

Abstract
The GABAA receptor γ2 subunit mutation, Q351X, associated with generalized epilepsy with febrile seizures plus (GEFS+), created a loss of function with homozygous expression. However, heterozygous γ2(+/−) gene deletion mice are seizure free, suggesting that the loss of one GABRG2 allele alone in heterozygous patients may not be sufficient to produce epilepsy. Here we show that the mutant γ2 subunit was immature and retained in the endoplasmic reticulum (ER). With heterozygous coexpression of γ2S/γ2S(Q351X) subunits and α1 and β2 subunits, the trafficking deficient mutant γ2 subunit reduced trafficking of wild-type partnering subunits, which was not seen in the hemizygous gene deletion control. Consequently, the function of the heterozygous receptor channel was reduced to less than the hemizygous control and to less than half of the wild-type receptors with a full gene dose. Pulse-chase experiments demonstrated that in the presence of the mutant γ2S(Q351X) subunit, wild-type α1 subunits degraded more substantially within 1 h of translation. We showed that the basis for this dominant-negative effect on wild-type receptors was due to an interaction between mutant and wild-type subunits. The mutant subunit oligomerized with wild-type subunits and trapped them in the ER, subjecting them to glycosylation arrest and ER-associated degradation (ERAD) through the ubiquitin proteosome system. Thus, we hypothesize that a likely explanation for the GEFS+ phenotype is a dominant-negative suppression of wild-type receptors by the mutant γ2S subunit in combination with loss of mutant γ2S subunit protein function.
Introduction
GABAA receptors are the major inhibitory neurotransmitter receptors in the CNS, and the α1β2γ2S receptor is the most abundant receptor isoform. The γ2 subunit is critical for receptor trafficking, clustering and synaptic maintenance (Essrich et al., 1998; Schweizer et al., 2003), and several GABAA receptor γ2 subunit missense mutations [γ2(R43Q) (Wallace et al., 2001), γ2(K289M) (Baulac et al., 2001), and γ2(R139G) (Audenaert et al., 2006)] and premature translation-termination codon (PTC)-generating mutations [γ2(Q1X) (Hirose, 2006), γ2(IVS6 + 2T→G) (Kananura et al., 2002), and γ2(Q351X) (Harkin et al., 2002)] have been associated with autosomal dominant idiopathic generalized epilepsies (IGEs). The missense mutations have been shown either to have reduced trafficking to the membrane surface with relatively normal function (Bianchi et al., 2002; Gallagher et al., 2004; Kang and Macdonald, 2004; Macdonald et al., 2006) or to traffic to the surface with impaired function (Feng et al., 2006, Bianchi et al., 2002). However, the pathophysiological mechanisms of GABAA receptor γ2 subunit nonsense mutations are not clear.
The γ2 subunit nonsense mutation, Q351X, has been associated with generalized epilepsy with febrile seizures plus (GEFS+) (Harkin et al., 2002). Phenotypes in the affected pedigree ranged from simple febrile seizures to the more severe seizures of the Dravet syndrome. Mutant mRNAs containing PTCs are often degraded by nonsense-mediated mRNA decay (NMD) (Maquat, 2004). However, the PTC in γ2(Q351X) subunits is located in the last exon of the GABRG2 gene. NMD only degrades mRNAs with PTCs at least 50–55 nt 5′ of an exon–exon junction, and therefore, does not degrade mRNAs with PTCs in the last exon (Holbrook et al., 2004). Thus, the γ2(Q351X) subunit mRNA should not be subject to NMD. Recent studies also suggest that PTCs can activate mRNA degradation due to an improperly configured 3′-UTR, the “faux 3′-UTR model,” even if the PTC is located in the last exon (Amrani et al., 2004). However, studies on NMD in other diseases have shown that NMD is often incomplete (Kuzmiak and Maquat, 2006). Thus, it is likely that γ2(Q351X) subunit mRNA and truncated mutant protein are present in neurons of affected heterozygous patients.
In the present study, we demonstrated that γ2(Q351X) subunit protein was produced but was immature and trafficking deficient, resulting in haploinsufficiency of the γ2 subunit. In addition, it had a dominant-negative effect on wild-type receptors by reducing their assembly, trafficking and surface expression. The dominant-negative effect was likely due to oligomerization of mutant and wild-type subunits, resulting in ER retention and glycosylation arrest of both wild-type and mutant subunits. Retained, immature wild-type subunits were rapidly degraded by ERAD through the ubiquitin-proteasomal system (UPS). As a result, the γ2 subunit mutation, Q351X, reduced mature surface GABAA receptors to an extent that was greater than that produced by hemizygous expression of the γ2 subunit. This combination of loss of surface expression and function of the mutant subunits and the dominant-negative effect of the mutant subunits on the wild-type subunits would result in considerable loss of inhibition and is the likely explanation for the GEFS+ associated with this mutation.
Materials and Methods
Expression vectors with GABAA receptor subunits.
The cDNAs encoding human α1, β2, γ2S, and enhanced cyan fluorescent protein (CFP)- or yellow fluorescent protein (YFP)-tagged GABAA receptor subunits (e.g., γ2SCFP, α1CFP, or γ2SYFP subunit) were as described previously (Kang and Macdonald, 2004; Kang et al., 2006). The γ2 subunit minigenes were generated by inserting the full-length intron 8 of the γ2 gene between exons 8 and 9 of the γ2 subunit cDNA constructs. Since intron 8 contains the sequences for the γ2L subunit, which is subject to alternative splicing inside the cells, the term γ2 subunit minigene was used instead of either γ2L or γ2S subunit minigene in Figure 1. Similarly, we are not sure whether truncated wild-type or mutant γ2S or γ2L subunits were produced with expression of the intron 8 minigenes, and therefore, we refer to γ2 and γ2(Q351X) subunits when the subunits were a product of minigene expression. We used γ2S or γ2L subunit when referring to the cDNA constructs for the rest of study. The ecliptic pHluorin (a pH-sensitive GFP variant)-tagged rat γ2L (γ2LpH) subunit was kindly provided by Dr. Stephen J. Moss (Tufts University School of Medicine, Boston, MA). The FLAG (DYKDDDDK) and HA (YPYDVPDYA) epitopes were inserted between amino acids 4 and 5 in the N terminus of the protein. Rat γ2 subunit siRNA and primers were all synthesized by Qiagen. The anti-γ2 subunit siRNA sequence (AAG AAA TCT GAT GAT GAC TAT) was complementary to an upstream sequence of the rat γ2 subunit N terminus that was interrupted in the YFP-tagged human γ2SYFP subunit sequence.
Figure 1.
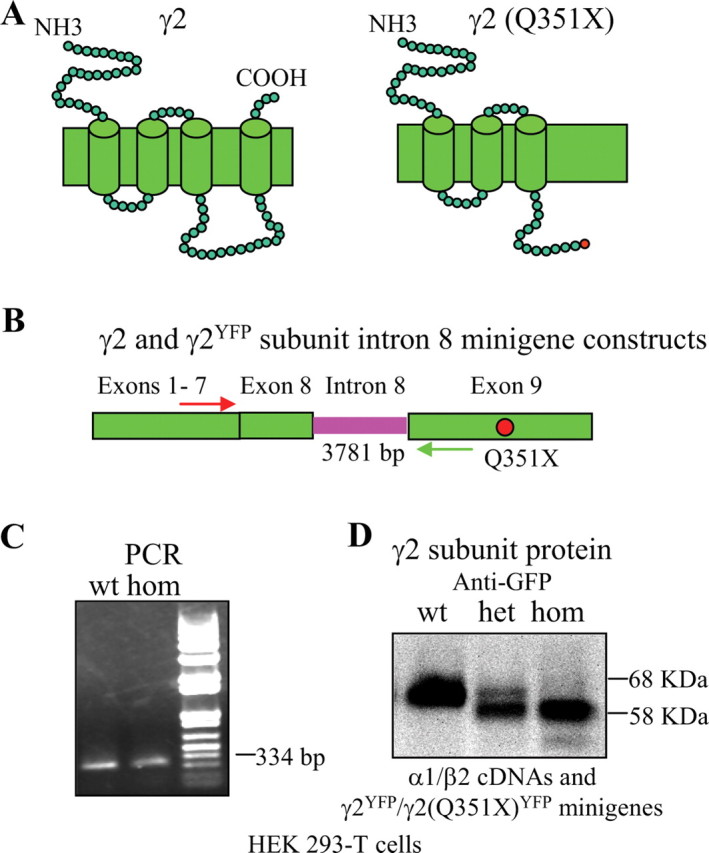
Mutant mRNA and truncated protein were produced from the γ2(Q351X) subunit intron 8 minigene in HEK 293-T cells. A, The γ2 subunit amino acid Q351 is located in the TM3–TM4 loop of the protein. B, The point mutation Q351X is located in the last (ninth) exon of the GABRG2 gene. Wild-type and mutant γ2 and γ2YFP subunit minigenes were constructed by including the entire intron 8 between exon 8 and 9 in γ2 and γ2YFP subunit cDNA constructs. Primers in flanking exons 7 and 9 were used to determine whether the γ2 subunit minigene was correctly spliced. C, HEK 293-T cells were transfected with γ2 or γ2(Q351X) subunit minigenes, and each construct displayed a band at 334 bp, suggesting correct splicing in the reverse-transcribed cDNA products from mRNAs of the γ2 subunit minigene. D, HEK 293-T cells were cotransfected with α1 and β2 subunit cDNAs and wild-type γ2YFP (1:1:1 ratio), heterozygous γ2YFP/γ2(Q351X)YFP (1:1:0.5:0.5 ratio), and homozygous γ2(Q351X)YFP (1:1:1 ratio) subunit minigenes. Total cell lysates were directly analyzed by SDS-PAGE.
Cell culture.
Hippocampi were dissected from the brains of embryonic day 18 Sprague Dawley rat pups. Dissociation of cells and cell culture and transfection procedures have been described previously (Kang et al., 2006).
Transfection.
