

Abstract
Background
Cyclin E is the normal inducer of S phase in G1 cells of Drosophila embryos. Stable G1 quiescence requires the downregulation both of cyclin E and of other factors that can bypass the normal regulation of cell cycle progression.
Results
High-level expression of cyclin A triggered the G1/S transition in wild-type embryos and in mutant embryos lacking cyclin E. Three types of control downregulated this activity of cyclin A. First, cyclin destruction limited the accumulation of cyclin A protein in G1. Second, inhibitory phosphorylation of cdc2, the kinase partner of cyclin A, reduced the S-phase promoting activity of cyclin A in G1. Third, rux, a protein with unknown biochemical function, limited cyclin A function in G1. Overexpression of rux blocked S phase induction by coexpressed cyclin A and promoted the degradation of cyclin A. Rux also prevented a stable cyclin A mutant from inducing S phase, indicating that inhibition does not require cyclin destruction, and drove the nuclear localization of cyclin A.
Conclusions
Cyclin A can drive the G1/S transition, but this function is suppressed by three types of control: cyclin A destruction, inhibitory phosphorylation of cdc2, and inhibition by rux. The partly redundant contributions of these three inhibitory mechanisms safeguard the stability of G1 quiescence until the induction of cyclin E. The action of rux during G1 resembles the action of inhibitors of mitotic kinases present during G1 in yeast, although no obvious sequence similarity exists.
Background
The control of growth and cell proliferation is essential to metazoan development. In general, when cells arrest proliferation they do so in G1; the importance of stable G1 quiescence is dramatized by the consequences of tumor suppressor gene mutations that erode this stability [1,2]. The characterization of a number of tumor suppressor mutations has demonstrated that the stability of G1 is critically dependent on the signal transduction pathways that regulate the expression of cyclin E in G1 [3–6]. Studies in mammalian cells have demonstrated that the complex between cyclin A and its catalytic partner, cyclin-dependent kinase 2 (cdk2), is an important S-phase kinase [7,8]. Here, we use Drosophila as a model to test whether the strict regulation of cyclin E activity can be bypassed by ectopic activation of cyclin A.
The first postmitotic quiescent (G1) state in Drosophila embryogenesis occurs in cycle 17 when cyclin E, encoded by the DmcycE gene, becomes limiting for progression to S phase. DmcycE mutant embryos arrest in G1, whereas premature expression of cyclin E results in a precocious advance into S phase [9–11]. Thus, in Drosophila embryos, as in mammalian cells, activation of cyclin E is a crucial step in the G1/S transition.
Cyclin A–cdk2 activity is detected in mammalian cells at the end of G1 and then rises during S phase [8,12,13]. Cyclin A–cdk2 can induce S phase in extracts from G1 cells [12], and expression of cyclin A in G1 can shorten the G1 period and drive early entry into S phase [14]. Most importantly, the injection of antibodies directed against cyclin A can prevent completion of S phase [7,8]. In Xenopus, cyclin A2 can promote S phase in interphase extracts depleted of cyclin E [15]. An S phase role for cyclin A in Drosophila was far from evident in earlier work. Endocycle S phases proceed in the absence of cyclin A [16–18], and ectopic expression of cyclin A in the embryo induced S phase only in the amnioserosa cells — which are thought to arrest (at least initially) in G2 [19]. However, recent results have suggested that cyclin A in Drosophila can drive G1 cells into S phase. The expression of cyclin A from a heat-shock promoter during eye development induced S phase in the G1 cells of the morphogenetic furrow [20]. Furthermore, in rux mutants, these furrow cells exhibited ectopic expression of cyclin A and premature entry into S phase. Because the rux phenotype was alleviated by reduction of DmcycA function, it seemed likely that the precocious S phase was driven by the ectopic cyclin A, and that rux might then function as a suppressor of DmcycA function [20,21].
Cyclin A has an established mitotic function in addition to its role in S phase. In Xenopus, it can induce nuclear envelope breakdown and chromatin condensation and can drive one mitotic cycle [22,23]. In Drosophila, genetic analysis of DmcycA has shown that cyclin A has mitotic functions that overlap with functions of cyclin B [24]. In vertebrate cells, cyclin A complexes with both cdk2 and cdc2, and it is thought that the S phase role of cyclin A is contributed by the cyclin A–cdk2 complex whereas the G2 role is provided by the cyclin A–cdc2 complex [7,8,13,25–27]. In Drosophila, however, cyclin A only complexes with cdc2, so that both S and G2 roles are presumed to be carried out by cyclin A–cdc2 (F.S., unpublished observations; [10,17]).
Here, we show that the ectopic expression of Drosophila cyclin A can trigger G1/S transitions in embryos even in the absence of cyclin E function, and that three safeguard mechanisms severely limit the efficiency of induction of S phase by ectopic cyclin A: rapid destruction of cyclin A in G1 cells, inhibition of cyclin A–cdc2 by rux, and inhibition of cyclin A–cdc2 by phosphorylation. By bypassing each of these safeguard mechanisms, we show that there is a substantial level of redundancy between them. Genes important for these safeguard mechanisms might behave as tumor suppressor genes in other systems. Additionally, our characterization of rux function suggests that it has widespread roles in regulating cell cycle progression and that it modulates the entry of cyclin A into the nucleus.
Results
Induction of S phase by cyclin A
Lehner et al. [19] expressed cyclin A from a heat-shock-inducible promoter, but S phase was not induced in G1 cells. We repeated these experiments using a different heat-shock protocol that produces higher levels of cyclin A but does not decrease viability, alter the S phase program or cause other visible abnormalities in nontransgenic embryos. By varying the length of the heat-shock, we were able to change the amount of cyclin A produced (see Figure 4s). To examine the effect of the expressed cyclin A on S phase, we labeled embryos for 30 minutes with bromodeoxyuridine (BrdU), beginning 20 minutes after the heat shock. The incorporated BrdU was then detected using indirect immunofluorescence and analyzed on a confocal microscope.
Figure 4.
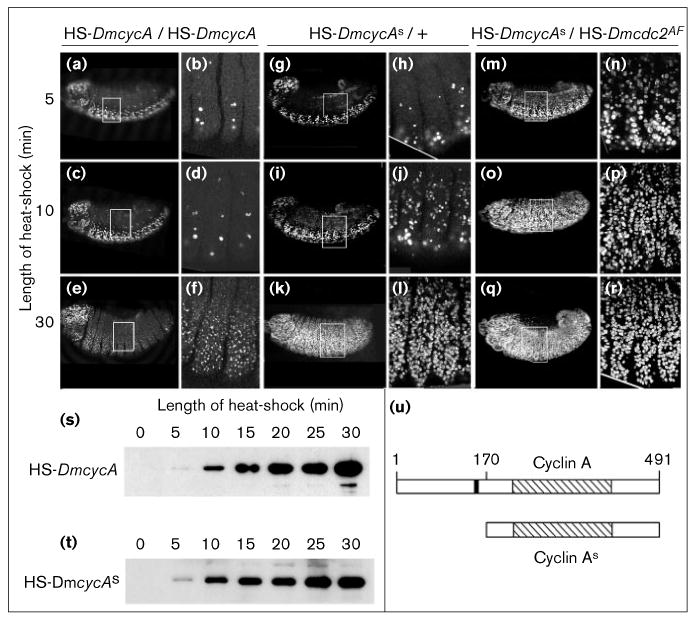
S-phase induction by cyclin As alone and in combination with cdc2AF. (a–r) Embryos were pulse-labeled with BrdU and incorporation visualized with indirect immunofluorescence. Optical sections of the epidermal cell layers are shown to detect S-phase induction in the epidermis. (a–f) A heat shock of more than 10 min is needed to induce S phase in the epidermis of embryos carrying two copies of the cyclin A transgene. (b,d,f) Higher magnifications of the embryos shown in (a,c,e), respectively. (g–l) In embryos carrying one copy of the cyclin As heat-shock transgene, S phase can be induced after a 10 min heat-shock. (m–r) Coexpression of cyclin As and cdc2AF can induce S phase after a 5 min heat-shock. (s,t) Expression of cyclin A and cyclin As from the heat-shock transgene after varying the heat-shock length. Cyclin A proteins are detected on immunoblots using a monoclonal antibody. Each lane contains the protein from one embryo, each carrying two copies of the respective transgene. Embryos were heat-shocked for the indicated times and fixed with methanol approximately 5 min after the heat-shock. The two blots were developed separately and the blot in (t) had to be exposed about 5-times longer to see the cyclin As signal. (u) Schematic presentation of the cyclin constructs. Cyclin A is the full-length Drosophila cyclin A. The cyclin box is striped and the destruction box is filled. Cyclin As lacks the first 169 amino acids, including the destruction box.
