

Abstract
Cells can respond to reductions in oxygen (hypoxia) by metabolic adaptations, quiescence or cell death (1). The nuclear division cycles of syncytial stage Drosophila melanogaster embryos reversibly arrest upon hypoxia. We examined this rapid arrest in real time using a fusion of green fluorescent protein and histone 2A. In addition to an interphase arrest, mitosis was specifically blocked in metaphase, much like a checkpoint arrest. Nitric oxide, recently proposed as a hypoxia signal in Drosophila, induced a reversible arrest of the nuclear divisions comparable with that induced by hypoxia. Syncytial stage embryos die during prolonged hypoxia, whereas post-gastrulation embryos (cellularized) survive (2, 3). We examined ATP levels and morphology of syncytial and cellularized embryos arrested by hypoxia, nitric oxide, or cyanide. Upon oxygen deprivation, the ATP levels declined only slightly in cellularized embryos and more substantially in syncytial embryos. Reversal of hypoxia restored ATP levels and relieved the cell cycle and developmental arrests. However, morphological abnormalities suggested that syncytial embryos suffered irreversible disruption of developmental programs. Our results suggest that nitric oxide plays a role in the response of the syncytial embryo to hypoxia but that it is not the sole mediator of these responses.
Oxygen is essential for the life of most multi-cellular organisms. Limitations in oxygen have profound physiological and health consequences in humans. Prolonged or severe decreases in physiological oxygen resulting from ischemia are a major contributor to morbidity and mortality in stroke and cardiac infarction (4). Cells in pretumorous growths are often deprived of oxygen as a result of insufficient and inefficient vascularization, and this hypoxia (low oxygen) is postulated to play a role in limiting tumor progression (5). The outcome of hypoxia can differ dramatically. For example, a turtle can survive months of hypoxic conditions, whereas the cells of the human brain begin to die following a few minutes of hypoxia (6). These outcomes presumably depend on the cellular responses to hypoxia, which range from metabolic adaptation to quiescence to apoptosis (1). Among the cellular responses to hypoxia is an arrest of the cell cycle (1, 5). Hypoxia is reported to arrest mammalian cells during G1, mid-S phase, and G2/M. Analysis of the response to hypoxia in Drosophila has similarly revealed an arrest of the cell cycle in mid-S phase and mitosis (2, 3). Despite the importance of these cellular responses, the signals mediating responses to hypoxia are ill defined, and the factors influencing the type of response are poorly understood. What allows some cells to survive when others die?
In this study, we examined the responses to hypoxia during Drosophila embryogenesis. The development of Drosophila from egg to larva is remarkably fast and dynamic. Development begins with 13 rapid mitotic divisions in a common cytoplasm, referred to as the syncytial stage. Next, cell membranes invaginate and form around the nuclei, referred to as cellularization. Following cellularization, the germ layers are formed during gastrulation, the body plan is defined, and tissue types begin to form. Finally, about 24 h after fertilized oocyte deposition, the embryo hatches into a larva. The ability to survive hypoxia is likely to be an important adaptive mechanism in an organism whose eggs develop on rotting fruit where microbial proliferation can deplete oxygen. It was previously reported that Drosophila embryos progress from being sensitive to hypoxia to being remarkably resistant (2). The early syncytial stage embryo is quite sensitive and can only survive a brief period of hypoxia. However, starting at about 2 h after cellularization, embryos survive almost complete anoxia. Cellularized, stage 9 embryos arrest upon depletion of oxygen, remain quiescent for a week, and successfully resume development upon restoration of oxygen.1
Recent work has suggested that the survival of Drosophila larvae and older embryos during hypoxia involves active responses that induce widespread changes in behavior, cell cycle progression, and development (2, 3, 7). These responses appear to be governed, in part, by nitric oxide (3). NO is a signaling molecule in mammals that contributes to neuronal signaling, the innate immune response, and regulation of blood vessel tone. In Drosophila, NO has also been shown to influence synaptogenesis during the development of the nervous system (8), to negatively regulate the cell cycle during imaginal disc growth (9), and to be involved with an S phase block and a change in larval behavior in response to hypoxia (3). NO involvement in the response to hypoxia in Drosophila has parallels with one of its roles in vertebrates in which it induces vasodilation and increased blood flow in response to local limitations in oxygen (10).
Exceptional features of the syncytial embryo such as the synchrony and speed of the nuclear divisions and the clear cytology provide an ideal system for characterization of the effect of hypoxia and NO on cell cycle progression. We examined the kinetics of the hypoxic arrest in real time, demonstrated a comparable arrest by NO, and characterized the reversal of these arrests at the level of metabolism, the cell cycle, and development. Rapid and reversible arrest of the nuclear divisions occurred at two stages: interphase and metaphase. The phenocopy of the hypoxia arrest by treatment with NO suggests a role for NO in the hypoxic response. Also, measurements of ATP levels and cyanide treatments of embryos reveal metabolic changes that could contribute to the cell cycle arrest in response to hypoxia. By following recovery after prolonged hypoxia, we showed that the failure of syncytial embryos to hatch after exposure to hypoxia was due to detrimental effects on development and did not reflect an inability of the embryo to recover metabolically or restore cell cycle progression.
Experimental Procedures
Embryo Collection, Fixation, and Staining
Sevelen was used as a wild-type stock. All embryo collections, fixations, and stainings were performed as described previously (11, 12). ≥95% of the embryos in the collections were of the proper age with a contamination of other unfertilized or older stage embryos (data not shown). Fixed images were acquired using an upright fluorescent microscope (Leica DMRD).
Hatching Assay
Hatching assays were performed by first washing treated embryos with a NaCl/Triton solution (0.7% NaCl, 0.03% Triton X-100). A known number of embryos were then placed on a grape juice agar plate, incubated for 24–30 h at 24 °C, and counted for the number of eggs that hatched into larvae.