Transfection of HEK 293-T cells for electrophysiological experiments was as previously described (Kang and Macdonald, 2004). The total amounts of cDNAs transfected in different conditions were normalized by adding the empty vector pcDNA. Hippocampal neurons and HEK 293-T cells for immunoblots were transfected with Lipofectamine or Fugene (Invitrogen). Cells were cotransfected with 2 μg of each subunit plasmid for each 60 mm2 dish and 1 μg for each 35 mm2 dish. The siRNA was prepared according to the manufacturer's instructions (Qiagen), and siRNAs (5 μg) were transfected with Oligofectamine or Lipofectamine (Invitrogen) to cells in each 35 mm2 dish on the same day of the transfection of receptor subunits and on the third or fourth day after the transfection of receptor subunits to maintain suppression of the endogenous rat γ2 subunit.
Electrophysiology.
Lifted whole-cell recordings were obtained from transfected HEK 293-T cells as described previously (Kang and Macdonald, 2004). Cells were voltage clamped at −50 mV, and E Cl was 0 mV. For mIPSC recordings, hippocampal neurons were voltage clamped at −60 mV, and tetrodotoxin (TTX) (1 μm) was added to block action potentials. d-(−)-2-Amino-5-phosphonovaleric acid (AP5; 40 μm) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μm) were added to block NMDA and AMPA receptor-mediated excitatory synaptic currents, and 2-hydroxysaclofen (100 μm) was added to block GABAB receptor currents. All chemicals were purchased from Sigma-Aldrich. The mIPSCs were abolished by addition of the GABAA receptor antagonist, bicuculline (10 μm). Analysis of mIPSC frequencies and amplitudes were made using the Mini Analysis program by Justin Lee, which is available online.
Live-cell confocal microscopy and fluorescence quantification.
Confocal microscopy and the measurements in COS-7 cells for each channel were performed as reported previously (Kang and Macdonald, 2004). Clusters in hippocampal neurons were counted by area (per 100 μm) as before (Kang et al., 2006). For labeling of rat γ2 subunits, neurons were fixed with 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde for 1 h and then permeabilized with 0.2% Triton X-100 for 10 min before staining.
Biotinylation, immunoprecipitation, and Western blot analysis.
Cell surface receptor biotinylation and Western blots were performed as reported previously (Kang and Macdonald, 2004). Membranes were incubated with primary mouse monoclonal antibodies against GABAA receptor α1 or β2/3 subunits or GFP (Millipore Bioscience Research Reagents). Monoclonal HA antibody was purchased from Covance, and rabbit polyclonal anti-γ2 subunit of GABA receptor antibody from Alamone Labs. After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibody [goat anti-mouse IgG (1:2000), goat anti-rabbit IgG (1:2000), Millipore Bioscience Research Reagents]. Digestion of protein lysates with Endo-H and PNGase F has been described previously (Gallagher et al., 2005). For immunoprecipitation, the wild-type γ2FLAG and γ2(Q351X)FLAG subunits were purified by incubating cell lysates (1 mg/1 ml) overnight with 50 μl of agarose-immobilized anti-FLAG M2 antibody (Sigma-Aldrich). The antibody resin was pelleted by centrifugation and washed three times with lysis buffer. FLAG-tagged subunits were liberated by incubation with FLAG peptide (Sigma-Aldrich) for 30 min on ice.
35S radiolabeling metabolic pulse-chase assays.
The protocol was modified from our previous study (Gallagher et al., 2007). Briefly, 48 h after transfection, the cells were placed in starving medium that lacked methionine and cysteine (Invitrogen) and incubated at 37°C for 30 min. The starving medium was then replaced by 1.5 ml of [35S] radionuclide methionine [100–250 μCi/ml (1 Ci = 37 GBq); PerkinElmer] labeling medium for 20 min at 37°C. The labeling medium was then changed to chase medium for a series of different time points. FLAG-tagged GABAA receptor subunits were then immunoprecipitated from radiolabeled lysates with anti-FLAG M2-agarose affinity gel by rotating at 4°C overnight. The immunoprecipitated products were eluted from the beads with FLAG peptide (Sigma-Aldrich). The immunopurified subunits were then analyzed by 12.5% SDS-PAGE and exposed on a digital PhosphorImager (GE Healthcare).
Data analysis.
Macroscopic currents were low-pass filtered at 2 kHz, digitized at 10 kHz, and analyzed using the pClamp9 software suite (Molecular Devices). Numerical data were expressed as mean ± SEM, except that the electrophysiology data for peak current amplitudes were plotted as mean ± SD. Proteins were quantified by ChemiImager AlphaEaseFC software, and data were normalized to either wild-type subunit proteins or loading controls. Pulse-chase experiments were quantified by using Quantity One software (Bio-Rad). Statistical significance, using Student's unpaired t test (GraphPad Prism), was taken as p < 0.05.
Results
The γ2 subunit minigene was completely spliced, and γ2(Q351X) subunit mRNA and protein were produced
The γ2 subunit nonsense mutation, Q351X, is located in the TM3–TM4 loop, and the PTC should result in loss of the C-terminal 78 aa, thus producing a truncated subunit (Fig. 1 A). The human γ2 subunit gene, GABRG2, has 9 exons and 8 introns, and the mutation (red dot) is in the last exon (Fig. 1 B). PTCs followed by an exon–exon junction >50–55 nt downstream generally activate NMD, but PTCs in the last exon should not activate NMD. To confirm that NMD was not activated by the γ2 subunit Q351X mutation, we made wild-type and mutant subunit minigene constructs that contained intron 8 inserted between exons 8 and 9 (Fig. 1 B) with and without a YFP epitope tag inserted into the N terminus of the mature peptide. We then expressed each minigene construct in HEK 293-T cells and used PCR with primer pairs in exon 7 and 8 that flanked the intron 8 insert (Fig. 1 B). We demonstrated that the intron 8 inserts in both the wild-type and mutant γ2 subunit minigenes were correctly spliced out (Fig. 1 C), suggesting that these minigenes were suitable for determining whether NMD was activated by the Q351X mutation.
We coexpressed YFP-tagged wild-type γ2YFP or mutant γ2(Q351X)YFP subunit minigenes with α1 and β2 subunit cDNAs in HEK 293-T cells. Whole-cell lysates were analyzed by Western blot and blotted with anti-GFP antibody (Fig. 1 D). In cells, transfected with the wild-type γ2YFP subunit minigene, there was a prominent band at ∼68 kDa (Fig. 1 D, wt). In cells cotransfected with the homozygous γ2(Q351X)YFP subunit minigene, a main band at ∼58 kDa was detected, consistent with the expected shift in molecular mass produced by a truncated subunit (Fig. 1 D, hom). With expression of heterozygous γ2YFP/γ2(Q351X)YFP subunit minigenes, two bands were detected. Surprisingly, the band with the molecular mass of the wild-type subunit was faint, and the band with the molecular mass of the mutant subunit was strong (Fig. 1 D, het).
Heterozygous α1β2γ2S/γ2S(Q351X) receptor currents were reduced compared with wild-type α1β2γ2S or hemizygous control α1β2γ2S(+/−) receptor currents
Based on our minigene results, truncated mutant γ2 subunits were produced. To determine the effect of the truncated γ2S(Q351X) subunit on expression of functional GABAA receptors, we used wild-type and mutant γ2S subunit cDNA constructs. We recorded from HEK 293-T cells following cotransfection of α1 and β2 subunits with wild-type γ2S subunits (1:1:1 cDNA ratio), a 50% concentration of hemizygous γ2S subunit (1:1:0.5 cDNA ratio) or heterozygous γ2S/γ2S(Q351X) subunits (1:1:0.5:0.5 cDNA ratio). We used “wild-type,” “hemizygous,” “heterozygous,” and “homozygous” subunit expression, receptors, and currents to indicate that we transfected cells with nonmutant (wild-type) cDNAs, half concentrations of nonmutant cDNAs (hemizygous), an equal mixture of nonmutant and mutant cDNAs (heterozygous), or mutant cDNAs (homozygous) and studied their expression and function. We used this notation to facilitate descriptions of the transfection techniques, and no genetic mechanism or specific assembly patterns were implied.
Currents were evoked by application of 1 mm GABA for 6 s (Fig. 2 A) or 28 s (data not shown). Hemizygous control receptor peak currents (2551 ± 1268 pA) (n = 16) were reduced to 61.3% of wild-type receptor currents (4161 ± 852 pA) (n = 17; p < 0.001) (Fig. 2 A,B). Heterozygous receptor peak currents (1449 ± 955 pA) (n = 15) were reduced to 34.9% of wild-type receptor currents (p < 0.0001) and to 56.9% of hemizygous control receptor currents (p = 0.012) (Fig. 2 A,B). Only very small currents were occasionally recorded from cells cotransfected with α1 and β2 subunits and “homozygous” γ2S(Q351X) subunits (1:1:1 cDNA ratio) (data not shown).
Figure 2.

Expression of heterozygous γ2S/γ2S(Q351X) subunits and α1 and β2 subunits resulted in reduced peak current amplitudes compared with expression of wild-type or hemizygous control α1, β2, and γ2S subunits. A, GABAA receptor currents were obtained from HEK 293-T cells cotransfected with wild-type α1 and β2 subunits and γ2S (1:1:1 cDNA ratio; wt, black trace), hemizygous control γ2S (1:1:0.5 cDNA ratio; hem, gray trace), or heterozygous γ2S/γ2S(Q351X) (1:1:0.5:0.5 cDNA ratio; het, green trace) subunits with application of 1 mm GABA applied for 6 s. Peak wt, hem, and het currents were marked with a black arrow, a gray arrow or a green arrow, respectively. B, The amplitudes of GABAA receptor currents from A were plotted. Values were mean ± SD (n = 15–16 cells from 6 different transfections) (***p < 0.001 vs wt, † p < 0.05 vs hem).