In 7.5–8.5 hour old wild-type (nontransgenic) embryos, BrdU incorporation was seen in the mitotic cells of the nervous system (Figure 1a) and in cells of the developing internal organs that undergo endoreduplication cycles (Figure 1b). Cells of the epidermis are in G1 and do not incorporate BrdU. In the internal tissues, S phases occur in a defined spatial and temporal order [28,29]; as shown in Figure 1b, BrdU incorporation could be seen in a few lagging cells of the central midgut, where most cells had completed S phase 17, and in cells throughout the anterior and posterior midgut where S phase 17 has recently started. No incorporation was seen in the salivary gland that had already completed S phase 17 and had not yet started S phase 18.
Figure 1.

Expression of cyclin A can induce S phase. Embryos were pulse-labeled with BrdU for 30 min to visualize DNA synthesis. Embryos were collected for 1 h, aged for 7.5 h and heat-shocked (HS) for the times indicated. After a recovery period of 20 min, embryos were labeled with BrdU for 30 min, then fixed. Incorporated BrdU was detected with indirect immunofluorescence and analyzed on a confocal microscope. Optical section are shown from (a,c,e) the epidermal cell layer and from (b,d,f) the internal cell layers of the developing organs. (a,b) Wild-type control embryos show normal BrdU incorporation in the central nervous system including the brain (Br), the anterior and posterior midgut (AMG and PMG, marked with arrows), the hindgut and malphigian tubules. In the central midgut (CMG, marked with a star) some cells finish S phase 17. No S phase can be seen in the epidermis. (c,d) Embryos carrying two copies of a HS-DmcycA transgene were heat-shocked for 15 min. Only a few cells in the epidermis were induced to go into S phase. In the internal tissues, the normal spatial and temporal program of S phases is disturbed and in all cells of the midgut and the salivary gland (marked with a triangle) S-phase induction can be seen. (e,f) A heat-shock of 30 min induces S-phase in cells of the epidermis. Cells of the ventral epidermis show a slightly stronger label than cells of the dorsal epidermis. In the internal tissues, a homogeneous S phase is induced. (g,h) Schematic drawings of the embryos in (e,f) Abbreviations: Br, brain; DE, dorsal epidermis; VE, ventral epidermis; SG: salivary gland; AMG, anterior midgut; CMG, central midgut; PMG, posterior midgut.
When cyclin A was expressed from the heat-inducible transgene (HS-DmcycA), the normal pattern of S phase was changed. After a 30 minute heat-shock, S phase was induced throughout the epidermis (Figure 1e). In the internal tissues, the normal pattern of S phase was eradicated and cells of different tissues that were normally in S phase at different times now incorporated BrdU at the same time (Figure 1f). Thus, the induction of cyclin A expression triggered premature S phase in epidermal cells and in endocycling tissues. These results are very similar to those obtained after ectopic expression of cyclin E from a heat-shock transgene [10,29]. Normally it is cyclin E, not cyclin A, that induces endocycle S phases, as a mutation in DmcycE blocks these S phases, while a mutation in DmcycA does not.
The induction of S phase by ectopic cyclin A suggests that cyclin A can replace cyclin E or that cyclin A induces cyclin E. To distinguish between these possibilities, we expressed cyclin A ubiquitously in DmcycE mutant embryos. Within an hour of inducing cyclin A, S phase was induced widely in the internal tissues that would endocycle in wild-type embryos (Figure 2b), but induction of S phase in the epidermal cells was delayed in the DmcycE mutant embryos compared with control embryos (compare Figure 2b,d,f). Thus, cyclin A is competent to drive a G1/S transition in the absence of cyclin E, but this induction is modified by DmcycE in some tissues. In contrast to cyclin A or cyclin E, expression of cyclin B from a heat-shock promoter did not induce S phase (data not shown).
Figure 2.

Cyclin A is competent to drive S phase in the absence of cyclin E. BrdU pulse labelling of control embryos that received no heat-shock (a,c,e) and embryos of the same genotypes after heat-shock treatment (b,d,f). BrdU pulse labelling was performed at the indicated times after heat-shock treatment and on embryos of equivalent age that received no heat shock treatment. (a,b) Embryos carrying two copies of HS-DmcycA. S-phase induction by cyclin A can be observed in DmcycE+ embryos in internal tissues (not shown) and the epidermis 1 h after heat-shock. (c–f) Embryos homozygous for DmcycEAR95 carrying two copies of HS-DmcycA. In embryos mutant for DmcycE, S-phase induction by cyclin A can be observed after 1 h in the endoreduplicating tissues (d), but epidermal S-phase induction is not seen until 180 min after S-phase induction (f).
To test whether cyclin A is able to induce S phase in a very different context, we expressed cyclin A in G1 cells of the developing Drosophila eye using the sevenless enhancer. Again S phase was induced by the induced expression of cyclin A (data not shown). This is in agreement with a recent report in which heat-shock-induced expression of cyclin A in the eye disk resulted in ectopic S phases [20]. From all these experiments, we conclude that cyclin A can trigger a G1/S transition and induce DNA replication in Drosophila
Cyclin A is unstable in G1
Although strong expression of cyclin A induced S phase throughout the embryo, milder heat-shock conditions resulted in a lower level of cyclin A expression (see Figure 4s), inducing S phase in the internal tissues but not in the epidermis (Figure 1c,d). To investigate whether cyclin A accumulated in cells of the epidermis under these conditions, we analyzed the expression of cyclin A under different heat-shock conditions. In wild-type embryos, cyclin A protein is detected in the germ-cells that are arrested in G2 (T. Su, personal communication) and in mitotic cells of the nervous system and brain (Figure 3a,b). After a mild heat shock of 15 minutes, there was an initial accumulation of cyclin A in the epidermis (Figure 3e) and in internal tissues (Figure 3f), yet only cells of the internal tissues entered S phase prematurely (Figure 1c,d). This indicates that the ectopic S phase in the epidermis requires higher levels of cyclin A protein.
Figure 3.
