

Abstract
The mechanisms through which hematopoietic cytokines accelerate revascularization are unknown. Here, we show that the magnitude of cytokine-mediated release of SDF-1 from platelets and the recruitment of nonendothelial CXCR4+VEGFR1+ hematopoietic progenitors, ‘hemangiocytes,’ constitute the major determinant of revascularization. Soluble Kit-ligand (sKitL), thrombopoietin (TPO, encoded by Thpo) and, to a lesser extent, erythropoietin (EPO) and granulocyte-macrophage colony-stimulating factor (GM-CSF) induced the release of SDF-1 from platelets, enhancing neovascularization through mobilization of CXCR4+VEGFR1+ hemangiocytes. Although revascularization of ischemic hindlimbs was partially diminished in mice deficient in both GM-CSF and G-CSF (Csf2−/−Csf3−/−), profound impairment in neovascularization was detected in sKitL-deficient Mmp9−/− as well as thrombocytopenic Thpo−/− and TPO receptor–deficient (Mpl−/−) mice. SDF-1–mediated mobilization and incorporation of hemangiocytes into ischemic limbs were impaired in Thpo−/−, Mpl−/− and Mmp9−/− mice. Transplantation of CXCR4+VEGFR1+ hemangiocytes into Mmp9−/− mice restored revascularization, whereas inhibition of CXCR4 abrogated cytokine- and VEGF-A–mediated mobilization of CXCR4+VEGFR1+ cells and suppressed angiogenesis. In conclusion, hematopoietic cytokines, through graded deployment of SDF-1 from platelets, support mobilization and recruitment of CXCR4+VEGFR1+ hemangiocytes, whereas VEGFR1 is essential for their angiogenic competency for augmenting revascularization. Delivery of SDF-1 may be effective in restoring angiogenesis in individuals with vasculopathies.
Each year, more than 100,000 individuals undergo lower-limb amputation because of peripheral vascular diseases1. Although initial trials with delivery of angiogenic factors such as VEGF-A were associated with short-term improvement, follow-up studies have yet to show long-term benefits. Therefore, identification of other proangiogenic factors that further accelerate and sustain revascularization of ischemic tissues in individuals with vasculopathies is urgently needed.
Among the known proangiogenic effectors, hematopoietic cytokines and cells may have a major role in supporting neoangiogenic processes2–4. Hematopoietic cells of myelomonocytic origin are recruited to ischemic vessels, delivering the proper stoichiometry of permissive factors necessary for the assembly and stabilization of nascent blood vessels3,5–11. Moreover, soluble hematopoietic cytokines also promote the proliferation, differentiation and motility of progenitors during hemangiogenesis7. The precise phenotype of these proangiogenic hematopoietic cells and the mechanism through which hematopoietic cytokines support neovascularization are not well defined.
Soluble progenitor-active cytokines, including granulocyte-macrophage colony-stimulating factor (GM-CSF, encoded by Csf2), granulocyte colony-stimulating factor (G-CSF, encoded by Csf3)12 and erythropoietin (EPO, encoded by Epo)13,14, promote angiogenesis in part through mobilization of endothelial progenitors. EPO has also been implicated in modulating tumor angiogenesis in the clinical setting15. Hematopoietic cytokines with both stem and progenitor cell activity, including soluble Kit-ligand (sKitL)16 and thrombopoietin (TPO)17, exert a proangiogenic effect on cultured endothelial cells. The mechanism by which hematopoietic cytokines support revascularization in vivo, however, remains unknown.
One factor that may mediate cross-talk between hematopoietic cytokines and vascular cells is the chemokine stromal-derived factor 1 (SDF-1, also known as CXCL12), which, through interaction with its receptor CXCR4 (also known as CD184), modulates hematopoiesis and angiogenesis18–20. SDF-1 regulates angiogenesis in part by recruiting endothelial progenitor cells21–24. In addition, subsets of VEGFR1+ hematopoietic progenitors may also promote tumor neoangiogenesis and metastasis9,25,26. VEGFR1+ hematopoietic progenitors are distinct from endothelial progenitors, and may contribute to revascularization by releasing angiogenic factors or by positioning themselves perivascularly to stabilize nascent neovessels3,5–11. Because VEGFR1+ cells also express CXCR4 (ref. 26), we hypothesized that hematopoietic cytokines, acting through elevation of plasma SDF-1 levels, support the mobilization and recruitment of a unique subset of proangiogenic hematopoietic CXCR4+VEGFR1+ cells, ‘hemangiocytes,’ to the neoangiogenic niche.