With coexpression of heterozygous γ2S/γ2S(Q351X) subunits and α1 and β2 subunits, surface expression of wild-type γ2S subunits was reduced due to ER retention
The current amplitude recorded with expression of heterozygous α1β2γ2S receptors was less than half that recorded from wild-type receptors and was less than that recorded from hemizygous control receptors. Was the reduced current recorded with expression of heterozygous α1β2γ2S receptors a result of loss of surface receptors due to the failure of assembly of γ2S(Q351X) subunits and/or due to reduction of surface α1β2γ2S receptors? To explore this question, we characterized the trafficking pattern of receptors with heterozygous expression of γ2S/γ2S(Q351X) subunits as would be seen in an autosomal dominant disease. We determined the surface expression of γ2S subunits by biotinylation and immunoblotting following cotransfection of HEK 293-T cells with α1 and β2 subunits and wild-type γ2SYFP, hemizygous control γ2SYFP, heterozygous γ2SYFP/γ2S(Q351X)YFP, and homozygous γ2S(Q351X)YFP subunits (Fig. 3 A, left). For all expression conditions, surface γ2SYFP/γ2S(Q351X)YFP subunit protein IDVs were normalized to wild-type levels (Fig. 3 A, right). Hemizygous control γ2SYFP subunit surface levels were reduced relative to control wild-type γ2SYFP subunit levels (0.68 ± 0.03, n = 7, p < 0.001). Heterozygous γ2SYFP/γ2S(Q351X)YFP subunit surface levels were also reduced relative to control wild-type γ2SYFP subunit levels (0.43 ± 0.04, n = 7, p < 0.0001) and were also reduced relative to the hemizygous control condition (p = 0.0002). Homozygous γ2S(Q351X)YFP subunit expression was reduced to such an extent that no significant surface protein was detected.
Figure 3.
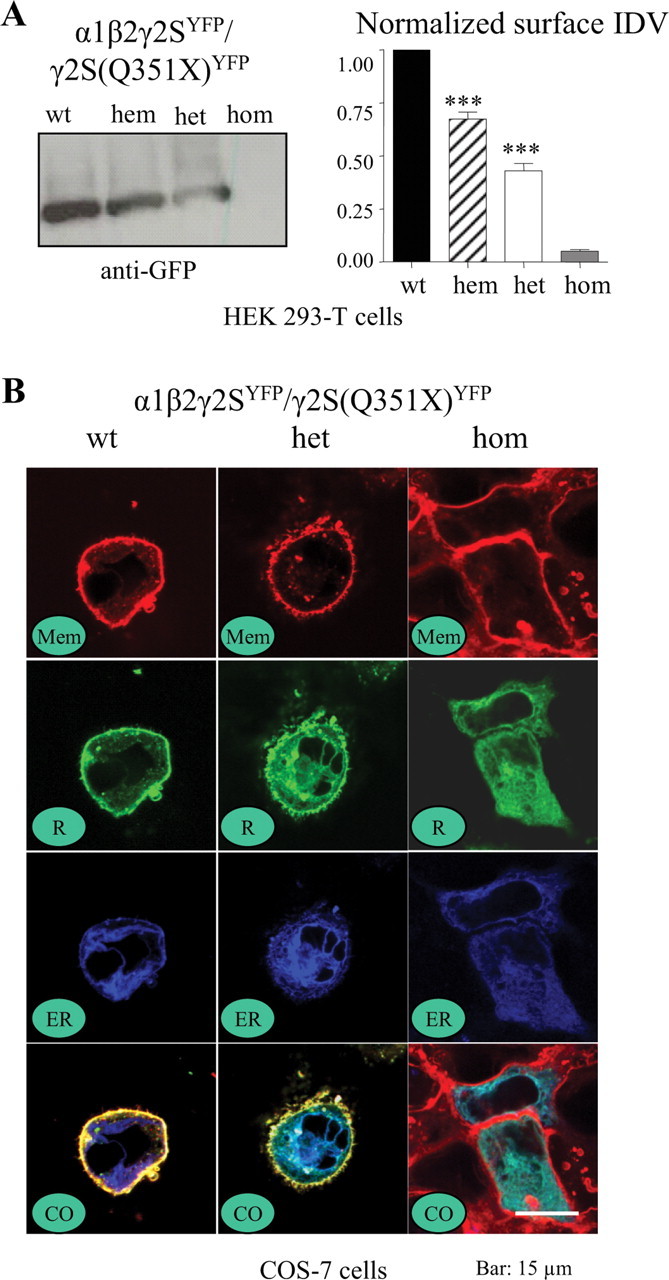
With heterozygous expression, surface γ2S subunit levels were reduced and γ2S subunits were retained in the ER. A, HEK 293-T cells were cotransfected with α1 and β2 subunits and γ2SYFP (1:1:1 cDNA ratio, wt), γ2SYFP (1:1:0.5 cDNA ratio, hem), γ2SYFP/γ2S(Q351X)YFP (1:1:0.5:0.5 cDNA ratio, het), or γ2S(Q351X)YFP (1:1:1 cDNA ratio, hom) subunits, and surface proteins were biotinylated, isolated, separated by SDS-PAGE, and probed with anti-GFP antibody. Surface proteins were normalized to the wild-type γ2SYFP subunit protein. Values are mean ± SEM (n = 7) (***p < 0.001 vs wt). B, COS-7 cells were cotransfected with α1 and β2 subunits and γ2SYFP (1:1:1 cDNA ratio, wt), γ2SYFP/γ2S(Q351X)YFP (1:1:0.5:0.5 cDNA ratio, het), or γ2S(Q351X)YFP (1:1:1 cDNA ratio, hom) subunits, and the images were obtained 48 h later under confocal microscopy. Mem stands for membrane, R for γ2SYFP subunit, ER for endoplasmic reticulum, and CO for colocalized images.
Because the mutant subunit did not appear to traffic to the cell surface, we determined the subcellular location of the mutant protein by cotransfecting COS-7 or HEK 293-T (data not shown) cells with γ2SYFP and/or γ2S(Q351X)YFP subunits and α1 and β2 subunits. The surface membrane was identified using the plasma membrane marker FM4–64 (red) (Fig. 3 B, Mem), and the ER was marked by pECFP-ER (blue) (Fig. 3 B, ER). The surface and intracellular distributions of wild-type γ2SYFP and γ2S(Q351X)YFP subunits (green) were determined (Fig. 3 B, R). All three signals were coregistered to demonstrate their relative subcellular localizations (Fig. 3 B, CO). Wild-type γ2SYFP subunit fluorescence was primarily on the surface and had a smooth distribution (Fig. 3 B, wt, CO). Heterozygous expression resulted in reduced surface expression and increased intracellular expression of γ2SYFP/γ2S(Q351X)YFP subunits (Fig. 3 B, het, CO). Homozygous expression resulted in no visible surface expression with substantial intracellular ER localization of γ2S(Q351X)YFP subunits (Fig. 3 B, hom, CO).
When coexpressed with α1 and β2 subunits, γ2L(Q351X) pHluorin-tagged subunits had minimal expression on the surface of rat hippocampal neurons
Because the data above were obtained using HEK 293-T and COS-7 cells, we extended the study to determine the surface expression of wild-type and mutant subunits in neurons. pHluorin-tagged rat γ2LpH and γ2L(Q351X)pH subunits were coexpressed with human α1 and β2 subunits in hippocampal neurons for 6 d. We used γ2L instead of γ2S subunits since γ2L subunits cannot traffic to the surface alone but γ2S subunits can. pHluorin should only generate fluorescence when at the cell surface and should produce minimal fluorescence at the acidic pHs (pH <6.5) that are characteristic of vesicular compartments (Miesenböck et al., 1998). After 6 d, fluorescence from γ2LpH and γ2L(Q351X)pH subunits was visible and appeared as puncta on neurons (Fig. 4 A; enlarged views for the red boxed areas in Fig. 4 A are shown in Fig. 4 B,C). Because pHluorin only fluoresces at the surface and γ2L subunits cannot traffic to the surface alone but can with β2 or α1 subunits, the puncta on neurons were assumed to be surface γ2L subunits assembled in receptors. Neurons cotransfected with wild-type α1β2γ2LpH subunits (Fig. 4 A, wt, B) had many more fluorescent puncta than neurons cotransfected with α1β2γ2L(Q351X)-pHluorin subunits (Fig. 4 A, mut, C), indicating that the majority of mutant receptors were trapped intracellularly in neurons.
Figure 4.
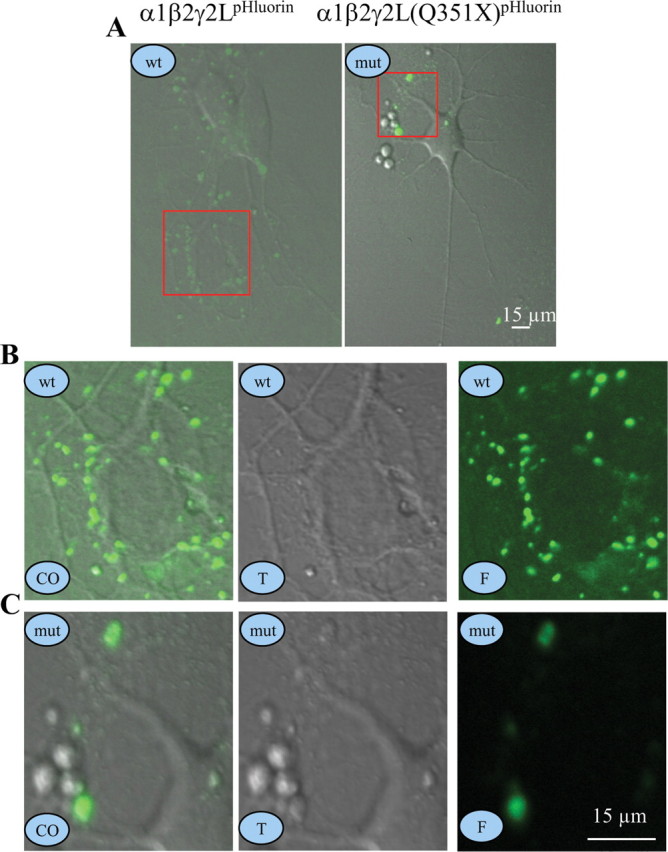
γ2L(Q351X)pHluorin subunit protein had minimal expression on the surface of rat hippocampal neurons. A–C, Rat hippocampal neurons were transfected with α1β2γ2LpHluorin (wt) or α1β2γ2L(Q351X)pHluorin (mut) subunits for 6 d and were imaged as puncta on the surface of neurons. A, Representative images of wt γ2LpHluorin or mut γ2L(Q351X)pHluorin subunits are presented. B, C, Enlarged views of the boxed areas for wt (B) and mut (C) receptors in A. In A–C, wt stands for wild-type γ2LpHluorin subunits, mut stands for mutant γ2L(Q351X)pHluorin subunits, CO stands for colocalized images, T stands for transmitted images, and F stands for fluorescent images.