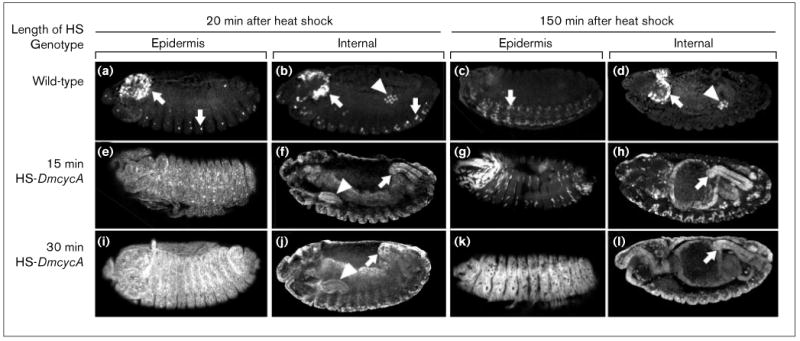
Cyclin A is unstable in G1, but it can induce its own stabilization. Immunostaining for cyclin A is shown at internal and superficial focal planes for embryos 20 min or 150 min after heat-shock. (a–d) The pattern of cyclin A expression in heat-shocked control embryos. As in undisturbed embryos of this stage, few cells express cyclin A: the mitotically active neuronal cells (arrows) are not arrested in G1 and are actively expressing cyclin A; the quiescent germ cells are arrested in G2 and retain maternally expressed cyclin A (T.T. Su, and P.H.O'F., unpublished observations). (e–h) Cyclin A is unstable in G1 cells. The cyclin A expressed from two copies of a HS-DmcycA transgene in response to a short heat-shock (15 min) accumulates throughout the embryo: staining is seen in the epidermis (e), and internal tissues such as the salivary gland (triangle) and the hindgut (arrow) (f). The cyclin A signal is lost in many cells of the embryo by 150 min following the heat-shock (compare (g) with (e)). Cells that exit G1 and enter S phase retain cyclin A at the 150 min time – for example, the internal tissues (h). The few cells of the epidermis that continue to stain for cyclin A at 150 min probably represent exceptional cells that were induced into S phase by the low level of cyclin A induction (see Figure 1c). This result suggests that cyclin A is considerably more stable in cells that have progressed to S phase than it is in G1 cells. (i–l) Cyclin A levels that induce G1 cells into S phase also induce stabilization of cyclin A. A 30 min heat-shock of embryos carrying two copies of a HS-DmcycA transgene induces high levels of cyclin A (i,j), and widespread S phase (Figure 1e). In contrast to the instability of cyclin A in the epidermis following a 15 min heat-shock (g), the cyclin A that was induced in response to this longer heat-shock persisted in most of the cells of the epidermis (k), as well as persisting in the internal tissues (l). There are a few cells that so not stain for cyclin A after 150 min (see black holes in the staining in panel (k)): these may represent cells that were not induced into S phase by the treatment. The difference between the middle and the bottom series of panels suggests that cyclin A can stabilize itself, and the conditions that lead to stabilization are the same as those that lead to induction of G1 cells into S phase (see Figure 1).
We also examined the stability of cyclin A by staining embryos for cyclin A 150 minutes after the heat-shock. In embryos that received a 30 minute heat-shock, almost all cells entered S phase (Figure 1e,f) and cyclin A was detectable at high levels in cells of the epidermis and in cells of the internal tissues (Figure 3k,l). In embryos that received a 15 minute heat-shock, which induced S phase in internal tissues and rarely in cells of the epidermis, high levels of cyclin A persisted in the internal tissues, but staining was only visible in a few cells of the epidermis (Figure 3g,h). These results show that cyclin A is unstable when cells are in G1 and that it is stabilized upon transit to S phase, whether the cells enter S phase of their own accord or are induced to do so by high levels of cyclin A. These data are consistent with observations made by Knoblich et al. [10]. They found that induction of cyclin E, which induced S phase, also allowed accumulation of cyclin A protein without a change in the levels of DmcycA mRNA. This showed that either increased translation or decreased degradation of cyclin A accompanied cyclin E induction [10].
Instability of mitotic cyclins in G1 has also been described for yeast and mammalian cells [30,31]. Here, cyclin proteolysis which is necessary for exit from mitosis continues throughout the G1 period (reviewed in [32]). Degradation is mediated by the ubiquitin–proteasome pathway and is dependent on a small conserved motif in the cyclin sequence, termed the destruction box [33,34]. Mitotic cyclins lacking the destruction box are resistant to destruction by this mechanism and, when expressed before mitosis, they block exit from mitosis [34].
To investigate whether the mitotic destruction machinery influences cyclin A activity in G1, we deleted the destruction box from the DmcycA gene (F.S and P.O.F. unpublished observations; [35]) and expressed the mutant construct (cyclin As; Figure 4u) during cell cycle 17. In this case, even a very mild induction of cyclin As — a 10 minute heat-shock with one copy of the HS-DmcycAs — during G1 resulted in S phase in the epidermis (Figure 4g–l). In comparison, two copies of HS-DmcycA and a heat-shock of more than 15 minutes were necessary to induce S phase in the epidermis (Figure 4a–f and Figure 1c,e).
The amount of accumulated cyclin A (Figure 4s) and cyclin As (Figure 4t) increased dramatically with increasing duration of the heat shock. For a given heat shock and with similar detection conditions, the level of staining obtained from embryos with the cyclin A transgene was much higher than the level obtained with the cyclin As transgene (the roughly comparable staining seen in Figure 4s,t is the result of longer exposure of the cyclin As blot). Thus, the cyclin As transgene is not expressed as efficiently as the cyclin A transgene. Titrations of the amount of protein accumulated after different heat shocks showed that ten-fold less cyclin As was required to induce S phase compared with cyclin A.
Cdc2 activity is suppressed in G1 by inhibitory phosphorylation
Cdk kinase activity is regulated both by phosphorylation and by cyclin binding (reviewed in [36]). Although inhibitory phosphorylation of cdc2 on the threonine at residue 14 (T14) and the tyrosine at position 15 (Y15) is generally thought to play a major role in controlling the transition from G2 into mitosis, it might also play a role in G1 to restrict the activity of cyclin A–cdc2. As the major target for phosphorylation is the cdk–cyclin complex rather than the free cdk, we would expect the level of inhibitory phosphorylation to depend on the presence of a cyclin partner. In G1 of cycle 17, cdc2 is present in the unphosphorylated form (Figure 5). At this embryonic stage, heat-shock-induced expression of cdc2 led to the accumulation of only the unphosphorylated form (data not shown). Induced expression of both cdc2 and cyclin A led to the accumulation of the phosphoisoforms of cdc2 (Figure 5), indicative of phosphorylation on T14 and Y15 [37].
Figure 5.
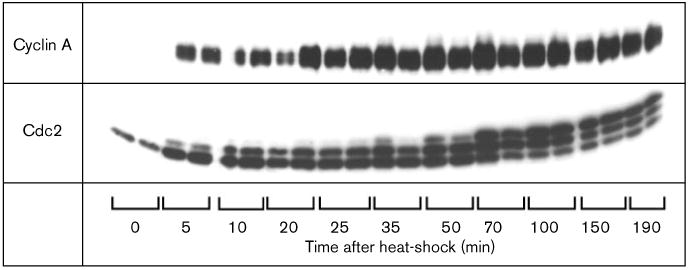
Expression of cyclin A results in inhibitory phosphorylation of cdc2. Total protein extracts from single embryos containing two copies of the HS-DmcycA transgene and one copy of the HS-Dmcdc2 gene were separated on SDS-PAGE and immunoblotted using a polyclonal anti-cyclin A and anti-cdc2 antibody respectively. Embryos were either not heat-shocked (0 time points) or were collected after a 30 min heat-shock at the times indicated. For each time point, two embryos were analyzed. Phosphorylation of cdc2 requires the binding of cyclin and neither cyclin A or cyclin B are normally expressed in the majority of the cells at this stage in development (0 min time points). Expression of cyclin A from the heat-shock promoter results in the appearance of slower migrating forms of cdc2 which are indicative of Y15 and/or T14 phosphorylated forms of cdc2.