Here, we show that hematopoietic cytokines support the release of SDF-1 from platelets, augmenting the mobilization of CXCR4+ VEGFR1+ hemangiocytes, thereby accelerating revascularization. Our studies suggest that delivery of SDF-1 may be effective in accelerating revascularization in individuals with vasculopathies.
Results
Ischemia increases plasma levels of hematopoietic cytokines
The physiological role of hematopoietic cytokines in the regulation of neovascularization is unknown. We showed that 3 d after hindlimb ischemia, there was a significant increase in the plasma levels of sKitL, TPO and, to a lesser extent, GM-CSF and EPO (Fig. 1a). These data suggest that ischemia may modulate neovascularization by direct release of these soluble hematopoietic cytokines.
Figure 1.

Hematopoietic cytokines promote ischemic revascularization. Hindlimb ischemia model was used to assess the role of hematopoietic cytokines in neoangiogenesis. (a) After unilateral femoral artery ligation, plasma TPO and sKitL were upregulated within 72 h. Plasma EPO and GM-CSF were increased to a lesser extent (n = 5 per group, *P < 0.05). (b) Intramuscular (i.m.) delivery of AdTPO (108 plaque-forming units (p.f.u.)) or rTPO (200 ng every 3 d) into the ischemic hindlimb accelerated blood perfusion compared to AdNull-treated controls. After ligation, hindlimb perfusion in untreated Thpo−/− mice was one-third that of wild-type mice (n = 5 per group, *P < 0.05). (c) Intravenous (i.v.) delivery of AdTPO or rTPO (200 ng every 3 d) to Thpo−/− mice induced a similar recovery of perfusion in ischemic hindlimb as did local delivery in wild-type mice (n = 5 per group, *P < 0.05). (d) Wild-type (Mmp9+/+) mice injected intravenously with AdNull, AdmKitL or AdsKitL (108 p.f.u. each) after ligation. Systemic delivery of sKitL or mKitL accelerated restoration of hindlimb blood flow (46% increase) compared to AdNull (left graph; n = 6 per group, *P < 0.05). Vessel density (CD31+ cells/muscle fiber) increased twofold in Mmp9+/+ mice injected with AdsKitL or AdmKitL compared to AdNull (right graph; n = 6 per group, *P < 0.05). (e) After ligation, wild-type mice were injected intravenously with 108 p.f.u. of AdNull, AdEPO or AdGM-CSF. AdEPO or AdGM-CSF accelerated blood flow by 30% 18 d after ligation compared to AdNull (n = 5, *P < 0.05). (f) Csf2−/−Csf3−/− mice were injected intravenously with 108 p.f.u. of AdNull or AdGM-CSF. Ischemic revascularization was slightly decreased in AdNull-treated Csf2−/−Csf3−/− mice, but accelerated in AdGM-CSF–treated Csf2−/−Csf3−/− mice (n = 5 per group, *P < 0.05). (g) At 2 weeks after femoral artery ligation, TPO and sKitL increased perfusion by 1.8- to 2-fold in ischemic hindlimb compared to controls. EPO and GM-CSF increased ischemic blood flow by 1.3-fold (n = 5 per group, *P < 0.05). (h) At 3 d after ligation, plasma TPO and sKitL increased 3.6-fold, whereas EPO and GM-CSF increased only 1.5-fold (n = 5 per group, *P < 0.05).
Neovascularization is impaired in cytokine-deficient mice
To examine the role of soluble cytokines in ischemic revascularization, we used mouse genetic models with impaired release of soluble hematopoietic cytokines, including Thpo−/−27, TPO receptor–deficient (Mpl−/−)28 mice and mice deficient in both GM-CSF and G-CSF (Csf2−/−Csf3−/−)29. We also studied Mmp9−/− mice5,30, in which the release of sKitL from membrane Kit-ligand (mKitL) is impaired, resulting in a severe defect in revascularization31.