With coexpression of heterozygous γ2SYFP/γ2S(Q351X)YFP subunits and α1 and β2 subunits in hippocampal neurons, mIPSCs had reduced peak current and reduced number of events
It is also important to characterize the functional properties of mutant heterozygous currents in neurons. However, since there were endogenous γ2 subunit-containing receptors in neurons, it would be difficult to characterize the phenotype of heterozygous receptors. We thus designed an siRNA to “knock down” endogenous γ2 subunits. Rat hippocampal neurons treated for 6–8 d with siRNA (5 μg) targeting the rat γ2 subunit gene had significantly reduced γ2 subunit protein fluorescence compared with the untreated control cultures as probed with anti-rat γ2 antibody and visualized with FITC (Fig. 5 A) or by measuring the γ2 subunit total protein (Fig. 5 B).
Figure 5.
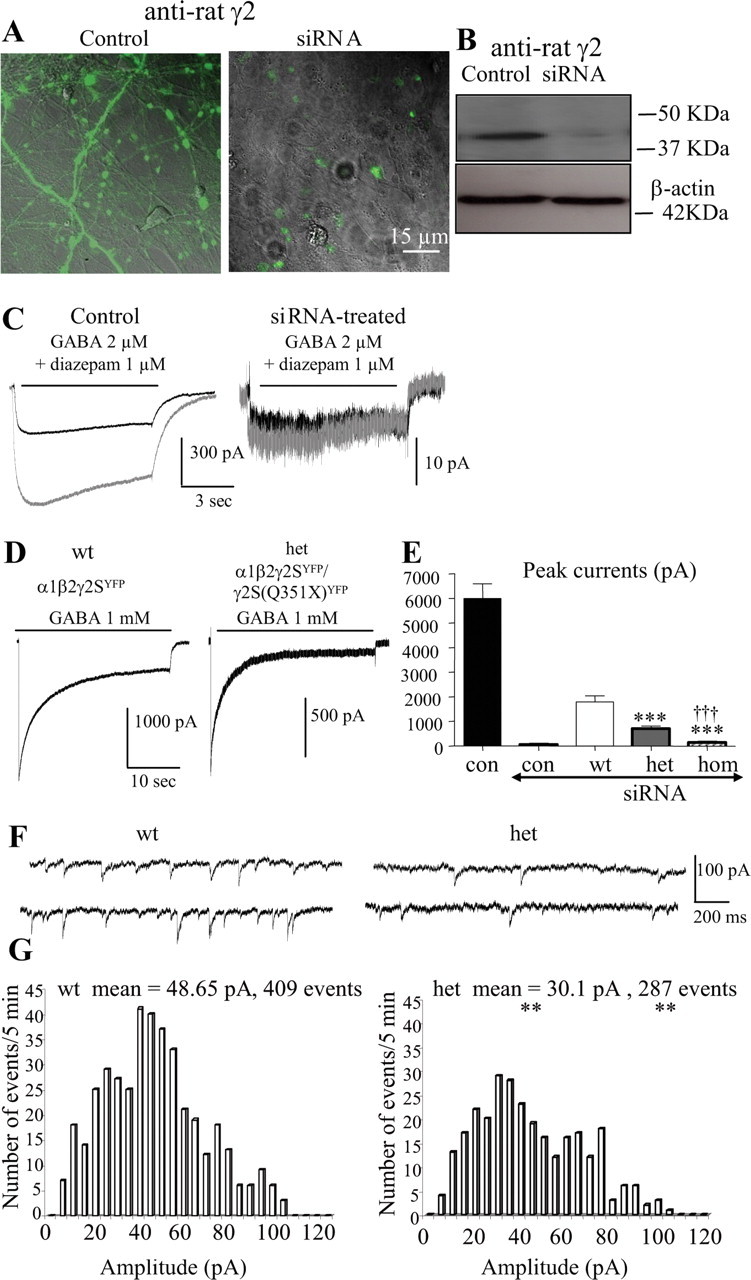
Heterozygous α1β2γ2S(Q351X) receptor current amplitudes were less than half of wild-type peak current amplitudes in hippocampal neurons. A, Rat hippocampal neurons in the absence or presence of 5 μg of siRNA targeting the rat γ2 subunit gene for 8 d were permeabilized and stained with anti-rat γ2 antibody and visualized with FITC. B, The whole-cell lysates from the sister dishes were probed with rabbit anti-rat γ2 antibody by Western blot. C, Whole-cell currents evoked by GABA (2 μm) and GABA (2 μm) plus diazepam (1 μm) were recorded from 16-d-old hippocampal neurons in the absence (Control) or presence of 5 μg of siRNA targeting the rat γ2 subunit gene for 6–8 d (siRNA). D, Wild-type α1β2γ2SYFP, heterozygous α1β2γ2SYFP/γ2S(Q351X)YFP, and homozygous α1β2γ2S(Q351X)YFP receptor currents from 15- to 16-d-old endogenous γ2 subunit silenced hippocampal neurons were evoked with GABA (1 mm) for 28 s. E, The peak current amplitudes of neurons untreated (con) or treated with siRNA (siRNA) and transfected with human α1β2γ2SYFP (wt), α1β2 γ2SYFP/γ2S(Q351X)YFP (het), and α1β2γ2S(Q351X)YFP (hom) were plotted (***p < 0.001 vs wt, †††hom vs het, data from 13–15 cells from 5 batches of cells). F, G, GABAergic mIPSCs for cells transfected with α1 and β2 subunits and wild-type γ2SYFP or heterozygous γ2SYFP/γ2S(Q351X)YFP subunits were recorded at a holding potential of −60 mV. G, Individual mIPSCs from a 5 min recording period were averaged for wild-type and heterozygous subunit-transfected cells. The amplitude distributions of mIPSCs obtained during a 5 min recording period were displayed in a histogram. The histograms were fitted using Mini Analysis program by Justin Lee (**p < 0.01 het vs wt, n = 6 for each group).
GABA (1 mm) evoked large currents (6003 ± 598 pA, n = 14) from untreated hippocampal neurons but evoked only small currents from siRNA-treated neurons (59.6 ± 11.7 pA, n = 9) (Fig. 5 E). Diazepam (1 μm) enhanced GABA (2 μm)-evoked currents in neurons untreated with siRNA (217% ± 15.3%, n = 5), but not in neurons treated with siRNA (110.6 ± 10.3%, n = 5) (Fig. 5 C), consistent with reduction of γ2 subunit-containing GABAA receptors in siRNA-treated neurons. Wild-type γ2SYFP, heterozygous γ2SYFP/γ2S(Q351X)YFP, and homozygous γ2S(Q351X)YFP subunit cDNAs and α1 and β2 cDNAs were then cotransfected into hippocampal neurons in addition to treatment with siRNA (5 μg) for 6–8 d. Neurons were voltage clamped at −50 mV, and GABA (1 mm) was applied for 28 s. GABA evoked large and rapidly desensitizing currents from wild-type receptors (1789.5 ± 234.1 pA, n = 11) and smaller currents from heterozygous receptors (Fig. 5 D,E) (695 ± 67.9 pA, n = 12) (p < 0.0001 vs wt). Only minimal currents were obtained from homozygous receptors (140 ± 21.2 pA, n = 7) (p < 0.0001 vs wt or het) (Fig. 5 E). The heterozygous current was less than half of the wild-type current, suggesting a dominant-negative effect of the mutant protein on the wild-type receptors in neurons.
We also recorded mIPSCs from the same groups of neurons. The cells were voltage clamped at −60 mV, and mIPSCs were recorded from neurons transfected with wild-type α1β2γ2SYFP and heterozygous α1β2γ2SYFP/γ2S(Q351X)YFP receptors (Fig. 5 F). The mIPSCs from neurons transfected with heterozygous receptors had a significantly smaller mean peak amplitude (30.1 vs 48.65 pA) and fewer events (287 vs 409 events) than mIPSCs from neurons transfected with wild-type receptors (Fig. 5 G).
With heterozygous expression, mutant γ2S(Q351X) subunits had immature glycosylation, and wild-type γ2S subunits had less total and less mature glycosylation compared with hemizygous expression of γ2S subunits
To explore the basis for the reduction of surface wild-type γ2S subunit proteins with coexpression of heterozygous γ2S/γ2S(Q351X) subunits and α1 and β2 subunits in HEK 293-T cells (Fig. 3), we determined the total expression and maturity of wild-type and mutant γ2S subunit proteins following coexpression of α1 and β2 subunits and wild-type γ2SYFP, heterozygous γ2SYFP/γ2S(Q351X)YFP, or homozygous γ2S(Q351X)YFP subunits (Fig. 6). As observed in previous studies, γ2 subunit protein bands in Western blots were often smeared rather than discreet (Connolly et al., 1996). With expression of wild-type subunits, the γ2SYFP subunit protein usually migrated as a single, broad main band at ∼68 kDa, and with homozygous subunit expression, the γ2S(Q351X)YFP subunit protein migrated as a main band at ∼58 kDa, consistent with the reduced molecular mass of the truncated subunit, and sometimes as a faint band ∼45 kDa, which was likely to be unglycosylated γ2S(Q351X)YFP subunit protein (Fig. 6 A). With heterozygous γ2SYFP/γ2S(Q351X)YFP subunit expression, subunit proteins migrated in two or three bands with a minor band at ∼68 kDa (wild-type subunit protein), a main band at ∼58 kDa (mutant subunit protein) and sometimes a band at ∼45 kDa (unglycosylated form of the mutant subunit protein). When normalized to the total γ2S subunit protein obtained with wild-type expression, total wild-type γ2SYFP subunit protein obtained with heterozygous subunit expression was reduced to less than half of total γ2SYFP subunit protein obtained with wild-type receptor expression.