Hypoxic, NO, and Cyanide Treatments
For all experiments, N2 and O2 gas was mixed to achieve desired oxygen levels. The mixture was humidified, and output oxygen was measured using an oxygen sensor (Reming Bioinstruments, model PROOX 110).
Different stage embryos were collected, dechorionated with 50% bleach, and resuspended in an aerated NaCl/Triton solution. A phosphate-buffered saline (PBS)2 solution was perfused with mixed N2/O2 air at least 15 min prior to treatment to equilibrate the solution to desired oxygen concentrations. Embryos were made hypoxic by replacing the NaCl/Triton solution with deoxygenated PBS + 0.05% Triton X-100. For NO and cyanide treatments, 10 mm s-nitroacetylpenicillamine (SNAP) or 0.2% sodium cyanide in PBS was used. Embryos were then rocked in an Eppendorf tube containing the specified PBS solution while lightly blowing N2/O2 gas mixture into the Eppendorf tube to replace the oxygenated air (for hypoxia treatments only). To return to normoxic conditions, the deoxygenated PBS was replaced with the NaCl/Triton solution and rocked gently. Prolonged exposures to hypoxia were performed as described previously (3).
Real Time Analysis of Living Drosophila Embryos
Embryos were collected from a transgenic line expressing a fusion protein of histone 2A and green fluorescent protein (GFP) (13). Embryos were dechorionated with 50% bleach, aligned on a glass coverslip, and placed under halocarbon oil or PBS to prevent dehydration. The GFP fluorescent signal was visualized in real time using an Olympus IX-70 microscope, and images were recorded using Delta Vision Version 2.10 software (Applied Scientific, Inc.). While monitoring chromosome dynamics live, embryos were made hypoxic by blowing a stream of N2/O2 gas onto specific embryos through a Pasteur pipette. Embryos were treated with NO donors or CN− by removing the PBS solution and replacing it with 10 mm SNAP or 0.2% sodium cyanide dissolved in PBS.
Preparation of Embryonic Extracts for ATP Determination
Embryonic extracts were made by resuspending 25–100 μl of embryos in 200–1000 μl of glycine buffer (O.2 m glycine, 50 mm MgCl2, 4 mm EDTA, pH 7.4) contained within a 1.5-ml Eppendorf tube. The embryo/buffer solution was then sonicated on ice with 1 s pulses from a microtip of a Branson Sonifier (model 185). Extracts were centrifuged for 5 min at 14,000 RPM (16,000 relative centrifugal force). The soluble supernatant was saved, and protein concentration was determined using the BCA assay (Pierce). Extracts were diluted to 2 mg/ml in glycine buffer and boiled for 45 s to precipitate protein. After a 15-min centrifugation, supernatants were transferred to fresh tubes, diluted, and measured for ATP concentration (see below).
Luciferase-Luciferin Assay for ATP Levels
The luciferase-luciferin assay for ATP levels was modified from a protocol provided with the luciferase-luciferin mix (Sigma catalog number L0633). Briefly, 20 μl of diluted extract with protein removed (see above) was mixed with 100 μl of 1 mg/ml luciferase-luciferin mix diluted in water. The mixture was placed in a small scintillation vial, and photon counts were measured using the “single-photon monitor” mode of a Beckman scintillation counter (model LS 1801). Amounts of ATP in extracts were calculated using a standard curve generated from known ATP standards. All data are represented as percentages of the “normoxic” control, which is a sample treated under similar conditions but kept under normal ambient oxygen conditions.
Software Image Processing and Graphing
All images and graphs were processed using Adobe Photoshop version 5.0 and Adobe Illustrator version 8.0. All graphs were plotted in Kaleidagraph version 3.0.5 (Abelbeck Software).
Results
Consistent with previous studies (2, 3), syncytial embryos (1–2 h old) are considerably more sensitive than cellularized (7–8 h old) embryos to the hypoxia treatments used during this study (Fig. 1). Older embryos in the original collection are sufficient to account for the low level of syncytial stage embryos (3 ± 1%) that survive 24 h of hypoxia. In contrast to this sensitivity of syncytial embryos, 82 ± 5% of cellularized embryos (7–8 h old) made hypoxic for 24 h survive to hatch into larvae (Fig. 1). The decline in hypoxia sensitivity begins as embryos pass two developmental milestones, cellularization, and the beginning of gastrulation (Refs. 2 and 3 and data not shown).
Fig. 1. Survival of syncytial and cellularized embryos after hypoxia.

Syncytial (1–2 h old) and cellularized (7–8 h) stage Drosophila embryos were made hypoxic and then assayed for survival. Embryos were incubated for 13.5 or 24 h in a saline solution perfused with <0.1% O2 in a O2/N2 mixture (a 200-fold reduction in O2 levels) and then placed on an agar plate under normal ambient oxygen levels (normoxic). Embryonic survival after hypoxia was scored as the number of embryos that hatched into larvae. The bar graph is the percentage of embryos exposed to hypoxia that hatched into larvae compared with control embryos without treatment. 27 ± 1 and 3 ± 1% of syncytial embryos survived to hatching after 13.5- and 24-h exposures to hypoxia, respectively. In contrast, 95 ± 1 and 82 ± 5% of older, cellularized embryos survived to hatching after 13.5- and 24-h hypoxic treatments. The error bars represent the standard error.
Hypoxia Arrests the Syncytial Nuclear Divisions in Metaphase and Interphase
A transgenic line expressing a fusion protein of histone 2A and green fluorescent protein (referred to as His-GFP; Ref. 13) was used to visualize chromosome condensation and movements. Visualization and imaging of embryos prior to hypoxia allowed precise staging of the cell cycle. Hypoxia was imposed without interruption of imaging by blowing a stream of gas (humidified nitrogen or air/nitrogen mixtures) on the embryos. This approach allowed us to determine how hypoxia influences individual events during mitosis and to define the detailed kinetics of arrest and release from hypoxia even over the short (7–8 min at 22 °C) time course of a normal mitosis (Fig. 2). We limited observations to embryos beyond the ninth mitotic cycle because the behavior of the His-GFP severely limits the quality of the imaging at earlier stages. Fig. 2 gives an example of a syncytial nuclear division from interphase through mitosis and into the following interphase. It is important to note that metaphase is short, lasting only 2–3 min (Fig. 2).