To investigate whether inhibitory phosphorylation on cdc2 limits the efficiency of cyclin A induction of S phase, we coexpressed a mutant form of cdc2 that lacks the inhibitory phosphorylation sites, cdc2T14AY15F (cdc2AF). In the absence of induced cyclin A expression, expression of cdc2AF or cdc2 from heat-shock transgenes had no effect on S-phase induction (data not shown), whereas the coexpression of cdc2AF with either cyclin A or cyclin As increased the efficiency of S-phase induction dramatically (Figure 4m–r and data not shown). Coexpression of wild-type cdc2 with cyclin A did not alter significantly the effectiveness of S-phase induction by cyclin A. S phase was not induced by coexpression of cdc2AF with cyclin B or cyclin B lacking the mitotic destruction box (data not shown). Together, these data show that cyclin A–cdc2 is able to induce S phase, a function of cdc2 not shared by the cyclin B–cdc2 complex. This S-phase function of cdc2 is substantially inhibited in G1 by the presence of kinases that phosphorylate cdc2 on inhibitory phosphorylation sites.
Rux acts an inhibitor of cyclin A in the embryo
Mutations in the rux gene cause premature cell cycle progress in two tissues. In eye discs lacking the function of the rux gene, cells enter S phase precociously, giving rise to a rough eye phenotype [21]. Detailed analysis of this phenotype reveals that rux is required for the acquisition and/or maintenance of G1 in this tissue. In the male gonad, rux function is needed for normal arrest of the cell cycle after meiosis II [38]. Both phenotypes of rux are dominantly suppressed by mutations in the DmcycA gene [21,38]. Loss of rux function also alters cyclin A protein accumulation in these tissues: rux-deficient eye disc cells exhibit ectopic accumulation of cyclin A, and rux-deficient premeiotic germ cells accumulate more cyclin A. An ectopic S phase in the eye disc can also be induced by the expression of cyclin A from a heat-shock transgene [20] or from a transgene in which DmcycA is under the control of the sevenless enhancer (data not shown), raising the possibility that the ectopic S-phase in rux mutants is caused by cyclin A.
The limitation of the rux phenotype to eyes and testis might reflect limited action of this gene, or might be a consequence of special sensitivity of these tissues to loss of rux function. Although rux is a non-lethal gene, strong mutations reduce overall viability. Given the interactions between rux and DmcycA, it seemed likely that, if rux were to have a function in G1 of cycle 17, this function might be exposed by expression of excess cyclin A. BrdU labeling and cyclin A staining did not indicate an abnormal phenotype in rux mutant embryos (data not shown). However, rux mutant embryos were more sensitive to induction of ectopic S phase by cyclin A (Figure 6a–f). For example, a 10 minute heat-shock of rux mutant embryos carrying a single copy of the HS-DmcycA transgene induced significant S phase in the epidermis (Figure 6d), whereas a similar heat-shock of a rux+ embryos carrying two copies of the transgene failed to induce S phase (Figure 4d). Thus, rux functions in the early embryo and this function is uncovered when the system is stressed by the ectopic expression of cyclin A.
Figure 6.
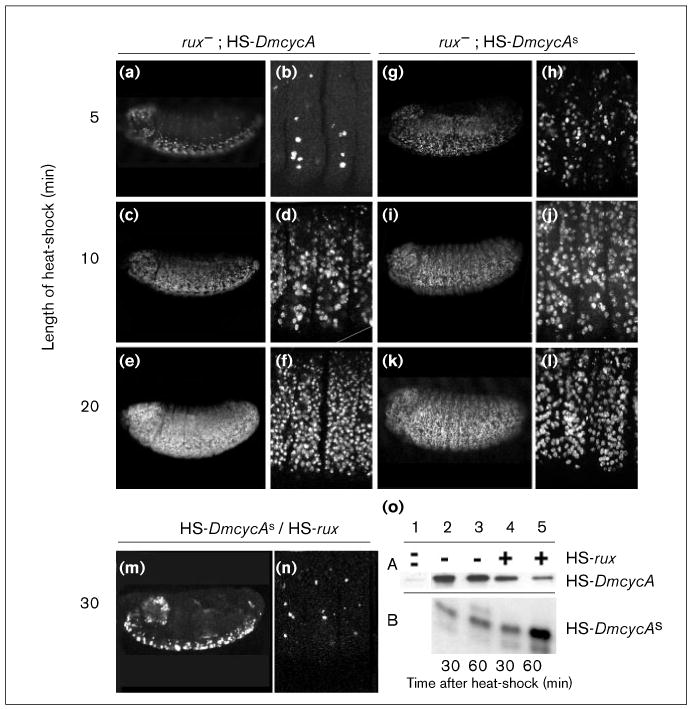
In rux mutant embryos, cyclin A and cyclin As are more effective in inducing S phase, but overexpression of rux can inhibit S phase function of cyclin As completely. (a–n) Embryos were pulse-labeled with BrdU and incorporation visualized with indirect immunofluorescence. Optical section were taken on a confocal microscope to determine S-phase induction in the epidermis. (a–f) In rux mutant embryos, induction of cyclin A from one copy of HS-DmcycA is sufficient to induce S-phase after a 10 min heat-shock (compare to Figure 4a–f). (g–l) In rux mutant embryos, S-phase induction by cyclin As is enhanced, causing S-phase induction after a 5 min heat-shock. (m,n) Overexpression of rux can block the ectopic S-phase induction of cyclin As completely. Normal S phases of the central nervous system are apparently not affected by rux overexpression. (o) Immunoblots of single embryos extracts collected 30 and 60 min after a 30 min heat-shock and developed for cyclin A (A) and cyclin As (B), respectively. Lane 1: control embryos that received no heat-shock show little endogenous cyclin A. Lanes 2,3: cyclin A and cyclin As accumulate after heat shock and levels appear stable 30 min and 60 min after the heat shock. Lane 4: coexpression of cyclin A or cyclin As with rux; 30 min after heat-shock, cyclin As accumulates to similar levels as seen in lane 2 and cyclin A is present already at lower levels. Lane 5: 60 min after coexpression with rux, cyclin A levels have diminished but cyclin As levels have increased.
Ubiquitous induction of rux from a HS-rux transgene had no obvious consequence on the patterns of BrdU incorporation following G1 17. This is consistent with results showing that this S phase is induced by cyclin E and that rux does not inhibit cyclin E function ([39]; N.Y. and P.H.O'F., unpublished observations). In contrast, we observed strong interactions between ectopic rux and ectopic cyclin A: induction of HS-DmcycA produced high levels of cyclin A, whereas coexpression with rux prevented the stable accumulation of cyclin A (Figure 7a,c and Figure 6o). The induction of rux also blocked the induction of S phase by HS-DmcycA (data not shown, but see results for cyclin As below). Although rux might counter cyclin A function by promoting its degradation, it might also inhibit cyclin A activity by a mechanism that is independent of its degradation. We therefore tested the ability of rux to inhibit the activity of the stable form of cyclin A. A comparison of the ability of cyclin As to induce BrdU incorporation in rux+ and rux− mutant embryos demonstrated that the rux mutant embryos were more sensitive to the ectopic expression (compare Figure 4h–j with Figure 6h–j). Thus, the endogenous rux activity can partially inhibit cyclin As. Joint overexpression of rux and cyclin As indicated that rux did not promote the destruction of cyclin As (in fact, cyclin As accumulated at higher levels: Figure 6o). Importantly, the induced rux completely blocked the ability of cyclin As to induce S phase (Figure 6m,n). We conclude that rux can inhibit the function of cyclin A in G1, preventing S-phase induction by cyclin A.