Figure 6.

Glycosylation arrest caused the reduction of mature wild-type γ2S subunit protein. A, HEK 293-T cells were cotransfected with α1 and β2 subunits and wild-type γ2SYFP (1:1:1 cDNA ratio, wt), heterozygous γ2SYFP/γ2S(Q351X)YFP (1:1:0.5:0.5 cDNA ratio, het), or homozygous γ2S(Q351X)YFP (1:1:1 cDNA ratio, hom) subunits. Total proteins were analyzed by SDS-PAGE and probed with anti-GFP antibody. B, C, The total cell lysates with wild-type γ2SYFP (B) or mutant γ2S(Q351X)YFP (C) subunit transfection were undigested (U) or digested with PNGase F (F) or Endo-H (H). D, Total cell lysates of hemizygous control α1β2γ2SYFP/pcDNA or α1β2γ2SHA/pcDNA (1:1:0.5:0.5 cDNA ratio) subunits and heterozygous α1β2γ2SYFP/γ2S(Q351X) or α1β2γ2SHA/γ2S(Q351X) subunits (1:1:0.5:0.5 cDNA ratio) were transfected into HEK 293-T cells and undigested (U) or digested with Endo-H (H) or PNGase F (F). E, The protein bands insensitive to Endo-H were quantified and presented as a fraction of their total undigested bands (*p < 0.05 vs wt; §§§ p < 0.001 vs hem; n = 5). Values are mean ± SEM.
To determine the maturation of γ2S and γ2S(Q351X) subunit proteins when coexpressed with α1 and β2 subunits in HEK 293-T cells, total wild-type γ2SYFP and homozygous mutant γ2S(Q351X)YFP subunit proteins were analyzed with and without digestion with Endo-H (H) or PNGase F (F). Endo-H removes high-mannose N-linked carbohydrates attached in the ER but not those attached in the trans-Golgi region. In contrast, PNGase F removes all carbohydrates attached in both ER and trans-Golgi regions. The undigested γ2SYFP subunit protein migrated primarily in one band at ∼68 kDa (Fig. 6 B, U), and the undigested γ2S(Q351X)YFP subunit protein migrated primarily in a band at ∼58 kDa (Fig. 6 C, U). With Endo-H digestion, the γ2SYFP subunit protein migrated in two strong bands (Fig. 6 B, H), a molecular mass shift that was consistent with a previous report (Connolly et al., 1996), and the γ2S(Q351X)YFP subunit protein migrated primarily in the same single band, suggesting that the majority of the mutant protein was retained inside the ER and only had immature ER glycosylation (Fig. 6 C, H). With PNGase F digestion, both γ2SYFP and γ2S(Q351X)YFP subunit proteins migrated in one band (Fig. 6 B,C, F in both panels) that was at the molecular mass of their Endo-H-sensitive bands. The Endo-H-insensitive γ2SYFP subunit upper band represented the protein trafficked beyond the ER to the trans-Golgi and surface membrane, and the Endo-H-sensitive γ2SYFP lower band represented the portion of wild-type γ2SYFP subunit that had not exited the ER. With the γ2S(Q351X)YFP subunit, Endo-H digestion shifted the main band to the same level obtained with PNGase F digestion, suggesting that when coexpressed with α1 and β2 subunits, γ2S(Q351X) subunit protein underwent ER, but not Golgi, glycosylation.
We also compared the total expression and Endo-H sensitivity of wild-type γ2S subunits in the hemizygous expression condition (hem; U, H, F) with those in the presence of mutant γ2S(Q351X) subunits (het; U, H, F). Hemizygous γ2SGFP or γ2SHA and heterozygous γ2SGFP/γ2S(Q351X) or γ2SHA/γ2S(Q351X) subunits were coexpressed with α1 and β2 subunits. To discriminate between the two types of subunit since the molecular masses of the unglycosylated wild-type subunit and mutant subunit were similar, only wild-type γ2S subunits were tagged. The smaller HA epitope was included to exclude alteration of trafficking due to YFP epitope tagging. Empty pcDNA vector was used to normalize the total amount of transfected cDNA in the hemizygous condition. In the presence of mutant γ2S(Q351X) subunits, both the total and Endo-H-insensitive amounts of wild-type γ2SYFP were reduced compared with the hemizygous condition (Fig. 6 D,E). The fraction of total γ2SYFP subunit protein that was Endo-H insensitive with wild-type subunit expression (0.40 ± 0.08) (Fig. 6 B,E) or hemizygous control subunit expression (0.52 ± 0.03) (Fig. 6 D,E) were not significantly different. However, with heterozygous subunit expression (Fig. 6 D), the Endo-H-insensitive fraction of γ2SYFP protein was reduced compared with wild-type (Fig. 6 B) and hemizygous control (Fig. 6 D) subunits (0.15 ± 0.04; p = 0.03 vs wt; p < 0.0001 vs hem) (Fig. 6 E). With homozygous subunit expression there was minimal Endo-H-insensitive γ2S(Q351X)YFP subunit protein (0.02 ± 0.007) (Fig. 6 C,E). Together, these experiments demonstrated that with homozygous or heterozygous expression, the γ2S(Q351X) subunit had immature glycosylation and did not traffic to the cell surface, and with heterozygous expression, the wild-type γ2 subunit also had less mature glycosylation and reduced trafficking to the cell surface.
The γ2S(Q351X) subunit oligomerized with and reduced the trafficking of α1 and β2 subunits
The above data demonstrated that although truncated γ2S(Q351X) subunits were expressed, they were retained in the ER and not trafficked to the cell surface and that coexpression of heterozygous γ2S/γ2S(Q351X) subunits with α1 and β2 subunits resulted in current that was less than half of wild-type receptor current and less than hemizygous receptor current. These findings suggested that mutant subunits may oligomerize with α1 and β2 subunits and/or impair their ability to assemble with γ2S subunits, resulting in fewer surface wild-type α1β2γ2S receptors. To determine whether γ2S(Q351X) subunits can assemble with α1 and β2 subunits, β2 subunits and fluorescent enhanced cyan fluorescent protein (CFP)-tagged α1 (α1CFP) subunits were coexpressed in COS-7 cells with or without γ2S or γ2S(Q351X) subunits (Fig. 7). If assembled, were the mutant receptors retained in the ER? If assembly did not occur, were α1β2 receptors formed and trafficked to the cell surface? Coexpression of α1CFP and β2 subunits alone (Fig. 7 A, left) or with γ2S subunits (Fig. 7 A, middle) resulted in localization of α1CFP subunits on the cell surface. In contrast, with coexpression of α1CFP, β2, and γ2S(Q351X) subunits, α1CFP subunits were localized primarily intracellularly (Fig. 7 A, right), suggesting that γ2S(Q351X) subunits oligomerized with α1 subunits and prevented them from oligomerizing with β2 subunits, and/or directly oligomerized with β2 subunits, and thus prevented assembly of α1β2 receptors.
Figure 7.
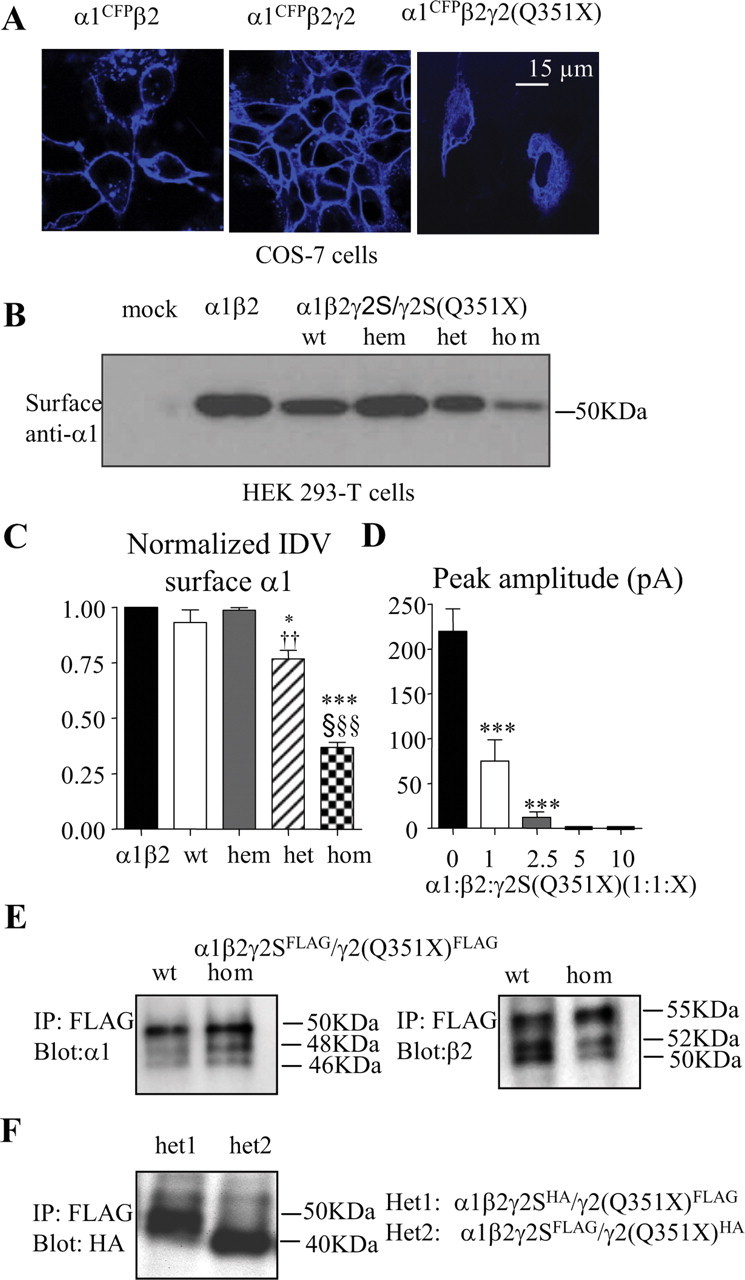
The truncated γ2S(Q351X) subunit impaired trafficking of α1β2 receptors to the cell surface due to its oligomerization with wild-type subunits. A, COS-7 cells were transfected with α1CFPβ2 (left), α1CFPβ2γ2S (middle), or α1CFPβ2γ2S(Q351X) subunits, and the confocal images were obtained 48 h later. B, HEK 293-T cells were transfected with empty pcDNA vector (mock) or with α1β2 (1:1 cDNA ratio), α1β2γ2S (1:1:1 cDNA ratio, wt), α1β2γ2S (1:1:0.5 cDNA ratio, hem), α1β2γ2S/γ2S(Q351X) (1:1:0.5:0.5 cDNA ratio, het), or α1β2γ2S(Q351X) (1:1:1 cDNA ratio) subunits, and surface proteins were prepared as described in the methods and visualized with anti-human α1 subunit antibody. C, Surface protein IDVs were normalized to α1β2 receptors (n = 4–8) (*p < 0.05, ***p < 0.001 vs wt; †† p < 0.01 vs hem; §§§ p < 0.001 vs α1β2). Values were mean ± SEM. D, HEK 293-T cells were transfected with α1β2 (1:1 cDNA ratio) or α1β2γ2S(Q351X) (1:1:1, 1:1:2.5, 1:1:5, 1:1:10 cDNA ratio) subunits, and the peak current amplitudes were plotted (***p < 0.0001 vs α1β2) [n = 19 for α1β2, n = 62 for α1β2γ2S(Q351X) (1:1:1), n = 16 for α1β2γ2S(Q351X) (1:1:2.5), n = 5 for α1β2γ2S(Q351X) (1:1:5 and 1:1:10)]. Values were mean ± SD. In A–D, the total amounts of cDNAs were normalized by the empty vector pcDNA. E, Total lysates from HEK 293-T cells expressing α1β2γ2SFLAG or α1β2γ2S(Q351X)FLAG (1:1:1 cDNA ratio) subunits were purified with agarose-immobilized anti-FLAG M2 antibody, and the conjugated subunits were liberated with FLAG-peptide and probed with anti-α1 (E, left) or anti-β2 (E, right). F, Total lysates from HEK 293-T cells expressing α1β2γ2SHA/γ2S(Q351X)FLAG (het1) or α1β2γ2SFLAG/γ2S(Q351X)HA (het 2) (1:1:0.5:0.5) were purified with FLAG M2 antibody as described in E and visualized with anti-HA antibody.