Fig. 2. Live, real time visualization of chromatin dynamics and cell cycle progression.

Syncytial stage embryos expressing a fusion protein of histone 2A and green fluorescent protein (His-GFP) were used to visualize chromatin in living embryos. A representative example of the syncytial, synchronous, nuclear divisions, progressing through mitosis with normal morphology and timing is shown. Decondensed DNA in interphase nuclei (A) began to condense as nuclei progressed into prophase (B). The condensed chromatin aligned along the mitotic spindle in metaphase (C). Normal metaphase persisted 2–3 min (C and D), followed by the separation and segregation of the DNA masses during anaphase (D). Finally, the chromatin decondenses in late telophase (E), and the cycle began again. The arrows indicate a single nucleus followed through mitosis (total length ∼8 min) The scale bar in E corresponds to 10 μm.
The nuclei of embryos made hypoxic in prophase progressed at normal rates through prophase and prometaphase and achieved a normal alignment of the chromatin on the metaphase plate (Fig. 3, A and B). However, progress to anaphase was blocked. The arrest in metaphase persisted for the duration of the hypoxic treatment (≤25 min). While blocked in metaphase, the nuclei adopted a hypercondensed morphology (Fig. 3, compare B and C). Upon restoration of oxygen, the nuclei delayed 2–8 min in metaphase and then exited mitosis with kinetics similar to controls. In contrast to a previous report (2), using real time observation we observed that the metaphase-anaphase transition is the only observed arrest point in mitosis. Nuclei made hypoxic in late metaphase/anaphase did not arrest in mitosis. However, progression through anaphase was slowed. Nonetheless, the nuclei reached telophase, the chromatin decondensed, and the nuclei took on an interphase-like morphology (data not shown).
Fig. 3. Hypoxia reversibly blocks the metaphase-anaphase transition.

His-GFP syncytial embryos were made hypoxic while visualizing chromosome dynamics in real time. Nuclei entering prophase (A) under normal oxygenated conditions were made hypoxic by blowing a stream of <0.1% O2 on the entire embryo. The hypoxic nuclei progressed normally into metaphase (B) and remained blocked in metaphase for the duration of the treatment (10 min), adopting a stereotypical hypercondensed DNA morphology (C). Normally, metaphase is 2–3 min. Upon reoxygenation with ambient air; the nuclei delayed 4 min in metaphase and then progressed with normal kinetics through anaphase (D) and into the next nuclear cycle. The arrows indicate a single nucleus during the treatment, and the scale bar in D corresponds to 10 μm.
The metaphase arrest is not unique to the syncytial cycles. Analysis of the later stage, cellularized embryos (stages 8–12) showed that hypoxia induced an identical type of arrest at metaphase. The kinetics of the arrest and its reversal resembled that of the syncytial embryos (data not shown).
As previously reported, hypoxia also causes interphase cells to arrest with an unusual nuclear morphology (2, 3). We examined the kinetics of this response. Interphase cells made hypoxic did not progress into mitosis and remained in interphase (data not shown). The interphase nuclei increased in volume, and chromatin became hypercondensed within 2–3 min of hypoxia. Like the metaphase arrest, this arrest was reversed by restoration of oxygen. Control embryos gassed with ambient air did not arrest.
Hypoxia blocked specific stages of the cell cycle, whereas other steps appeared normal or slowed. The specificity and reversibility of the arrest in metaphase suggest that the arrest behaves like a checkpoint. By observing the response to oxygen deprivation in live embryos, the specificity of the metaphase arrest in mitosis is more clearly illustrated.
NO Causes an Arrest of the Syncytial Nuclear Divisions
NO, which usually acts to signal between cells (10), has been implicated in the S phase arrest in cellularized embryos and larvae (3). We tested its effect on mitosis in the syncytial embryo. His-GFP embryos were treated with a NO producing agent, SNAP, in a saline solution while visualizing cell cycle stage as described above. Treatment of syncytial embryos with 10 mm SNAP caused a reversible arrest of interphase (data not shown) and metaphase (Fig. 4) with similar kinetics and morphology to that seen with hypoxia. Fig. 4 shows an embryo treated with the NO donor, SNAP, as the nuclei entered prophase. Nuclei progressed normally into metaphase but remained blocked in metaphase for the duration of the SNAP exposure (10 min). The nuclei arrested in metaphase adopted a hypercondensed chromatin morphology similar to hypoxic arrested metaphase nuclei (Fig. 4C). The effects of NO were reversible; nuclei exited mitosis normally within 3 min after removal of SNAP (Fig. 4D). Similar effects in blocking the metaphase-anaphase transition were also observed in older, cellularized embryos exposed to NO donors (data not shown).
Fig. 4. NO treatment arrests the syncytial nuclear division in metaphase.

His-GFP syncytial embryos were used to monitor the effect of NO treatment on the progression of the nuclear divisions in the absence of hypoxia in real time (see Fig. 3). When the nuclei were in prophase, as indicated by condensed chromatin (A), the embryos were treated with 10 mm SNAP, an NO donor, in saline solution. Nuclei progressed normally into metaphase (B) but remained blocked in metaphase with hypercondensed DNA (C) for the duration of the 10 min treatment. The embryos were then washed with saline solution to remove the NO-donor. Nuclei exhibited a 3-min delay and then exited mitosis normally. Anaphase was synchronous and with normal morphology and timing after the wash (D). The arrows follow a single nucleus through the treatment. The scale bar in D corresponds to 10 μm.