Figure 7.

Rux causes cyclin A to be degraded and cyclin As to accumulate in the nucleus. Optical sections of whole embryos stained with a polyclonal antibody to cyclin A (αA) or stained with propidium iodide to visualize DNA. Embryos were 7.5–8.5 h old and heat-shocked for 30 min. After 60 min, embryos were fixed. Cyclin A proteins were detected using a polyclonal cyclin A antibody and a secondary FITC-labeled antibody. Optical sections are shown that focus on epidermal cells of thoracic and abdominal segments. (a) Embryo carrying two copies of HS-DmcycA Most cells shown are in G1 and accumulate cyclin A mainly in the cytoplasm. The inset shows a region of the first thoracic segment with some cells accumulating higher levels of cyclin A. These cells also have bigger nuclei and are believed to be in G2. Immediately above those cells are a few cells with no cyclin A staining. Presumably, those cells have just completed mitosis. It can be seen that the cells at the top of the inset also have some, although weak, nuclear staining of cyclin A. (b) Embryos with one copy of HS-DmcycAs. The expression of cyclin As appears almost uniform throughout the cells, including the nucleus. (c) This embryo has two copies of the HS-DmcycA and one copy of the HS-rux transgene. In almost all cells, cyclin A is expressed at very low levels with some residual expression seen in the cytoplasm. The stronger staining cells are likely neuroblasts in which cyclin A localization and accumulation is apparently not affected by the expression of rux. (d) In embryos that carry one copy of HS-DmcycAs and one copy of HS-rux, cyclin As shows distinct nuclear accumulation.
Rux causes a change in cyclin A subcellular localization
In wild-type embryos of the cellular blastoderm stage (cell cycle stages 14–16), cyclin A is localized predominantly to the cytoplasm in S and G2. In prophase, it enters the nucleus and is then degraded during mitosis [16]. In G1 cells, cyclin A is unstable and little staining of cytoplasmic cyclin A can be observed (data not shown). Expression of cyclin A from HS-DmcycA during G1 resulted in a G1/S transition and cyclin A accumulation which appeared to be predominantly cytoplasmic (Figure 7a). In embryos lacking rux function, no change in the localization of endogenous or heat-shock-induced cyclin A protein was observed (data not shown). Heat-shock-induced expression of rux did not alter the general staining pattern of cyclin A in the wild-type embryo (data not shown). However, coexpression of rux and cyclin A prevented the accumulation of cyclin A (Figure 7c).
The subcellular localization of cyclin As was strikingly different from that of cyclin A, being distributed homogeneously in the cytoplasm and the nucleus. This suggests that the stabilization of cyclin A allows some accumulation in the nucleus. Upon coexpression with rux, the cyclin As staining is strongly nuclear. Thus, while overexpression of rux prevents the accumulation of cyclin A, it promotes the localization of cyclin As to the nucleus.
Discussion
In both vertebrates and flies, the entry into S phase from G1 depends on the activation of E2F and cyclin E [4,6,9,10,40]. However, some studies suggested that cyclin A has some ability to induce G1 cells to enter S phase. Here, we show that heat-shock-induced cyclin A expression can trigger S phase in G1 cells and that this activation of S phase can occur without cyclin E activity. These observations emphasize that the faithful regulation of the G1/S transition by the E2F–cyclin E pathway relies on effective silencing of alternative routes to triggering S phase.
In Drosophila, the introduction of a G1 phase in cell cycle 17 involves the downregulation of cyclin E [10,29,41,42]. Cyclin A is also downregulated in the tissues that enter quiescence [16], and we show here that ectopic cyclin A activity can drive G1 quiescent cells into S phase. We show that, at the time of cell cycle 17, there are at least three constraints that restrict the ability of cyclin A to drive S phase. These constraints on the activity of cyclin A are likely to be generally important for the stability of G1.
Cyclin A expression can drive S phase even in the absence of DmcycE function
Induction of expression of cyclin A from a transgene under the control of the hsp70 promoter induced S phase in cells that otherwise would have been in G1 of cell cycle 17. Induction of cyclin A in a DmcycE mutant background demonstrated that cyclin A induction is able to bypass the S phase requirement for cyclin E. S phase in the internal tissues followed shortly after induction of cyclin A, while induction of S phase in the epidermis was delayed. The different kinetics may be the result of the cyclin E independent activation of E2F in the internal cells that are scheduled to undergo endocycle S phases [9]. As E2F activity is required for S phase, the delay in the response of the epidermis might reflect inefficient induction of E2F by cyclin A [43]. In any case, it appears that cyclin A induction of S phase can occur in the absence of cyclin E. The DmcycE allele used in this study is a null mutation and maternal cyclin E disappears or is inhibited prior to the time of this experiment ([9–11,41,42], P.H.H.O'F., J. Vidwans and N.Y., unpublished observations). As Drosophila cyclin A interacts only with cdc2, while cyclin E interacts with cdk2, this experiment demonstrates not only that one cyclin can substitute for the other, but also that one kinase can substitute for the other.
As high levels of cyclin A were required to induce S phase in quiescent G1 cells, the high expression might be overwhelming controls that ordinarily act to preclude this action. Analysis of cyclin A levels suggested that rapid degradation of cyclin A during G1 might be one such constraint, while phosphorylation of cdc2 suggested that inhibitory phosphorylation would be another, and the genetic interactions of rux and DmcycA suggested that inhibition by rux might be third. We tested the importance of these by bypassing each of the constraints and examining the effect on the efficiency of cyclin A induction of S phase.
Cyclin A turnover
Experiments in budding yeast first demonstrated that the rapid degradation of cyclins that occurs at mitosis continues during G1 [30]. Recent work has shown that rapid turnover of G2 cyclins also occurs during G1 in mammalian cells [31]. Knoblich et al. [10] had previously demonstrated that cyclin E expression induces cyclin A protein accumulation in cell cycle 17 without an increase in DmcycA mRNA. Here, we show that cyclin A is rapidly degraded in G1 cells and stabilized once cells enter S phase (Figure 3). Together, the findings in yeasts, flies and vertebrate cells suggest that G1 destruction of G2 cyclins is general.
A transgene encoding a stable form of cyclin A lacking the mitotic destruction box is more effective at inducing S phase than a transgene encoding the normal cyclin A. This increased effectiveness might be because the particular transgene is unusually highly expressed, because the increased stability of the protein ultimately allows it to accumulate to higher levels, or because the changed properties of the product make it a more effective inducer of S phase. Comparisons of protein levels demonstrated that lower levels of cyclin As are required to induce S phase compared with cyclin A. This increased potency of cyclin As can be explained if, for example, active cyclin A is a complex that forms slowly. In this case, the complex would have a greater opportunity to form if the cyclin is stable. Its persistence per se might alter the level of posttranslational modifications, complex formation or subcellular distribution, factors that might be important for the S phase function of cyclin A. At present, we cannot discriminate among these possibilities, but the results argue that rapid destruction of cyclin A during G1 limits its activity.