We then compared α1 subunit surface protein detected using biotinylation following coexpression of α1 and β2 subunits or of α1 and β2 subunits with wild-type γ2S, hemizygous γ2S, heterozygous γ2S/γ2S(Q351X), or homozygous γ2S(Q351X) subunits (Fig. 7 B). Surface α1 subunit levels were high with expression of α1β2 (taken as 1) and α1β2γ2S (0.93 ± 0.06, n = 5) subunits and with coexpression of hemizygous γ2S subunits with α1 and β2 subunits (0.99 ± 0.01, n = 4) (Fig. 7 C). Surface α1 subunit protein levels were reduced slightly but significantly with heterozygous γ2S/γ2S(Q351X) subunit expression (0.77 ± 0.04 n = 6, p = 0.038 vs wt; p = 0.002 vs hem) when compared with either wild-type or hemizygous γ2S subunit expression and were reduced substantially with homozygous γ2S(Q351X) subunit expression (0.37 ± 0.02, n = 8, p < 0.0001 vs wt) when compared with all other conditions (Fig. 7 C). However, there was a quantitative discrepancy between the loss of surface α1 subunit and the peak current in the heterozygous condition. This probably suggests that there are other unknown mechanisms involved in the receptor trafficking and function in the presence of the mutant protein. In addition, it is unknown about the ratio of α1β2 versus α1β2γ2S receptors on the surface in the heterozygous condition. α1β2 and α1β2γ2S receptors have different channel conductance and kinetics, thus resulting in different current amplitudes. The discrepancy could also be due to the fact that both Western blot and whole-cell recording are poorly quantitative techniques.
We then compared the whole-cell current amplitudes from receptor channels formed by expression of α1 and β2 subunits with or without the γ2S(Q351X) subunit. Currents recorded with α1β2γ2S(Q351X) subunit transfection (75.1 ± 23.6 pA, n = 62) were much smaller than those recorded with α1β2 subunit transfection (219.9 ± 25 pA, n = 19; p < 0.0001) (Fig. 7 D) or with α1β2γ2S subunit transfection (4161 ± 852 pA, n = 17) (Fig. 2 A,B). Two types of currents were obtained following α1β2γ2S(Q351X) subunit transfection and both were small and ran-down rapidly (data not shown). In addition, incremental increases in the α1β2γ2S(Q351X) subunit cDNA ratios from 1:1:1 to 1:1:10 resulted in a γ2S(Q351X) subunit cDNA concentration-dependent loss of the small current (Fig. 7 D) although the total cDNA amounts were normalized by adding the empty vector pcDNA.
What was the basis for γ2S(Q351X) subunits causing retention of wild-type α1, β2, and γ2S subunits? Does the γ2S(Q351X) subunit oligomerize with α1 and β2 subunits? To determine this we coexpressed FLAG-tagged γ2S (γ2SFLAG) and γ2S(Q351X)FLAG subunits with α1 and β2 subunits in HEK 293-T cells and purified the γ2SFLAG and γ2S(Q351X)FLAG subunits from the total lysates with FLAG-conjugated beads. The protein that was pulled down was probed with either anti-α1 or anti-β2 antibody. In both γ2S (wt) and γ2S(Q351X) (hom) protein complexes, α1 (Fig. 7 E, left) or β2 subunits (Fig. 7 E, right) were detected, suggesting that the γ2S(Q351X) subunit oligomerized with both α1 and β2 subunits. Similarly, we cotransfected wild-type and mutant γ2S subunits tagged with either the FLAG or HA tag (i.e., γ2SFLAG and γ2S(Q351X)HA or γ2SHA and γ2S(Q351X)FLAG subunits) with α1 and β2 subunits. We then pulled down the total protein with anti-FLAG antibody and blotted with HA. In each heterozygous condition, the γ2SHA or γ2S(Q351X)HA subunit protein was detected, suggesting that the mutant γ2S (Q351X) subunit also oligomerized with the wild-type γ2S subunit (Fig. 7 F).
α1 subunits were retained in the ER and had accelerated degradation in the presence of γ2S(Q351X) subunits
Reduction of α1 and β2 subunits on the surface produced by coexpression with γ2S(Q351X) subunits could be caused by reduction of total cellular subunit proteins due to accelerated degradation or by ER retention and failure of subunit maturation. We thus determined total expression and glycosylation of wild-type subunits by Western blot and Endo-H digestion. In HEK 293-T cells following coexpression of α1 and β2 subunits with wild-type γ2S, hemizygous control γ2S, heterozygous γ2S/γ2S(Q351X), or homozygous γ2S(Q351X) subunits, total α1 subunit proteins were undigested (U) or digested (H) with Endo-H (Fig. 8 A). GAPDH was used as the gel loading control, and α1 subunit protein levels were expressed as α1 subunit/GAPDH IDV ratios normalized to wild-type receptor (Fig. 8 B). With heterozygous γ2S/γ2S(Q351X) subunit expression, total α1 subunit protein (85.7 ± 14.28) was slightly, but not significantly, reduced compared with wild-type α1 subunit expression (p = 0.50, n = 5) but was reduced compared with hemizygous control γ2S subunit expression (138.79 ± 10.22) (p = 0.02). With homozygous γ2S(Q351X) subunit expression, total α1 subunit protein was reduced (24.49 ± 6.12) compared with that with either wild-type γ2S (p = 0.002) or heterozygous γ2S/γ2S(Q351X) (p = 0.004) subunit expression (Fig. 8 B). These data demonstrated that there was a small decrease in total α1 subunit protein with heterozygous γ2S/γ2S(Q351X) subunit expression, and a large decrease in total α1 subunit protein with homozygous γ2S(Q351X) subunit expression.
Figure 8.