The data show that NO can provoke an arrest of the cell cycle in the syncytial embryo that resembles the arrest caused by hypoxia. The possible role of NO as a mediator of the hypoxic response is presented under “Discussion.”
Effects of Hypoxia on ATP Levels
We sought to determine whether the arrest of embryogenesis protects the embryo from a drop in ATP levels during hypoxia. Generally, oxidative phosphorylation uses molecular oxygen as the final electron acceptor in the coupling of electron transport and the conversion of ADP into ATP. Pronounced reductions in oxygen levels should slow the generation of ATP, leading to a drop in ATP levels in the absence of compensating responses. Levels of ATP were monitored as a measure of the ability of Drosophila embryos to accommodate reduced oxygen availability. Using a modification of a luciferase-based assay, total ATP levels were measured in embryonic cell extracts from different stage embryos under various treatment conditions.
Syncytial (1–2 h old) and cellularized embryos (>3.5 h old) were made hypoxic with ≤0.1% O2 for different lengths of time, and total ATP levels were determined (Fig. 5A). ATP levels decreased within 5 min of treatment at all stages examined. However, a difference between the rates of ATP decline was observed between cellularized and syncytial stage embryos. In syncytial stage embryos, ATP levels dropped to 65 ± 4% of control embryos within 5 min and continued to decline to 38 ± 2% (Fig. 5A) at 30 min of hypoxia (the minimal level observed is 29 ± 1%; see Fig. 7). In contrast, in cellularized, older embryos, ATP levels appeared to level off at 75% of controls within 15 min (Fig. 5A) and did not further decline after prolonged hypoxic treatments of >90 min (data not shown). These results suggest that cellularized embryos are better equipped to metabolically adjust to hypoxia than syncytial embryos.
Fig. 5. ATP levels in response to hypoxic treatment.

Embryos were treated with hypoxia and assayed for total ATP levels during or after hypoxic treatment. The amount of ATP levels are represented as the percentage of control, untreated embryos and plotted over time of hypoxic exposure or recovery. A, syncytial (1–2 h old) and cellularized, older embryos (4–8 h) were made hypoxic with <0.1% O2 and assayed for total ATP levels. ATP levels showed declines for both syncytial and cellularized stages, but at different rates, reaching 38 ± 2 and 73 ± 2% after 30 min of hypoxia, respectively. B, syncytial embryos were made hypoxic with different mixtures of O2/N2 to achieve final oxygen concentrations of <0.1%, 1.0%, and 2.0% O2. ATP levels declined for <0.1 and 1.0% O2 (38 ± 2 and 61 ± 2% after 30 min, respectively) but did not significantly decline upon exposure to 2.0% O2 (94 ± 2% after 30 min). C, recovery of ATP levels in syncytial stage embryos after 30 min of hypoxic exposure was tested. Embryos were made hypoxic for 30 min with <0.1% O2 and then transferred to oxygenated saline solution to recover. ATP levels declined to 38 ± 2% of controls during the hypoxic treatment (see also A and B) and recovered immediately after reoxygenation, returning to 86 ± 2% after 5 min. Error bars represent standard error.
Fig. 7. Recovery of ATP levels after prolonged hypoxia.

ATP levels were assayed during recovery from prolonged, lethal exposures to hypoxia. Syncytial embryos were exposed to <0.1% O2 for 13.5 h and then transferred to an oxygenated saline solution to recover for 3 h. ATP levels were assayed immediately after the hypoxic exposure (left column) and after the 3 h recovery (right column). ATP levels declined to 29 ± 1% after 13.5 h of hypoxia and recovered to 93 ± 6% of controls 3 h after reoxygenation. Error bars represent standard error.
Because both syncytial and cellular embryos arrest in metaphase with similar kinetics, it is unlikely that the dissimilar drop in ATP levels directly causes the metaphase arrest in both stages. It remains possible that a decline in ATP levels arrests syncytial embryos, whereas a distinct mechanism mediates the similar metaphase arrest of the later stage embryos. If the decrease in ATP levels caused the cell cycle and developmental arrest of syncytial embryos, the decline in ATP levels and the responses ought to be tightly correlated during this stage.
To test whether there is a correlation in ATP levels and the observed effects of hypoxia, we compared the threshold in oxygen concentrations at which responses (cell cycle and developmental arrests) are triggered to concentrations that cause ATP decline in syncytial embryos. Treatment of syncytial embryos with 2% O2, failed to arrest the cell cycle or development, and survival was not decreased. Conversely, exposure to 1% O2 arrested the cell cycle and development at all stages tested (data not shown). Likewise, embryonic and larval survival was decreased. Thus, a transition in response to oxygen deprivation occurred between 1 and 2% O2. ATP levels were determined for embryos exposed to different O2 levels. ATP levels declined in response to <0.1 and 1% but not to 2% O2 treatments (Fig. 5B). These results suggest a parallel between the oxygen requirements for sustaining ATP levels and for continued development and cell cycle progression in the syncytial embryo. However, a more detailed analysis of the correlation would be necessary to examine the parallel in the rates of the decline of ATP and the timing of the metaphase block at different O2 concentrations. At the level of the present analysis, the correlation is unclear. That is, at 5 min after imposition of hypoxia, the ATP levels declined about 12% in 1% O2, whereas the drop was 35% in <0.1% O2, yet both of these treatments resulted in a metaphase arrest. However, our ability to score a difference between the rate at which nuclei arrest is limited. Metaphase arrest is observed about 4 min after the beginning of the hypoxic treatment. We do not know when during the 4 min between the imposition of hypoxia and the metaphase block, oxygen concentration is sensed and elicits an arrest. We can, however, conclude that if the decline in ATP mediates the response to oxygen, cells would have to respond to a remarkably small decline in ATP (i.e. considerably less than the 12% decline seen at 5 min).