Although platelets and megakaryocytes in Thpo−/− and Mpl−/− mice are present at <90% of wild-type levels, these mice do not manifest any spontaneous bleeding diathesis19. Ischemic revascularization is impaired in Thpo−/− (Fig. 1b) and Mpl−/− mice (Supplementary Fig. 1 online) as compared to wild-type controls. Either intramuscular or intravenous injection of TPO into wild-type (Fig. 1b) or Thpo−/− mice (Fig. 1c) rapidly restored revascularization. Thpo−/− mice treated systemically with recombinant TPO (rTPO) showed a fivefold increase in vascular density and less muscular necrosis in both gastrocnemius and adductor muscles of the ischemic hindlimbs (Supplementary Fig. 1). In addition, adenoviral delivery of sKitL (AdsKitL), mKitL (AdmKitL), GM-CSF (AdGM-CSF) or EPO (AdEPO) enhanced vascular recovery in wild-type mice as compared to AdNull-treated controls (Fig. 1d,e). Csf2−/−Csf3−/− mice showed a mild defect in ischemic revascularization, which was effectively rescued by systemic administration of AdGM-CSF (Fig. 1f) or AdEPO (data not shown).
Mice treated with various hematopoietic cytokines showed a gradation of improved ischemic revascularization. TPO and sKitL conferred 1.8- and 2-fold increases in the rate of vascular recovery, respectively, whereas EPO or GM-CSF enhanced the rate of ischemic revascularization by only 1.3-fold (Fig. 1g). Plasma elevation of cytokines also followed a similar trend, with levels of TPO and sKitL 2.3-fold higher than those of EPO and GM-CSF 3 d after induction of hindlimb ischemia (Fig. 1a,h). These data show that hindlimb ischemia induces a graded increase in plasma levels of hematopoietic cytokines.
Elevation of plasma TPO promotes tumor neovascularization
To examine the role of TPO in pathologic angiogenesis, we transplanted syngeneic tumor cell lines, including murine Lewis lung carcinoma (LLC), B16 melanoma, B6RV2 lymphoma and EL-4 thymoma, subcutaneously into Thpo−/−, Mpl−/− or wild-type mice (Supplementary Fig. 2 online). The growth rate of transplanted tumors was reduced in Thpo−/− mice as compared to wild-type controls. There was increased apoptosis by TUNEL assays and reduced neovessel density at tumor sites in Thpo−/− mice transplanted with LLC as compared to wild-type controls. Tumor vessels in Thpo−/− mice were disorganized, with focal areas of hemorrhage. Transplanted tumors in Thpo−/− or Mpl−/− mice also showed a decreased propensity to metastasize to the lungs and kidneys. Intravenous delivery of AdTPO into Thpo−/− mice restored tumor growth and vessel density to wild-type levels, supporting the idea that TPO promotes angiogenesis through a systemic effect, likely via mobilization and recruitment of marrow-derived proangiogenic cells.
sKitL restores revascularization in Mmp9−/− mice
To understand the mechanism through which plasma elevation of hematopoietic cytokines supports neovascularization, we studied Mmp9−/− mice, which have an impairment in the release of sKitL from the membrane-bound form (mKitL)30. We hypothesized that defective ischemic revascularization in Mmp9−/− mice results from the diminished systemic release of sKitL, which would attenuate mobilization of CXCR4+VEGFR1+ hemangiocytes. We tested this hypothesis in Mmp9−/− mice31 to assess the role of sKitL in supporting mobilization of hemangiocytes to ischemic limbs.
Four weeks after femoral artery ligation in Mmp9−/− mice, blood flow recovery was attenuated to 50% of the wild-type level (Fig. 2a), resulting in diminished vessel density (Fig. 2b), swelling, atrophy, necrosis and, subsequently, autoamputation of the ischemic hindlimbs (Fig. 2c). Histological examination showed marked necrosis and diminished vessel density in the hindlimb musculature of Mmp9−/− mice, whereas the extent of neovascular recovery in wild-type mice was normal (Fig. 2b,c). To determine whether the defect in ischemic revascularization resulted from impaired release of sKitL, we showed that plasma elevation of sKitL in Mmp9−/− mice was diminished after ligation as compared to Mmp9+/+ mice (Fig. 2d). This finding led us to hypothesize that intramuscular delivery of sKitL, but not mKitL, in Mmp9−/− mice may reverse the revascularization defect, as we expected that mKitL would not elevate the plasma level of sKitL in Mmp9−/− mice. sKitL introduced either intramuscularly into the ischemic limbs (Fig. 2e,f) or injected systemically (Fig. 2g) was sufficient to restore neovessel density (Fig. 2g) and to effectively reverse the revascularization defect (Fig. 2e,g) in Mmp9−/− mice. In contrast, direct administration of AdmKitL locally into the ischemic limbs of Mmp9−/− mice did not increase plasma sKitL levels (Fig. 2h) and showed no significant effect on augmenting perfusion recovery (Fig. 2e,g) or vessel density (Fig. 2g), resulting in muscle necrosis (Fig. 2f,h). Injection of AdmKitL into wild-type mice, however, resulted in plasma elevation of sKitL (Fig. 2h) and effectively restored revascularization (Fig. 2e). These data suggest that systemic plasma elevation of sKitL is essential for acceleration of ischemic revascularization in wild-type mice and for reversal of the angiogenic defect in Mmp9−/− mice.