Inhibitory phosphorylation of cdc2 during G1
Cdc2 kinase activity can be regulated by inhibitory phosphorylation on residues T14 and Y15 by Wee- and Myt-like kinases [44,45]. In G1, cdc2 is normally not phosphorylated because of the lack of cyclins — which must bind to cdc2 for it to be phosphorylated. When we expressed cyclin A in G1, we identified phosphoisoforms of cdc2 that correspond to inhibited, T14- or Y15-phosphorylated forms of cdc2. Thus, inhibitory phosphorylation, which is normally viewed as a regulatory event governing G2, can have a role in G1. Indeed, coexpression of cyclin A and cdc2AF, a variant lacking the inhibitory phosphorylation sites, enhanced the S-phase-inducing activity of cyclin A. Thus, we conclude that inhibitory phosphorylation restricts activity of cyclin A in G1.
Human cdc25A and cdc25B, which can promote progress of the cell cycle from G1 to S, appear to function as oncogenes [46] This suggests that inhibitory phosphorylation contributes to the stability of G1 in humans. It is not clear, however, whether the cyclin A-associated kinase is a relevant target of inhibitory phosphorylation in mammalian cells as it is in Drosophila.
Rux promotes nuclear localization of cyclin A but inhibits its function in G1 of cycle 17
Studies of rux mutant phenotypes suggested that rux behaves as an inhibitor of DmcycA function in the testis and in the eye [21,38]. We examined rux mutant embryos to determine whether it has a zygotic function in the embryo. Although we could detect no cell cycle defect in rux mutant embryos, ectopic cyclin A is more effective in inducing S phase in the mutant background. Reciprocally, overexpression of rux completely blocked S phase induction by cyclin A. These results show that rux activity can inhibit cyclin A function and that endogenous rux activity at the time of cycle 17 contributes to the restraints limiting cyclin A activity.
In addition to the inhibition of cyclin A function, we found that the overexpression of rux promoted the degradation of cyclin A and the nuclear localization of cyclin As. As the biochemical activity of rux is unknown, any of the observed consequences of rux overexpression might be secondary. However, we found that rux expression blocks the induction of S phase by cyclin As, which demonstrates that rux has an ability to inhibit cyclin A function that is separate from its ability to promote its degradation. Additionally, we coexpressed cyclin As and cdc2AF with or without rux (data not shown); rux was again able to suppress induction of S phase by cyclin As. As rux inhibition could not be bypassed by either stabilizing cyclin A or blocking inhibitory phosphorylation, we argue that rux has a mode of action that is not mediated by either degradation or inhibitory phosphorylation.
At present, we cannot resolve whether rux has independent activities that promote cyclin A degradation and nuclear localization, or whether these are secondary to other actions of rux. For example, we have shown that cyclin A can promote its own stabilization. As rux inhibits cyclin A activity, it should inhibit the ability of cyclin A to stabilize itself. Such an inhibition of stabilization might account for, or at least contribute to, the apparent enhancement of degradation in response to rux. Alternatively, or additionally, if a rate limiting step in the degradation of cyclin A were to occur in the nucleus, rux-enhanced degradation of cyclin A might be promoted by enhanced nuclear localization.
In Drosophila, cyclin A is cytoplasmic during interphase but enters the nucleus during prophase [16]. In human cells, cyclin A is predominantly nuclear from S phase onwards [47] and colocalizes with replication proteins RP-A and PCNA and with replication origins [48,49]. Expression of cyclin A in G1 cells of Drosophila embryos results in a predominantly cytoplasmic localization, but the cyclin As protein is distributed homogeneously throughout the cell. There are at least two possibilities that could account for the difference in the distribution of cyclin A and cyclin As. If full-length cyclin A is degraded by a destruction system localized to the nucleus, then it would fail to accumulate in this compartment even if it could transit the nuclear membrane. Alternatively, cyclin As might lack a cytoplasmic retention signal that prevents cyclin A from entering the nucleus. Such a retention sequence might be a cytoplasmic anchor sequence like that described for human cyclin B1 [50].
The expression of rux changes the subcellular localization of cyclin A: full-length cyclin A is degraded and cyclin As is present only in the nucleus. In premeiotic spermatocytes, full-length cyclin A is normally cytoplasmic but translocates into the nucleus when the rux gene dose is increased [38]. Additionally, when we induced rux expression in cell cycle 14 or 15 embryos, full-length cyclin A translocated to the nucleus of G2 cells (data not shown). Thus, rux promotes nuclear entry of cyclin A as well as cyclin As. Our failure to detect nuclear accumulation of cyclin A in G1 cells might then reflect degradation of nuclear cyclin A in G1. Perhaps a key component of cyclin A destruction is localized in the nucleus. In yeast, mutations in a nuclear import receptor block clb2 destruction [51], suggesting that cyclin destruction might occur in the nucleus. If this were also true for Drosophila, the rux effect on the nuclear translocation of cyclin A would target cyclin A for degradation.
The action of rux
Rux acts as an inhibitor of cyclin A function. On the basis of previous work, it is reasonable to expect that rux, or a rux-dependent factor, might bind to cyclin A–cdc2 and inhibit its activity. Rux is not homologous to known cyclin-dependent kinase inhibitors, however, and we have failed to detect binding of rux to cyclin A or cyclin A–cdc2 in vitro (F.S., unpublished observations). It is possible that rux itself is not the inhibitor, but somehow is required for or promotes the association of a cyclin-dependent kinase inhibitor with cyclin A–cdc2, thus inhibiting its kinase function. Binding of this inhibitor may promote nuclear uptake of the complex, explaining the nuclear localization of cyclin A upon rux expression. Alternatively, rux could promote entry of cyclin A into the nucleus, where the inhibitor is located. Other explanations are possible and more experiments are needed to define the nature of rux inhibition of cyclin A.
At a functional level, rux shares several features with rum1 from Schizosaccharomyces pombe [52] and SIC1 from Saccharomyces cerevisiae [53]. Sic1 protein is present in G1 and inhibits G2 cyclins (Clbs) but not the G1 cyclins (Clns) [54]. The p25rum1 protein — present in G1 but absent in G2 — acts as an inhibitor of the mitotic cdc13–cdc2 kinase complex. In addition, like rux, it prevents the accumulation of mitotic cyclins in G1 [55]. Formally, rux has activities similar to these yeast inhibitors: all stabilize G1 by inhibiting the activity of G2 cyclins; and all are dispensable, consistent with a role in safeguard mechanisms that ensure the stability of G1 while being dispensable for progress of the cycle. The results presented here suggest that sophisticated and partially redundant safeguard controls make important contributions to the creation of a stable quiescent arrest of the cell cycle. The dissection of events that accompany the introduction of G1 into the embryonic cell cycle should reveal the steps necessary to remodel the cycle for such periods of quiescence.
Materials and methods
Plasmid constructs for germ-line transformation
The construct for the HS-DmcycAs transgene was built by digesting the DmcycA cDNA with EcoRV and EcoRI and ligating it into the vector pSF407, which contains a Xenopus β-globin leader as the 5′ untranslated region, resulting in the vector pSF447. This vector was digested with Asp718 and NotI and the DmcycA fragment cloned into pBD403, which contains the hsp70 promoter, resulting in the vector pSF484. For the HS-DmcycBs transgene, a fragment from the DmcycB cDNA was amplified by the polymerase chain reaction using the oligonucleotides SF42 (ATATCCATGGGCATAAGTCGTCCCATCGCA) and SF43 (GCTCTAGAGCACTATTTCCTCTGGCTCT). The resulting fragment was cut with NcoI and XbaI and exchanged with the cDNA contained in the vector pBtor resulting in pSF254 that contains the coding region for a cyclin B protein lacking the first 46 amino acids. A BstBI–XbaI fragment was excised from this vector and replaced with a BstBI–XbaI fragment from the DmcycB cDNA, resulting in pSF283 from which the insert was moved into a Bluescript based vector thereby creating pSF463. Then the insert was excised with Asp718/NotI and cloned into pBD403, resulting finally in the vector used for germ-line transformation, pSF483.