The kinetics of ATP recovery was assayed after 30 min of <0.1% O2 treatment (Fig. 5C). ATP levels quickly (within 5 min) recovered to maximal levels. ATP levels are fully recovered by the time cell cycle progression is renewed. These data cannot discount the possibility that the restoration of ATP levels is responsible for the reversal of the arrest (see “Discussion”).
Inhibition of Oxidative Phosphorylation Is Sufficient to Arrest the Nuclear Divisions
Because ATP declines in response to hypoxia, a failure to maintain ATP levels (and other metabolites) could contribute directly or indirectly to the hypoxic responses (i.e. cell cycle arrest). Results from a previous study have shown that treatment of syncytial embryos with dinitrophenol caused a mitotic arrest, among other effects (7). To test whether an inhibition of oxidative phosphorylation in the absence of hypoxia is sufficient to arrest the syncytial nuclear divisions, an inhibitor of oxidative phosphorylation, CN−, was used to decrease ATP levels.
Cyanide was titrated to a concentration that effectively reduced ATP levels with similar kinetics to that of hypoxia (Fig. 6). Dechorionated embryos treated with 0.02% CN− were assayed for ATP levels compared with control, untreated embryos. With 0.02% CN− treatment, ATP levels declined with similar kinetics to hypoxia (56% for CN− to 52% for hypoxia after 15 min of treatment; Figs. 5 and 6). Using the His-GFP syncytial embryos, the effect of CN− treatment on the syncytial nuclear divisions was determined. As in the case of hypoxia and NO addition, administering 0.02% CN− at prophase and interphase blocked the nuclear divisions in metaphase and interphase, respectively. Hence, inhibiting oxidative phosphorylation in the absence of hypoxia is sufficient to arrest the cell cycle. The reversibility was not tested because CN− is an irreversible inhibitor.
Fig. 6. ATP levels upon treatment with an inhibitor to oxidative phosphorylation and an NO donor.
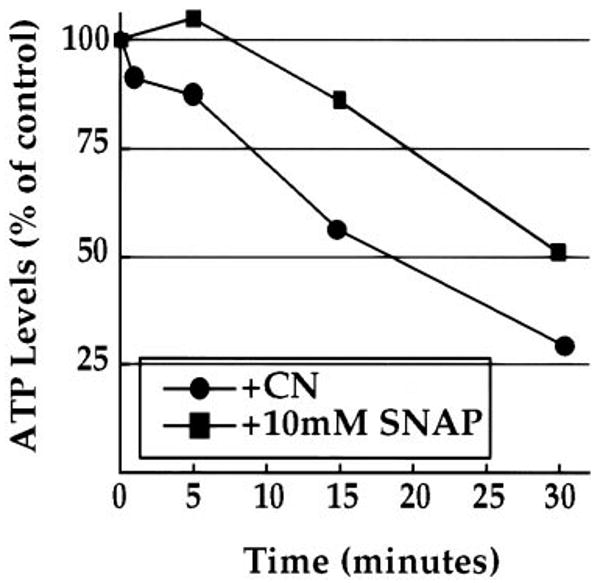
ATP levels were assayed in syncytial embryos treated with cyanide (CN−), an irreversible inhibitor of oxidative phosphorylation, or the NO donor, SNAP. Fig. 6 is a plot of ATP levels (represented as the percentage of the normoxic, untreated control embryos) versus time of treatment. During 0.2% CN− treatment, ATP levels declined with an intermediate rate compared with exposure to <0.1 and 1.0% O2 (see Fig. 5B), decaying to 30% of untreated controls after 30 min. During treatment with 10 mm of SNAP, ATP levels remained high after 5 min (105% of untreated controls) and then began to decline after 15 min (86% of untreated controls).
In summary, hypoxia, NO, and inhibition of oxidative phosphorylation are each sufficient to arrest the syncytial nuclear division in interphase and metaphase. It was also shown that hypoxia and CN− cause a decline in ATP levels. Treatment of syncytial stage embryos with the NO donor, SNAP also caused a decline in ATP, but the decline was delayed (Fig. 6). There was an approximately 5–10-min lag before the decline in ATP levels. Because the arrest of the cell cycle by NO occurs within a minute, there is discordance between ATP decline and the biological response upon treatment with the NO donor. This disparity suggests that NO has an ATP-independent mode of controlling cell cycle progress (see “Discussion”).
Recovery and Subsequent Developmental Failure Following Prolonged Hypoxia
As shown above, normal cell cycle, development and ATP levels are rapidly restored following short, sublethal exposures to hypoxia. We examined these same parameters after prolonged hypoxia to investigate the basis for the unique vulnerability of syncytial embryos exposed to hypoxia. Fig. 1 shows that 13.5- and 24-h exposures to <0.1% O2 reduce the survival of syncytial stage embryos to 23 ± 1% and 3 ± 1% of control embryos, respectively. The same treatments do not significantly reduce the survival of older embryos.
To assay the recovery of ATP metabolism after a lethal exposure to hypoxia, syncytial embryos were made hypoxic for 13.5 h and shifted to an oxygenated saline solution for 3 h. ATP levels dropped to 29 ± 1% of control embryos after 13.5 h of hypoxia but returned to 93 ± 6% of the normoxic controls within 3 h of oxygen restoration (Fig. 7). Similar results were observed with 24-h hypoxic treatments of syncytial embryos (data not shown). We conclude that ATP levels recover rapidly following hypoxia and that a persistent failure of ATP metabolism does not contribute to the embryonic lethality of prolonged hypoxia.