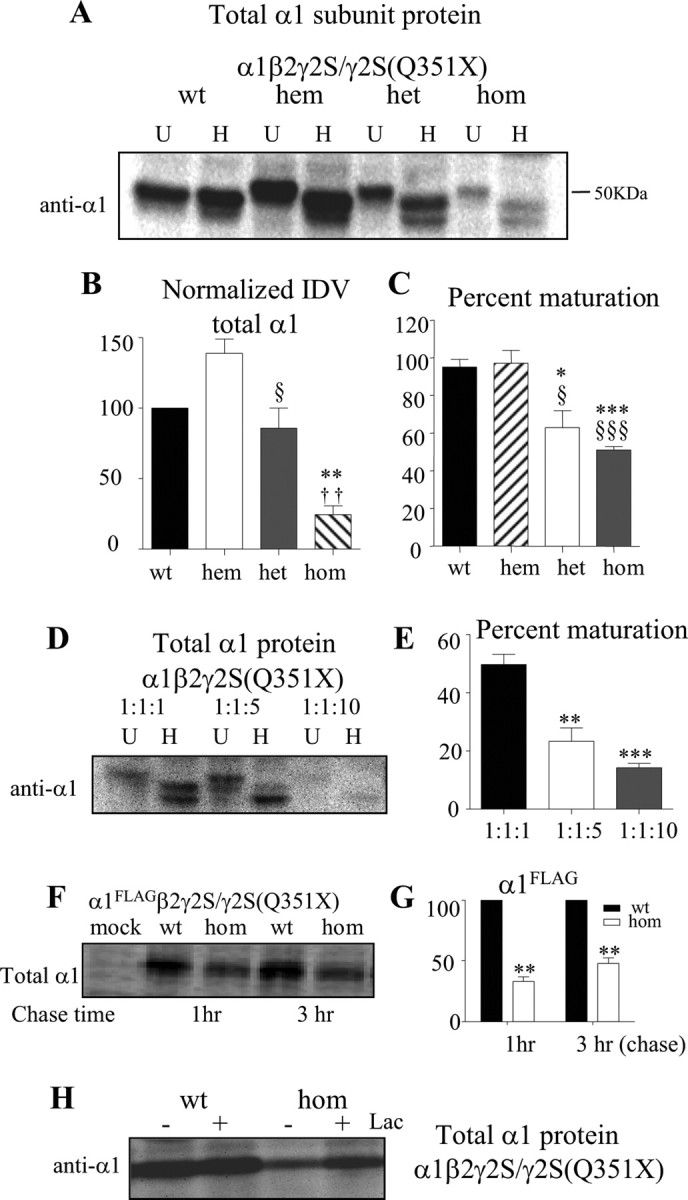
The α1 subunit was subject to glycosylation arrest and enhanced degradation in the presence of the γ2S(Q351X) subunit. A, HEK 293-T cells were cotransfected with α1 and β2 subunits and wild-type γ2S (1:1:1 wt or 1:1:0.5 hem cDNA ratio), heterozygous γ2S/γ2S(Q351X) (1:1:0.5:0.5 hem cDNA ratio), and homozygous γ2S(Q351X) (1:1:1 cDNA ratio) subunits. The cell lysates were either undigested (U) or digested with Endo-H (H) and analyzed by SDS-PAGE. B, Total undigested α1 subunit protein IDVs were quantified, and the data were normalized to GAPDH IDVs (n = 5). C, The fractions of Endo-H-digested α1 subunit proteins were quantified and expressed as percentage maturation [taken as 100% if there was only a single 48.4 kDa present and a percentage of the 48.4 kDa band (A, top lanes in H) over 48.4 plus 46 kDa bands (A, bottom lanes in H) if there were two bands present]. In B and C, *p < 0.05, **p < 0.01, ***p < 0.001 versus wt; †† p < 0.01 versus het 0.5; § p < 0.05, §§ p < 0.01, §§§ p < 0.001 versus hemizygous control (n = 5). D, HEK 293-T cells were cotransfected with α1, β2, and γ2S(Q351X) subunits using cDNA ratios of 1:1:1, 1:1:5, and 1:1:10. The lysates were either undigested (U) or digested with Endo-H (H) and analyzed by 10% SDS-PAGE. E, The fractions of Endo-H-digested α1 subunit protein IDVs were quantified as described in C. In A–E, the total amounts of cDNAs were normalized by the empty vector pcDNA. **p < 0.01, ***p < 0.001 versus α1β2γ2S(Q351X) (1:1:1) (n = 4). F, G, HEK 293-T cells containing 35S methionine-radiolabeled α1FLAGβ2γ2S (wt) and α1FLAGβ2γ2S(Q351X) (hom) receptors were lysed, immunopurified with FLAG M2 antibody, and analyzed by SDS-PAGE. After 20 min of labeling, the cells were chased for the indicated times (n = 4). The same amount of total protein (800 μg) from each sample was used for immunopurification (F). The graph plots the percentage radioactivity normalized to α1FLAG subunit IDV of α1FLAGβ2γ2S receptors at the given chase time (G) (**p < 0.01 vs α1FLAGβ2γ2S). H, The total lysates from cells expressing α1β2γ2S and α1β2γ2S(Q351X) receptors with or without treatment by lactacystin (Lac, 10 μm) for 6 h were analyzed by SDS-PAGE and blotted with anti-α1 antibody.
The reduction of α1 subunit protein was likely due to ER retention and subsequent ERAD. We therefore studied the effect of expression of the γ2S(Q351X) subunit on maturation of α1 subunit protein by characterizing its glycosylation pattern (Fig. 8 A,C) by using digestion with Endo-H. With coexpression of wild-type subunits, Endo-H digestion shifted the α1 subunit molecular mass from 50 to 48.4 kDa and also to 46 kDa depending on different transfection methods (data not shown). The presence of a 46 kDa band with expression of wild-type subunits was likely caused by the excess protein production partially overwhelming the cellular glycosylation machinery with a high efficient transfection system. The 48.4 kDa band represented the mature subunit trafficked beyond the trans-Golgi region, and the 46 kDa band represented the immature subunit protein in the ER (Gallagher et al., 2005). With Endo-H digestion, most α1 subunits expressed with either wild-type or hemizygous control γ2S subunits migrated in the 48.4 kDa band with only a very small portion migrating in the immature 46 kDa band (48.4 kDa/total = 95 ± 4% for wt and 97 ± 6.9 for hem) (Fig. 8 A,C). However, the ratio of 48.4 kDa/total bands was decreased with heterozygous γ2S/γ2S(Q351X) subunit expression (62.9 ± 9) and was decreased even more with homozygous γ2S(Q351X) subunit expression (51 ± 1.9) (Fig. 8 C). These results suggested that increased mutant γ2 subunits decreased maturation of α1 subunits, consistent with ER retention. To further confirm the dominant-negative effect of the γ2S(Q351X) subunit on α1 subunits, we transfected HEK 293-T cells with α1β2γ2S(Q351X) subunits at cDNA ratios of 1:1:1, 1:1:5, and 1:1:10 (Fig. 8 D). With increasing γ2S(Q351X) subunit cDNA concentration, there was a progressive reduction of total α1 subunit protein (data not shown) and reduction of mature relative to immature α1 subunit protein (Fig. 8 E).
What is the basis for the reduction of the wild-type partnering subunits? We determined how rapid wild-type α1 subunits were degraded when expressed with the mutant γ2S(Q351X) subunits, and whether the reduction of wild-type α1 and β2 subunits was through ubiquitin-proteosome degradation. HEK 293-T cells were cotransfected with α1 and β2 subunits and wild-type γ2S or mutant γ2S(Q351X) subunits. For pulse-chase experiments, α1FLAG subunits were radiolabeled with 35S and purified with FLAG M2 antibody-conjugated beads. Wild-type α1FLAG subunits were substantially reduced in the presence of mutant γ2S(Q351X) subunits compared with the presence of wild-type γ2 subunits with either a 1 h (100 vs 33.19 ± 3.7%) or 3 h (100 vs 47.81 ± 4.7%) chase (Fig. 8 F,G), suggesting that degradation of wild-type α1FLAG subunits occurred rapidly. The proteasomal inhibitor, lactacystin (10 μm, 6 h), increased α1FLAG subunit levels when coexpressed with either wild-type γ2S or mutant γ2S(Q351X) subunits (Fig. 8 H). However, total α1 subunits increased to a greater extent with mutant γ2S(Q351X) subunits (176.4 ± 10.29%, n = 5) than with wild-type γ2S subunits (122.2 ± 3.6%, n = 5), suggesting that α1 subunits were subject to enhanced proteasomal degradation in the presence of γ2S(Q351X) subunits.
Discussion
The γ2 subunit mutation, Q351X, produced a truncated subunit protein with loss of function
The γ2 subunit mutation, Q351X, is in the last exon of a 9 exon gene, and thus, should not activate NMD but should allow translation of a truncated protein. Using an intron 8 minigene construct, we demonstrated that truncated γ2(Q351X) subunits were produced and that whole-cell levels were actually higher than those of wild-type γ2 subunits. However, despite the presence of large amounts of mutant subunits, they only attained immature, core glycosylation, were retained in the ER and were not trafficked to the cell surface when coexpressed with α1 and β2 subunits. As a result, currents recorded with coexpression of α1 and β2 subunits and heterozygous γ2S/γ2S(Q351X) subunits were smaller than those produced with coexpression of wild-type α1, β2, and γ2S subunits. Thus, the γ2 subunit mutation, Q351X, is a loss-of-function mutation.
The γ2S(Q351X) subunit had a dominant-negative effect on partnering wild-type α1 and β2 subunits and on γ2S subunits leading to reduction in surface expression of α1β2γ2S and α1β2 receptors
Because the γ2 subunit mutation, Q351X, is a loss-of-function mutation, this autosomal dominant disease could result from γ2 subunit hemizygosity. However, the heterozygous, haploinsufficient γ2 subunit knock-out mouse had a phenotype of hyper-anxiety, not epilepsy (Crestani et al., 1999), suggesting a relatively mild functional compromise of GABAAergic inhibition. Our data also suggested that the functional impairment produced by a nonfunctional, truncated γ2S(Q351X) subunit in the heterozygous condition was more severe than in the haploinsufficient condition. With coexpression of heterozygous γ2S/γ2S(Q351X) subunits and α1 and β2 subunits, currents and surface expression of α1β2γ2 receptors were reduced compared with the hemizygous control condition. Our study suggested that with heterozygous expression of γ2S/γ2S(Q351X) subunits, the reduction of whole-cell current and surface α1β2γ2 receptors relative to hemizygous control receptors was due to a dominant-negative effect of γ2S(Q351X) subunits. We demonstrated that γ2S(Q351X) subunits oligomerized with α1, β2, and γ2S subunits to form oligomers that were trapped in the ER, thus impairing assembly and forward trafficking of α1β2γ2S receptors to the cell surface and resulting in reduced whole-cell current.
The dominant-negative effect of γ2S(Q351X) subunits on α1 and β2 subunits was supported by both electrophysiological and Western blot results. With coexpression of γ2S(Q351X) subunits and α1 and β2 subunits, only very small currents that “ran down” rapidly were recorded. These small currents were abolished by increasing the concentration of γ2S(Q351X) subunit cDNA. This, suggested that the small current was likely to be produced by “escape” of α1β2 receptors from trapping by γ2S(Q351X) subunits, and the increasing amount of mutant protein increasingly reduced surface expression α1β2 receptors. With coexpression of γ2S(Q351X) subunits and α1 and β2 subunits, the wild-type partnering α1 and β2 subunits had reduced maturation and were retained in the ER. Similarly, the biochemical data demonstrated that an incremental increase of γ2S(Q351X) subunits progressively diminished the total expression of α1 subunits. Finally, α1, β2, and γ2S subunits were all coimmunoprecipitated with mutant γ2(Q351X) subunits. However, it is unclear whether oligomerization comes before glycosylation arrest, since both different glycosylated forms of α1 and β2 subunits appeared to be pulled down by γ2S(Q351X)FLAG subunits. The dominant-negative effect of γ2S(Q351X) subunits on γ2S subunits was supported by the observation that with heterozygous expression, less than one half of the total wild-type γ2S subunit was found with α1β2γ2S receptor expression, and total wild-type γ2S subunit levels were less than those of mutant subunits. In addition, wild-type and mutant γ2S subunits were demonstrated to coimmunoprecipitate. Together, these data suggest that the γ2S(Q351X) subunit accumulated intracellularly and oligomerized with wild-type α1, β2, and γ2S subunits, thus impairing assembly and trafficking of wild-type receptors.