Fly stocks
Control embryos are of the genotype y w67, the parental strain used for germ-line transformation. The fly-stocks with the transgenes HS-DmcycA, HS-DmcycB and HS-cdc2, allowing the expression of DmcycA cDNA, DmcycB cDNA and Dmcdc2 cDNA under the control of a heat-shock promoter, have been described [24,56] The fly stock carrying the HS-cdc2AF transgene was made by P. Leopold, L. Connely and P.H.H.O'F. (unpublished). For the HS-DmcycAs and HS-DmcycBs fly stocks, germ-line transformation was performed by standard P-element transformation into y w67 host strains using the plasmids pSF484 (for HS-DmcycAs) and pSF483 (for HS-DmcycBs). The HS-rux fly stock was kindly provided by B. Thomas (NCI, Bethesda).
Egg collection, heat-shock procedure and BrdU labeling
Embryos were collected on grape-juice agar plates for 1 h and aged for 7 h. Heat-shocks were performed by floating the agar plates for 30 min. directly on the surface of a water bath placed in a 37°C incubator. After heat-shock, the plates were returned to room temperatures. After various recovery times from the heat-shock (see text), the embryos were washed into a basket, dechorionated with 50% bleach, washed extensively with water, dehydrated for 30 sec with isopropanol, permeabilized with octane for 180 sec and placed into Schneider's Medium containing 1 mg/ml BrdU for 20 min. Immediately after the BrdU pulse, the embryos were devitellinized and fixed in 1:1 mixture of methanol and heptane, washed twice with methanol and then fixed for 15 min. with 8% formaldehyde. To detect incorporated BrdU, the embryos were hydrolyzed in 2 N HCl for 40 min., neutralized in 0.1 M Na2B4O7 and washed in PBS containing 0.2 % Tween 20 several times. Incorporated BrdU was observed by immunofluorescence. To block unspecified binding, embryos were incubated in 5% normal goat serum (Jackson Labs) before adding mouse monoclonal anti-BrdU (Beckton Dickinson) primary antibody. For conventional microscopy studies, rhodamine-conjugated goat anti-mouse (Jackson Labs) secondary antibodies were used to visualize BrdU incorporation. For confocal microscopy studies, a FITC-conjugated goat anti-mouse primary antibody was used, the embryos treated with RNAse A (1 mg/ml) for one hour and DNA stained with 1 μg/ml propidium-iodide.
Cyclin A antibodies
Cyclin A protein was expressed and isolated from bacteria. The purified protein was used to immunize rabbits (Caucalito) and polyclonal serum was affinity purified against the bacterially expressed cyclin A protein coupled to cyanogen-bromide activated Sepharose 4B.
Immunoblotting
Heat-shocked embryos were recovered at room temperature for the times indicated, dechorionated, and solubilized in SDS-gel sample buffer. After SDS-PAGE, gels were immunoblotted onto PVDF-membrane (Millipore), blocked with 5% milk in PBS containing 0.2% Tween 20 and incubated with affinity purified cyclin A antibody. After an incubation with horseradish peroxidase labeled secondary antibody (Amersham), the blots were developed using the ECL system (Amersham) and Hyperfilm (Amersham) to detect the signal.
Immunofluorescence detection of cyclin A
Embryos were fixed in 4% formaldehyde for 10 min, devitellinized and blocked for 1 h with 5% normal goat serum in PBS with 0.2% Tween 20 (PBT). Affinity purified cyclin A antibody applied over night in PBT, embryos washed 3 times with PBT and then incubated for 2 h with pre-absorbed FITC labeled goat-anti-rabbit antibodies (Jackson Laboratory) in PBT containing 1 mg/ml RNAse A. Embryos were washed three times with PBT and embedded in Vectashield mounting medium (Vector Laboratories).
Acknowledgments
We thank Barbara Thomas for the HS-rux flies, sharing unpublished observations and discussion, and Edan Foley and Peter Follette for comments on the manuscript. Supported by NIH grant ROI GM37193 (P.H.H.O'F). F.S was supported from H.F.S.P. and the Max-Planck Society.
References
- 1.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 2.Gutmann DH. Tumor suppressor genes as negative growth regulators in development and differentiation. Int J Dev Biol. 1995;39:895–908. [PubMed] [Google Scholar]
- 3.Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 4.La Thangue NB. DP and E2F proteins: components of a heterodimeric transcription factor implicated in cell cycle control. Curr Opin Cell Biol. 1994;6:443–450. doi: 10.1016/0955-0674(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 5.Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. 1995;15:2612–2624. doi: 10.1128/mcb.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 7.Girard F, Strausfeld U, Fernandez A, Lamb NJ. Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell. 1991;67:1169–1179. doi: 10.1016/0092-8674(91)90293-8. [DOI] [PubMed] [Google Scholar]
- 8.Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–971. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duronio RJ, O'Farrell PH. Developmental control of the G1 to S transition in Drosophila: cyclin E is a limiting downstream target of E2F. Genes Dev. 1995;(9):1456–1468. doi: 10.1101/gad.9.12.1456. [DOI] [PubMed] [Google Scholar]
- 10.Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, Lehner CF. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell. 1994;77:107–120. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 11.Richardson H, O'Keefe LV, Marty T, Saint R. Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development. 1995;121:3371–3379. doi: 10.1242/dev.121.10.3371. [DOI] [PubMed] [Google Scholar]
- 12.D'Urso G, Marraccino RL, Marshak DR, Roberts JM. Cell cycle control of DNA replication by a homologue from human cells of the p34cdc2 protein kinase. Science. 1990;250:786–791. doi: 10.1126/science.2173140. [DOI] [PubMed] [Google Scholar]
- 13.Rosenblatt J, Gu Y, Morgan DO. Human cyclin-dependent kinase 2 is activated during the S and G2 phases of the cell cycle and associates with cyclin A. Proc Natl Acad Sci USA. 1992;(89):2824–2848. doi: 10.1073/pnas.89.7.2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson PK, Chevalier S, Philippe M, Kirschner MW. Early events in DNA replication require cyclin E and are blocked by p21CIP1. J Cell Biol. 1995;130:755–769. doi: 10.1083/jcb.130.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehner CF, O'Farrell PH. Expression and function of Drosophila cyclin A during embryonic cell cycle progression. Cell. 1989;56:957–968. doi: 10.1016/0092-8674(89)90629-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sauer K, Knoblich JA, Richardson H, Lehner CF. Distinct modes of cyclin E/cdc2c kinase regulation and S-phase control in mitotic and endoreduplication cycles of Drosophila embryogenesis. Genes Dev. 1995;9:1327–1339. doi: 10.1101/gad.9.11.1327. [DOI] [PubMed] [Google Scholar]
- 18.Smith AV, King JA, Orr-Weaver TL. Identification of genomic regions required for DNA replication during Drosophila embryogenesis. Genetics. 1993;135:817–829. doi: 10.1093/genetics/135.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehner CF, Yakubovich N, O'Farrell PH. Exploring the role of Drosophila cyclin A in the regulation of S phase. Cold Spring Harb Symp Quant Biol. 1991;56:465–475. doi: 10.1101/sqb.1991.056.01.053. [DOI] [PubMed] [Google Scholar]