To test for restoration of cell cycle progression following prolonged hypoxia, syncytial embryos were made hypoxic for 13.5 h, returned to normoxic conditions for 3 h, and then fixed and assessed for their cell cycle and developmental state by visualizing the DNA (not shown) and microtubules (Fig. 8). Embryos fixed early during hypoxia and at the end of hypoxia showed the same distribution of metaphase and interphase arrest types with hypercondensed DNA morphology. There was no evidence of progression of the cell cycle or development during this prolonged hypoxia. Upon oxygen restoration, some embryos (<30%) failed to reinitiate nuclear cycles or did so abnormally. These embryos remained either uncellularized with aberrant DNA masses and spindle morphology or remained arrested with no morphological defects (Fig. 8A). In contrast, the majority of the embryos (>70%) reinitiated mitotic cycles and reached a cell density comparable with control embryos not subjected to the 13.5 h of hypoxia (Fig. 8, B–D). Although <30% of the embryos demonstrated defective cell divisions after prolonged hypoxia, this cannot account for the >75% decrease in survival. These data suggest that most syncytial stage embryos still remain competent to re-enter the cell cycle even after a lethal, prolonged period of hypoxia.
Fig. 8. Cell cycle progression and development after prolonged hypoxia.
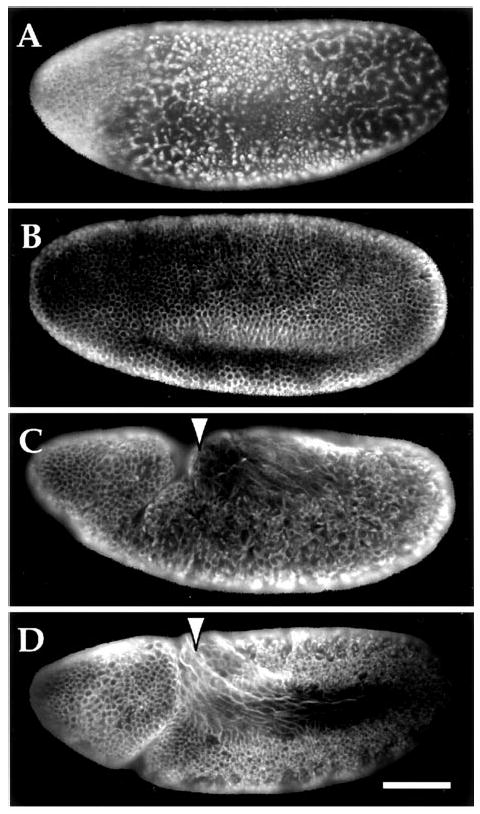
Syncytial embryos were made hypoxic for 13.5 h with <0.1% O2 and allowed to recover in oxygenated saline solution for 3 h. Embryos were then fixed and stained with antibody specific to α-tubulin to monitor cell cycle progression and morphology. Examples of classes of observed phenotypes are shown (A–C), including an untreated control with normal morphology and cell density (D). A shows an example of an embryo that has failed to cellularize or undergo any scorable gastrulation events. In addition, the nuclear division cycles appear to be delayed or blocked, asynchronous, and display aberrant mitotic spindle morphology. B, this embryo is an example of a class of embryos that has attained proper cell density and demonstrates normal morphology and cellularization. However, development was delayed and gastrulation events or germband extension are not detectable (compare with D). C, this embryo is typical of a class that appears to have advanced the most during recovery. Almost normal cell density was achieved and gastrulation has begun; however, there is aberrant morphology and perturbed germband extension (see arrow). D, an untreated control embryo that has cellularized, begun to gastrulate, and germband extend (see arrow). The scale bar in D corresponds to 65 μm.
The ability of embryos to recommence development following prolonged hypoxia was tested by examining their progression to later stages of development, including cellularization, gastrulation, and germband extension. Upon reperfusion of oxygen, some embryos (<30%) reinitiated mitotic division but exhibited early developmental defects, failing to cellularize either partially or completely (Fig. 8A). Another group of embryos (<30%) exhibited normal morphologies but appeared to have been delayed. They became cellularized but not germband extended (Fig. 8B). Another group (∼30%) had extended germ-bands and appeared to be at stages comparable with the control embryos (Fig. 8, C versus D); however, these embryos displayed variable abnormalities in morphology and cell density. It is not clear whether the few embryos that survived arose from only one of these classes. We conclude that a lethal exposure to hypoxia does not preclude further development of syncytial embryos, but the ensuing development is highly abnormal.
In summary, at least a significant fraction of syncytial embryos exposed to prolonged hypoxia are competent to restore their ATP levels, re-enter the cell cycle, and resume development. Based on the observed morphological defects, we infer that defects in development are responsible for much of the lethality of the prolonged hypoxia. In contrast, older embryos exposed to prolonged hypoxia show few morphological defects upon restoration of oxygen, which are presumably corrected as these embryos develop into normal flies (data not shown).
Discussion
Despite the key role that oxygen plays in the life of most organisms, little is known about the cellular mechanisms and factors governing survival of oxygen deprivation. We provide evidence that hypoxia and NO elicit a number of responses in syncytial and cellularized Drosophila embryos, including checkpoint-like arrests of the cell cycle and development.
Hypoxia Reversibly Blocks the Metaphase-Anaphase Transition
The gap phases in the cell cycle seem like natural times for cells to arrest. Indeed, cells arrest at these stages when quiescence is induced by nutritional or growth factor deprivation. However, hypoxia appears to act at additional stages (3, 5). Studies using mammalian cell lines have reported situations in which hypoxia causes cells to accumulate with G1, S, or G2/M DNA content. In Drosophila embryos, hypoxia arrests cells within S phase and within mitosis during syncytial stages (2, 7). Using real time methods, we have characterized the effects of hypoxia on each step of mitosis.