Reduction of the total wild-type subunits was rapid and likely due to ERAD through the UPS
ER quality control only allows correctly folded and assembled molecules to mature and traffic to later compartments of the secretory pathway (Ellgaard and Helenius, 2003). Misfolded wild-type and mutant proteins like the cystic fibrosis transmembrane conductance regulator are substrates for proteasomal degradation (Jensen et al., 1995; Ward et al., 1995). We recently demonstrated that the misfolded mutant GABAA receptor α1(A322D) subunit was also degraded through the UPS (Gallagher et al., 2007). Here we show that in the presence of mutant γ2S(Q351X) subunits, wild-type partnering α1 subunits were subject to glycosylation arrest and degraded at least partially through the proteasomal system. We assume that the other wild-type partnering β2 subunit was subject to the similar cellular fate, but the magnitude of reduction for each subunit may be different due to different protein metabolic kinetics demonstrated in our pulse-chase experiments (data not shown). A dominant-negative effect of mutant subunits on wild-type subunits has been observed with other ion channel proteins including calcium channels (Page et al., 2004) and potassium channels (Gong et al., 2004). The basis for the dominant-negative effect of mutant proteins on wild-type proteins in these channels involves multimerization of wild-type and mutant proteins (Aizawa et al., 2004; Gong et al., 2004) and the unfolded protein response including ERAD (Page et al., 2004; Mezghrani et al., 2008). The same mechanism of suppression of wild-type subunit maturation has also been reported in autosomal dominant retinitis pigmentosa (Rajan and Kopito, 2005), thus suggesting that the promoted ERAD of wild-type protein may be a unifying mechanism underlying many dominant-negative diseases (Mezghrani et al., 2008). However, the lysosome pathway may also contribute to the reduction of the wild-type receptors in the presence of mutant γ2S(Q351X) subunits during receptor trafficking, endocytosis, and recycling, but this needs to be clarified.
Heterozygous expression of nonfunctional γ2S(Q351X) subunits and hemizygous γ2S subunits have different molecular pathologies
The dominant-negative effect of γ2S(Q351X) subunits with heterozygous expression may explain why hemizygous GABRG2 (+/−) gene-deletion and heterozygous γ2S(Q351X) mutation conditions are pathologically different. Although the mutant subunit is trafficking deficient, it can oligomerize with partnering wild-type subunits, thus preventing their maturation and promoting their degradation. This negative effect is not present in the hemizygous GABRG2 gene-deletion animal. In contrast, there is a small compensatory subunit increase in the hemizygous condition, resulting in slightly more than the half of the wild-type subunit expression. The underlying mechanisms could be related to the availability of chaperone proteins or other unknown reasons. The reason that γ2S(Q351X) subunits can oligomerize with wild-type subunits is probably due to their intact N termini, which have been demonstrated to mediate subunit assembly in both GABAA and nACh receptors (Verrall and Hall, 1992; Klausberger et al., 2001).
α1β2 receptors were formed due to the decrease in available γ2S subunits
Our data on hemizygous control receptors and studies of the heterozygous γ2 subunit knock-out mouse (Crestani et al., 1999) both suggested that α1β2 receptors are formed and trafficked to the surface with reduced expression of the γ2 subunit. This suggests that α1β2 receptors will be formed if γ2 subunits are reduced or unavailable, resulting in a slight compensation for the loss of GABAAergic inhibition due to the decrease in available γ2S subunits.
γ2S(Q351X) subunits result in both “loss of function” and “dominant-negative suppression”
This study is the first report of dominant-negative suppression of a naturally occurring mutant GABAA receptor subunit protein. The mutant protein could have a “gain of toxic function” in addition to the loss of the function of the mutant allele itself. For example, the presence of the mutant protein caused more degradation of the wild-type subunits. This effect could be due to the direct interaction of the mutant and wild-type subunits or partially to the mutant protein triggering ER stress or the unfolded protein response. This response is to reduce the burden on the ER resulting from unfolded or trafficking deficient molecules through a series of cellular processes including inhibition of transcription, rescue of unfolded protein by increasing the expression of chaperones and ERAD (Hampton, 2000; Rao et al., 2004). Future study focusing on transcription, recycling, and degradation may further elucidate the underlying mechanisms of this trafficking deficient mutant in epileptogenesis.
Footnotes
This work was supported by a Citizens United for Research in Epilepsy research grant to J.-Q.K. and National Institutes of Health Grants R01 NS33300 and NS51590 to R.L.M. We give special thanks to Dr. Gregory Mathews for assistance with the mIPSC analysis.
References
- Aizawa Y, Ueda K, Wu LM, Inagaki N, Hayashi T, Takahashi M, Ohta M, Kawano S, Hirano Y, Yasunami M, Aizawa Y, Kimura A, Hiraoka M. Truncated KCNQ1 mutant, A178fs/105, forms hetero-multimer channel with wild-type causing a dominant-negative suppression due to trafficking defect. FEBS Lett. 2004;574:145–150. doi: 10.1016/j.febslet.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–118. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, Suls A, Van Dyck T, Lagae L, Van Broeckhoven C, Macdonald RL, De Jonghe P. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–690. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Song L, Zhang H, Macdonald RL. Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci. 2002;22:5321–5327. doi: 10.1523/JNEUROSCI.22-13-05321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ. Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J Biol Chem. 1996;271:89–96. doi: 10.1074/jbc.271.1.89. [DOI] [PubMed] [Google Scholar]
- Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, Belzung C, Fritschy JM, Lüscher B, Mohler H. Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat Neurosci. 1999;2:833–839. doi: 10.1038/12207. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Lüscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Kang JQ, Song L, Dibbens L, Mulley J, Macdonald RL. δ subunit susceptibility variants E177A and R220H associated with complex epilepsy alter channel gating and surface expression of α4β2δ GABAA receptors. J Neurosci. 2006;26:1499–1506. doi: 10.1523/JNEUROSCI.2913-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MJ, Song L, Arain F, Macdonald RL. The juvenile myoclonic epilepsy GABAA receptor α1 subunit mutation A322D produces asymmetrical, subunit position-dependent reduction of heterozygous receptor currents and α1 subunit protein expression. J Neurosci. 2004;24:5570–5578. doi: 10.1523/JNEUROSCI.1301-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MJ, Shen W, Song L, Macdonald RL. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the alpha 1 subunit. J Biol Chem. 2005;280:37995–38004. doi: 10.1074/jbc.M508305200. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Ding L, Maheshwari A, Macdonald RL. The GABAA receptor alpha1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc Natl Acad Sci U S A. 2007;104:12999–13004. doi: 10.1073/pnas.0700163104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Keeney DR, Robinson JC, Zhou Z. Defective assembly and trafficking of mutant HERG channels with C-terminal truncations in long QT syndrome. J Mol Cell Cardiol. 2004;37:1225–1233. doi: 10.1016/j.yjmcc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Hampton RY. ER stress response: getting the UPR hand on misfolded proteins. Curr Biol. 2000;10:R518–R521. doi: 10.1016/s0960-9822(00)00583-2. [DOI] [PubMed] [Google Scholar]
- Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH, Richards MC, Williams DA, Mulley JC, Berkovic SF, Scheffer IE, Petrou S. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–536. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res. 2006;70(Suppl 1):S206–S217. doi: 10.1016/j.eplepsyres.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, Rebstock J, Heils A, Steinlein OK. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. The GABAA receptor γ2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of α1β2γ2S receptors in the endoplasmic reticulum. J Neurosci. 2004;24:8672–8677. doi: 10.1523/JNEUROSCI.2717-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci. 2006;26:2590–2597. doi: 10.1523/JNEUROSCI.4243-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Sarto I, Ehya N, Fuchs K, Furtmuller R, Mayer B, Huck S, Sieghart W. Alternate use of distinct intersubunit contacts controls GABAA receptor assembly and stoichiometry. J Neurosci. 2001;21:9124–9133. doi: 10.1523/JNEUROSCI.21-23-09124.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12:306–316. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ, Feng HJ. GABAA receptor mutations associated with generalized epilepsies. Adv Pharmacol. 2006;54:147–169. doi: 10.1016/s1054-3589(06)54007-4. [DOI] [PubMed] [Google Scholar]
- Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- Mezghrani A, Monteil A, Watschinger K, Sinnegger-Brauns MJ, Barrère C, Bourinet E, Nargeot J, Striessnig J, Lory P. A destructive interaction mechanism accounts for dominant-negative effects of misfolded mutants of voltage-gated calcium channels. J Neurosci. 2008;28:4501–4511. doi: 10.1523/JNEUROSCI.2844-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Page KM, Heblich F, Davies A, Butcher AJ, Leroy J, Bertaso F, Pratt WS, Dolphin AC. Dominant-negative calcium channel suppression by truncated constructs involves a kinase implicated in the unfolded protein response. J Neurosci. 2004;24:5400–5409. doi: 10.1523/JNEUROSCI.0553-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan RS, Kopito RR. Suppression of wild-type rhodopsin maturation by mutants linked to autosomal dominant retinitis pigmentosa. J Biol Chem. 2005;280:1284–1291. doi: 10.1074/jbc.M406448200. [DOI] [PubMed] [Google Scholar]
- Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- Schweizer C, Balsiger S, Bluethmann H, Mansuy IM, Fritschy JM, Mohler H, Lüscher B. The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol Cell Neurosci. 2003;24:442–450. doi: 10.1016/s1044-7431(03)00202-1. [DOI] [PubMed] [Google Scholar]
- Verrall S, Hall ZW. The N-terminal domains of acetylcholine receptor subunits contain recognition signals for the initial steps of receptor assembly. Cell. 1992;68:23–31. doi: 10.1016/0092-8674(92)90203-o. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]