- 20.Dong XZ, Zavitz KH, Thomas BJ, Lin M, Campbell S, Zipursky SL. Control of G(1) in the developing Drosophila eye: rca1 regulates Cyclin A. Gene Develop. 1997;(11):94–105. doi: 10.1101/gad.11.1.94. [DOI] [PubMed] [Google Scholar]
- 21.Thomas BJ, Gunning DA, Cho J, Zipursky L. Cell cycle progression in the developing Drosophila eye: roughex encodes a novel protein required for the establishment of G1. Cell. 1994;77:1003–1014. doi: 10.1016/0092-8674(94)90440-5. [DOI] [PubMed] [Google Scholar]
- 22.Roy LM, Swenson KI, Walker DH, Gabrielli BG, Li RS, Piwnica-Worms H, Maller JL. Activation of p34cdc2 kinase by cyclin A. J Cell Biol. 1991;(113):507–514. doi: 10.1083/jcb.113.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swenson KI, Farrell KM, Ruderman JV. The clam embryo protein cyclin A induces entry into M phase and the resumption of meiosis in Xenopus oocytes. Cell. 1986;47:861–870. doi: 10.1016/0092-8674(86)90801-9. [DOI] [PubMed] [Google Scholar]
- 24.Knoblich JA, Lehner CF. Synergistic action of Drosophila cyclins A and B during the G2–M transition. EMBO J. 1993;12:65–74. doi: 10.1002/j.1460-2075.1993.tb05632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Resnitzky D, Hengst L, Reed SI. Cyclin A-associated kinase activity is rate limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol Cell Biol. 1995;15:4347–4352. doi: 10.1128/mcb.15.8.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai LH, Harlow E, Meyerson M. Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus E1A-associated p33 kinase. Nature. 1991;353:174–177. doi: 10.1038/353174a0. [DOI] [PubMed] [Google Scholar]
- 27.Hamaguchi JR, Tobey RA, Pines J, Crissman HA, Hunter T, Bradbury EM. Requirement for p34cdc2 kinase is restricted to mitosis in the mammalian cdc2 mutant FT210. J Cell Biol. 1992;117:1041–1053. doi: 10.1083/jcb.117.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith AV, Tl OW. The regulation of the cell cycle during Drosophila embryogenesis: the transition to polyteny. Development. 1991;112:997–1008. doi: 10.1242/dev.112.4.997. [DOI] [PubMed] [Google Scholar]
- 29.Duronio RJ, O'Farrell PH. Developmental control of a G1–S transcriptional program in Drosophila. Development. 1994;120:1503–1515. doi: 10.1242/dev.120.6.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amon A, Irniger S, Nasmyth K. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 1994;77:1037–1050. doi: 10.1016/0092-8674(94)90443-x. [DOI] [PubMed] [Google Scholar]
- 31.Brandeis M, Hunt T. The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. EMBO J. 1996;15:5280–5289. [PMC free article] [PubMed] [Google Scholar]
- 32.King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 33.Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- 34.Glotzer M. Cell cycle. The only way out of mitosis. Curr Biol. 1995;5:970–972. doi: 10.1016/s0960-9822(95)00190-4. [DOI] [PubMed] [Google Scholar]
- 35.Sigrist S, Jacobs H, Stratmann R, Lehner CF. Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B and B3. EMBO J. 1995;14:4827–4838. doi: 10.1002/j.1460-2075.1995.tb00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 37.Edgar BA, Sprenger F, Duronio RJ, Leopold P, O'Farrell PH. Distinct molecular mechanism regulate cell cycle timing at successive stages of Drosophila embryogenesis. Genes Dev. 1994;8:440–452. doi: 10.1101/gad.8.4.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonczy P, Thomas BJ, Di Nardo S. roughex is a dose-dependent regulator of the second meiotic division during Drosophila spermatogenesis. Cell. 1994;77:1015–1025. doi: 10.1016/0092-8674(94)90441-3. [DOI] [PubMed] [Google Scholar]
- 39.Thomas BJ, Zavitz KH, Dong X, Lane ME, Weigmann K, Finley R, et al. roughex downregulates G2 cyclins in G1. Genes Dev. 1997;11:1289–1298. doi: 10.1101/gad.11.10.1289. [DOI] [PubMed] [Google Scholar]
- 40.Duronio RJ, O'Farrell PH, Xie JE, Brook A, Dyson N. The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev. 1995;9:1445–1455. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- 41.deNooij JC, Letendre MA, Hariharan IK. A cyclin-dependent kinase inhibitor, dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell. 1996;87:1237–1247. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- 42.Lane ME, Sauer K, Wallace K, Jan YN, Lehner CF, Vaessin H. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell. 1996;87:1225–1235. doi: 10.1016/s0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- 43.Duronio RJ, Brook A, Dyson N, O'Farrell PH. E2F-induced S phase requires cyclin E. Genes Dev. 1996;(10):2505–2513. doi: 10.1101/gad.10.19.2505. [DOI] [PubMed] [Google Scholar]
- 44.Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science. 1995;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- 45.Piwnica-Worms H, Atherton-Fessler S, Lee MS, Ogg S, Swenson KI, Parker LL. p107wee1 is a serine/threonine and tyrosine kinase that promotes the tyrosine phosphorylation of the cyclin/p34cdc2 complex. Cold Spring Harb Symp Quant Biol. 1991;56:567–576. doi: 10.1101/sqb.1991.056.01.064. [DOI] [PubMed] [Google Scholar]
- 46.Galaktionov K, Lee AK, Eckstein J, Draetta G, Meckler J, Loda M, Beach D. CDC25 phosphatases as potential human oncogenes. Science. 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 47.Pines J, Hunter T. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J Cell Biol. 1991;115:1–17. doi: 10.1083/jcb.115.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardoso MC, Leonhardt H, Nadal-Ginard B. Reversal of terminal differentiation and control of DNA replication: cyclin A and Cdk2 specifically localize at subnuclear sites of DNA replication. Cell. 1993;74:979–992. doi: 10.1016/0092-8674(93)90721-2. [DOI] [PubMed] [Google Scholar]
- 49.Sobczak TJ, Harper F, Florentin Y, Zindy F, Brechot C, Puvion E. Localization of cyclin A at the sites of cellular DNA replication. Exp Cell Res. 1993;206:43–48. doi: 10.1006/excr.1993.1118. [DOI] [PubMed] [Google Scholar]
- 50.Pines J, Hunter T. The differential localization of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 1994;(13):3772–3781. doi: 10.1002/j.1460-2075.1994.tb06688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loeb JD, Schlenstedt G, Pellman D, Kornitzer D, Silver PA, Fink GR. The yeast nuclear import receptor is required for mitosis. Proc Natl Acad Sci USA. 1995;92:7647–7651. doi: 10.1073/pnas.92.17.7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreno S, Nurse P. Regulation of progression through the G1 phase of the cell cycle by the rum1+ gene. Nature. 1994;367:236–242. doi: 10.1038/367236a0. [DOI] [PubMed] [Google Scholar]
- 53.Mendenhall MD. An inhibitor of p34CDC28 protein kinase activity from Saccharomyces cerevisiae. Science. 1993;259:216–219. doi: 10.1126/science.8421781. [DOI] [PubMed] [Google Scholar]
- 54.Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–44. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 55.Correa-Bordes J, Nurse P. p25rum1 orders S phase and mitosis by acting as an inhibitor of the p34cdc2 mitotic kinase. Cell. 1995;83:1001–1009. doi: 10.1016/0092-8674(95)90215-5. [DOI] [PubMed] [Google Scholar]
- 56.Stern B, Ried G, Clegg NJ, Grigliatti TA, Lehner CF. Genetic analysis of the Drosophila cdc2 homolog. Development. 1993;117:219–232. doi: 10.1242/dev.117.1.219. [DOI] [PubMed] [Google Scholar]