Following the undisturbed progression through prophase, hypoxic nuclei arrest in metaphase with a hypercondensed morphology (Fig. 3C). Many important steps of mitosis (nuclear envelope breakdown, mitotic spindle assembly and breakdown, centrosome migration, chromosome movements, and cytokinesis) proceed normally or are only slightly delayed in the absence of oxygen. In contrast, the events of the metaphase-anaphase transition, such as sister chromatid segregation and separation, are specifically blocked. We have followed metaphase-arrested cells in living embryos for more than 20 min, and the arrest persisted. Analysis of fixed embryos shows that metaphase arrested cells are still present after 13.5 h of hypoxia. We conclude that a step required for the metaphase/anaphase transition is efficiently blocked during hypoxia.
The metaphase block is reversible; upon oxygen restoration the nuclei re-entered the cycle after ≥2 min (Fig. 3). The delay in onset of anaphase increases with the duration of hypoxia (Fig. 3, B and C). Presumably, recovery and completion of metaphase events occur during this time. Despite increased recovery time, syncytial embryos re-enter the cell cycle after a prolonged (≥13.5 h) and lethal exposure to hypoxia (Fig. 8).
In addition to a metaphase arrest, a previous study of fixed preparations had suggested that a prophase arrest occurred during hypoxia (2). In contrast, by following cells from the moment of oxygen depletion, we show that prophase cells continue to metaphase with no obvious delay. We attribute the presence of prophase-like cells in the fixed preparations of this previous study to interphase cells, which upon hypoxia assume a hypercondensed DNA morphology similar to prophase nuclei.
The remarkable speed of the metaphase arrest allows us to estimate how quickly the embryo responds to hypoxia. Prophase nuclei ordinarily progress through metaphase into anaphase in 4 min. Because prophase nuclei arrest at metaphase when made hypoxic, we conclude that nuclei respond within this 4 min between the end of prophase and the beginning of anaphase. The failure of metaphase nuclei to arrest could be explained if the time required to respond to hypoxia is equivalent to the length of metaphase or if the oxygen-requiring step is at the beginning of metaphase.
The specificity and reversibility of the arrest in metaphase suggests that the arrest behaves like a checkpoint. Checkpoints prevent progression of the cell cycle during stress by blocking at precise stages such as the metaphase-anaphase transition or S phase (14). Most cell cycle checkpoints involve signaling networks that sense a stress (i.e. DNA or mitotic spindle damage) and modulate the activity of certain cell cycle regulators. For example, the spindle assembly checkpoint is thought to sense damage to or improper formation of the mitotic spindle and block the metaphase-anaphase transition by stabilizing substrates whose degradation is normally required to exit mitosis (15). We suggest that arrest at the metaphase-anaphase transition acts as a checkpoint protecting the cell from segregation errors that might occur if cells progress through anaphase in the absence of oxygen. This interpretation is supported by the slowed and abnormal anaphase that occurs in the few hypoxic cells that escape the checkpoint (data not shown).
Nitric Oxide Mimics Hypoxia
NO exposure of cellularized Drosophila embryos and larvae induces responses characteristic of hypoxia, and inhibition of NO generation suggests that it mediates at least some of the responses to hypoxia (3, 9). To extend the tests of the ability of NO to phenocopy hypoxia, we asked whether NO would induce a metaphase arrest.
Using NO donors and inhibitors, we tested whether NO can mimic hypoxia and is required for the normal responses to hypoxia. Exposure to a nitric oxide donor, SNAP, arrested syncytial embryos with kinetics, specificity, and morphology that resemble the hypoxic arrest (Fig. 4). NO provokes a response that accurately mimics the response to hypoxia. Pretreatment of syncytial embryos with an inhibitor to nitric oxide, 2-phenyl-4,4,5,5-tetramethylimidazoline-1oxyl-3-oxide (PTIO), suggests a requirement for NO in the hypoxic interphase arrest. His-GFP, syncytial embryos were microinjected with PTIO as a pretreatment and assayed live for their response to hypoxic treatments. When the nuclei were synchronously in interphase, the embryos were exposed to hypoxia. During hypoxia, interphase nuclei of PTIO-treated embryos progressed slowly to assume a more compact shape and assemble a mitotic spindle (data not shown). This terminal mitotic-like state suggests that PTIO partially bypasses the block in interphase by hypoxia and suggests that NO mediates at least part of the hypoxia effect on the cell cycle in syncytial embryos as it does in older embryos.
The role of NO in the metaphase arrest is less clear. NO can induce a metaphase arrest, but we have been unable to bypass the hypoxia-induced metaphase arrest with inhibitors of NO. Furthermore, as discussed below, cyanide induces a similar metaphase arrest, suggesting that progress through mitosis is sensitive to disruption of metabolism (Fig. 6). Our finding that NO induces a mitotic block prior to any decline in ATP (Fig. 6) suggests that NO can induce this arrest in the absence of a decline in metabolism. Nonetheless, because we were unable to completely bypass the block caused by hypoxia there presumably are other contributors to the block. Perhaps nitric oxide does not ordinarily mediate the metaphase arrest or if it does, it might act redundantly with another effector.
In addition to NO, previous findings with dinitrophenol treatment and our analysis of cyanide show that poisoning metabolism induces cell cycle arrest similar to that caused by hypoxia (Fig. 6 and Ref. 7). Perhaps the perturbation of metabolism might be another contributor to the hypoxic arrest. Because reductions in ATP levels during hypoxia are very small at the time of the initial arrest of the cell cycle, it seems implausible that ATP levels make an important contribution to the arrest signal. Nonetheless, we cannot fully exclude this possibility, nor can we argue against the possibility that reduction in metabolic activity might create some signal other than ATP that might contribute to the response. One could experimentally test the role of energy metabolism in the hypoxic response if it were possible to impose hypoxia without disrupting energy metabolism. The Drosophila embryo is not amenable to this, and we were not able to reverse effects of hypoxia by introducing ATP into the embryo (data not shown). Nonetheless, the effects of cyanide and the hypoxia-induced ATP decline suggest that reduced energy metabolism could contribute to the observed cell cycle arrests. We favor an interpretation that decreased oxidative phosphorylation acts in coordination with or redundantly to signaling systems such as that activated by NO to induce the responses during hypoxia.
