways of bicyclo[1.1.0]butanes and the facile removal of the nitrogen protecting group, this synthetic strategy allows for the rapid assembly of a diverse set of molecular scaffolds17 from a common pool of functionalized bicyclo[1.1.0]butanes.
Scheme 3.
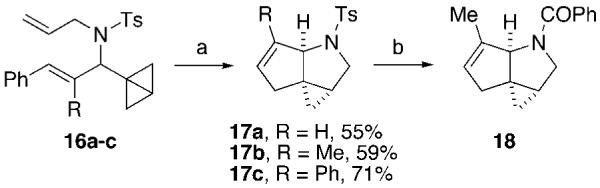
Synthesis of 3-Azatricyclo[6.1.0.01,5]nonanes via Tandem Isomerization-RCMa
a Reagents and conditions: (a) [Rh(=)2Cl]2, Ph3P, PhMe, 110 °C then metathesis catalyst,16 60 °C; (b) i. Na/naphthalene, THF, -78 °C; ii. PhCOCl, DMAP, Et3N, CH2Cl2,0 °C, 53%.
In summary, we have developed an efficient rhodium-catalyzed cycloisomerization of N-allylated bicyclo[1.1.0]butylalkylamines. Depending on the nature of the Rh(I) phosphine ligands, these reactions provide pyrrolidines and azepines with high levels of stereo- and regiocontrol. A related transformation is also feasible for allylic ethers, providing substituted furans and oxepanes.
Supplementary Material
Acknowledgment
This work was supported by NIGMS (NIH P50 GM067082). We thank Dr. Steve Geib for X-ray crystallographic analyses.
References
- (1).(a) Hoz S. In: The Chemistry of the Cyclopropyl Group. Rappoport Z, editor. Wiley; New York: 1987. [Google Scholar]; (b) Wiberg KB, Lampman GM, Ciula RP, Connor DS, Schertler P, Lavanish J. Tetrahedron. 1965;21:2749. [Google Scholar]
- (2).(a) Newton MD, Schulman JM. J. Am. Chem. Soc. 1972;94:767. [Google Scholar]; (b) Pomerantz M, Hillenbrand DF. J. Am. Chem. Soc. 1973;95:5809. [Google Scholar]
- (3).Wipf P, Walczak MAA. Angew. Chem., Int. Ed. 2006;45:4172. doi: 10.1002/anie.200600723. [DOI] [PubMed] [Google Scholar]
- (4).(a) Wender PA, Gamber GG, Williams TJ. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]; (b) Namyslo JC, Kaufmann DE. Chem. Rev. 2003;103:1485. doi: 10.1021/cr010010y. [DOI] [PubMed] [Google Scholar]; (c) Rubin M, Rubina M, Gevorgyan V. Chem. Rev. 2007;107:3117. doi: 10.1021/cr050988l. [DOI] [PubMed] [Google Scholar]
- (5).(a) Paquette LA. Acc. Chem. Res. 1971;4:280. [Google Scholar]; (b) Bishop KC., III Chem. Rev. 1976;76:461. [Google Scholar]
- (6).(a) Sakai M, Masamune S. J. Am. Chem. Soc. 1971;93:4610. [Google Scholar]; (b) Sakai M, Westberg HH, Yamaguchi H, Masmune S. J. Am. Chem. Soc. 1971;93:4611. [Google Scholar]
- (7).(a) Gassman PG, Reitz RR. J. Organomet. Chem. 1973;52:C51. [Google Scholar]; (b) Gassman PG, Atkins TJ, Williams FJ. J. Am. Chem. Soc. 1971;93:1812. doi: 10.1021/ja00733a701. [DOI] [PubMed] [Google Scholar]; (c) Gassman PG, Nakai T. J. Am. Chem. Soc. 1972;94:5497. [Google Scholar]
- (8).(a) Noyori R, Suzuki T, Kumagai Y, Takaya H. J. Am. Chem. Soc. 1971;93:5894. doi: 10.1021/ja00766a065. [DOI] [PubMed] [Google Scholar]; (b) Noyori R. Tetrahedron Lett. 1973;14:1691. [Google Scholar]; (c) Noyori R, Kawauchi H, Takay H. Tetrahedron Lett. 1974;15:1749. [Google Scholar]; (d) Takaya H, Suzuki T, Kumagai Y, Hosoya M, Kawauchi H, Noyori R. J. Org. Chem. 1981;46:2854. [Google Scholar]
- (9).(a) Dauben WG., Jr J. Am. Chem. Soc. 1972;94:3669. doi: 10.1021/ja00779a058. [DOI] [PubMed] [Google Scholar]; (b) Dauben WG, Kielbania AJ, Jr., Raymond KN. J. Am. Chem. Soc. 1973;95:7166. [Google Scholar]
- (10).For recent reviews, see:Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861. doi: 10.1021/cr0200217.Merlic CA, Zechman AL. Synthesis. 2003:1137.Davies H. Org. React. 2001;57:2.Padwa A. J. Organomet. Chem. 2001;617-618:3.Doyle MP, Forbes DC. Chem. Rev. 1998;98:911. doi: 10.1021/cr940066a.
- (11).(a) See Supporting Information for additional optimization conditions. (b) Determined by 1H NMR analysis of a crude reaction mixture. The relative configuration was determined by NOESY (2) or X-ray analysis of the corresponding ketone (3). For details, see Supporting Information.
- (12).(a) Miyashita A, Takahashi M, Takaya H. J. Am. Chem. Soc. 1981;103:6257. [Google Scholar]; (b) Miyashita A, Watanabe Y, Takaya H. Tetrahedron Lett. 1983;24:2595. [Google Scholar]
- (13).Isomerization of N-sulfonamides (no allyl substituents) under conditions favoring formation of 12 or 13 afforded only diene products.
- (14).(a) Ikto N, Takamura N, Young SD, Ganem B. Tetrahedron Lett. 1981;22:4163. [Google Scholar]; (b) Shi W, Xiao F, Wang J. J. Org. Chem. 2005;70:4318. doi: 10.1021/jo050173c. [DOI] [PubMed] [Google Scholar]
- (15).May JA, Stoltz BM. J. Am. Chem. Soc. 2002;124:12426. doi: 10.1021/ja028020j. [DOI] [PubMed] [Google Scholar]
- (16).Trnka TM, Grubbs RH. Acc. Chem. Res. 2001;34:18. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- (17).(a) Wipf P, Stephenson CRJ, Walczak MAA. Org. Lett. 2004;6:3009. doi: 10.1021/ol0487783. [DOI] [PubMed] [Google Scholar]; (b) Dolle RE, Le Bourdonnec B, Morales GA, Moriarty KJ, Salvino JM. J. Comb. Chem. 2006;8:597. doi: 10.1021/cc060095m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.