Homeostatic Regulation of ATP Improves as Embryogenesis Progresses
Hypoxia is predicted to compromise energy metabolism. Induced quiescence might reduce energy consumption and thereby protect the embryo from the consequences of the decreased energy metabolism. We examined the ability of the embryo to maintain ATP levels in the face of hypoxia. ATP levels decline during hypoxia and recover rapidly upon restoration of oxygen, but the rate and the extent of ATP decline in cellularized embryos was much less than in syncytial embryos. We conclude that late embryos have a more robust ability to accommodate the reductions in oxidative phosphorylation that occur during hypoxia. Although it was intriguing to speculate that this difference might contribute to the improved ability of the older embryos to survive hypoxia, our characterization of the recovery and eventual death of embryos does not support this idea (see below).
How Do Syncytial Embryos Die?
For a rapidly developing embryo to survive hypoxia, it must recover metabolically and developmentally. For unknown reasons, cellularized embryos accommodate and survive prolonged hypoxia significantly better than syncytial embryos (Fig. 1 and Refs. 2 and 3).
Syncytial stage embryos develop rapidly with little zygotic transcription; they may not be able to compensate metabolically as well as older embryos. Indeed, early embryos show a greater decline in ATP levels during hypoxia (Fig. 5). However, despite a decline in ATP levels to less than 30% of controls after 13.5 and 24 h of hypoxia, the syncytial embryos are not metabolically crippled. Upon oxygen restoration, ATP levels are quickly restored (Fig. 5). Metabolically, the syncytial embryo recovers.
Initially, we speculated that the extraordinarily fast nuclear divisions of the syncytial embryos (8–20 min) would not permit arrest and/or recovery from the hypoxic arrest. This is not true: hypoxia reversibly arrests the nuclear divisions just as efficiently as it arrests the slower cell cycles of late stage embryos. Also, embryos re-enter the cell cycle and continue to develop after prolonged hypoxia. However, these recovered embryos exhibit defects of varying severity marked by failure of or aberrations in cellularization and gastrulation (Fig. 8). We conclude that the decrease in survival of syncytial embryos is not from an inability to overcome the arrest of embryogenesis. Rather, prolonged hypoxic treatments decrease the ability of the embryo to faithfully execute the processes of development, resulting in lethality.
Many cellular processes such as energy metabolism and the cell cycle are slowed or blocked by hypoxia. Abnormal development of the syncytial embryo could result from the prolonged period of virtual suspended animation during hypoxia. The syncytial embryo could be particularly sensitive to this slowing of development. Older, cellularized embryos might be more resistant to hypoxia because patterning signals have already begun to be interpreted and the cell membranes might limit the inappropriate diffusion of molecular signals. This hypothesis remains to be tested.
Acknowledgments
We thank Renny Feldman, Simon Prochnik, and Devin Parry for critical reading of the manuscript. We also thank Robert Saint for providing the His-GFP transgenic fly stock. Finally, we especially thank James Wingrove for support, help, and assistance during all stages of the project.
Footnotes
This work was supported by National Institutes of Health Grants GM 37193 and GM 60988 (to P. H. O.), by a National Science Foundation predoctoral fellowship (to J. A. U.), and by a Ford Foundation predoctoral fellowship for minorities (to P. J. D.)
J. A. Wingrove and P. H. O'Farrell, unpublished observations.
The abbreviations used are: PBS, phosphate-buffered saline; SNAP, sodium nitroacetylpenicillamine; CN−, cyanide; GFP, green fluorescent protein; PTIO, 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide.
References
- 1.Guillemin K, Krasnow MA. Cell. 1997;89:9–12. doi: 10.1016/s0092-8674(00)80176-2. [DOI] [PubMed] [Google Scholar]
- 2.Foe VE, Alberts BM. J Cell Biol. 1985;100:1623–1636. doi: 10.1083/jcb.100.5.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wingrove JA, O'Farrell PH. Cell. 1999;98:105–114. doi: 10.1016/S0092-8674(00)80610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson JD, Fauci AS, Martin JB, Kasper DL, Braunwalk E, Isselbacher KJ, Hauser SL, Longo DL. Harrison's Principle of Internal Medicine. 12th. McGraw-Hill Book Co.; New York: 1991. p. 2568. [Google Scholar]
- 5.Green SL, Giaccia AJ. Cancer J Scientific American. 1998;4:218–223. [PubMed] [Google Scholar]
- 6.Hochachka PW, Buck LT, Doll CJ, Land SC. Proc Natl Acad Sci U S A. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zalokar M, Erk I. J Microscop Biol Cell. 1976;25:97–107. [Google Scholar]
- 8.Truman JW, De Vente J, Ball EE. Development. 1996;122:3949–3958. doi: 10.1242/dev.122.12.3949. [DOI] [PubMed] [Google Scholar]
- 9.Kuzin B, Roberts I, Peunova N, Eni, Kolopov G. Cell. 1996;87:639–649. doi: 10.1016/s0092-8674(00)81384-7. [DOI] [PubMed] [Google Scholar]
- 10.Bredt DS, Snyder SH. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 11.Su TT, Sprenger F, DiGregorio PJ, Campbell SD, O'Farrell PH. Genes Dev. 1998;12:1495–1503. doi: 10.1101/gad.12.10.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashburner M, editor. Drosophila: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. p. 434. [Google Scholar]
- 13.Clarkson M, Saint R. DNA Cell Biol. 1999;18:457–462. doi: 10.1089/104454999315178. [DOI] [PubMed] [Google Scholar]
- 14.Elledge SJ. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 15.Zachariae W, Nasmyth K. Genes Dev. 1999;13:2039–2058. doi: 10.1101/gad.13.16.2039. [DOI] [PubMed] [Google Scholar]