Figure 2.
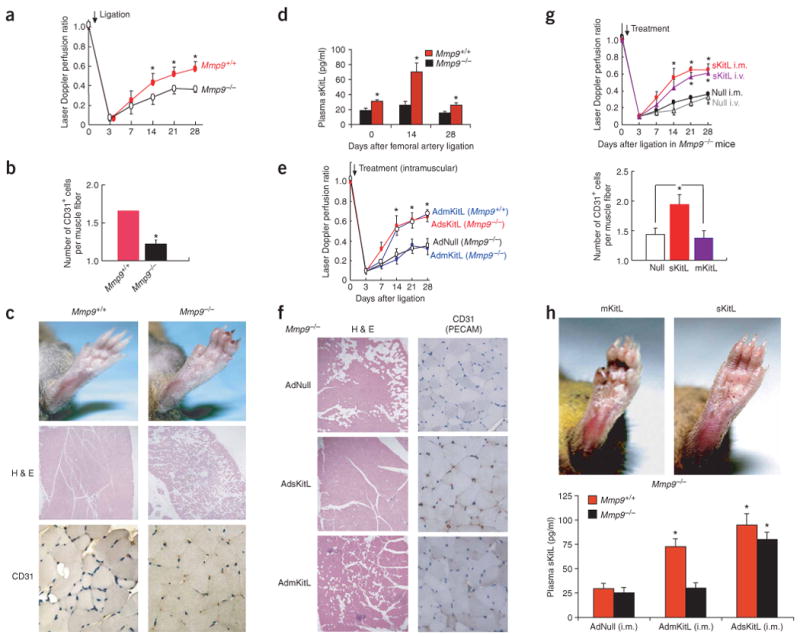
sKitL but not mKitL restores ischemic revascularization in angiogenesis-defective Mmp9−/− mice. (a) The Laser Doppler perfusion ratio was impaired in Mmp9−/− mice compared to Mmp9+/+ controls (n = 12 per group, *P < 0.05). (b) Vessel density (CD31+ cells/muscle fiber) decreased significantly in Mmp9−/− mice compared to Mmp9+/+ controls (n = 12 per group, *P < 0.005). (c) At 28 d after ligation, ischemic footpads showed profound swelling, ulceration and necrosis in Mmp9−/− compared to Mmp9+/+ mice (upper panels). H&E staining of Mmp9+/+ mice showed restoration of angiomyogenesis (middle panels; original magnification, ×100). Mmp9−/− mice showed loss of viable tissues followed by necrosis and adipose replacement. CD31 (PECAM-1) staining of hindlimb muscle sections showed decreased vessel density in Mmp9−/− mice (bottom panels; original magnification, ×400). (d) Hindlimb ischemia induced elevation of sKitL in plasma in Mmp9+/+ but not in Mmp9−/− (n = 6 per group, *P < 0.05). (e) Adenoviral delivery of mKitL (AdmKitL, single dose of 108 p.f.u.) augmented blood flow in Mmp9+/+ but not Mmp9−/− mice (n = 5 per group; P < 0.05). Adenoviral delivery of sKitL (AdsKitL, single dose of 108 p.f.u.) accelerated ischemic revascularization in Mmp9−/− mice. Control mice were injected with 108 p.f.u. of AdNull. (f) H&E (left panels; original magnification, ×100) and CD31 (right panels; original magnification, ×400) staining of lower limb tissue of Mmp9−/− mice injected with AdNull (upper panels), AdsKitL (middle panels) and AdmKitL (bottom panels) 28 d after ligation. Loss of viable tissues with adipose replacement and muscle necrosis was seen after treatment with AdNull or AdmKitL, but not in the AdsKitL-treated group, in which there was restoration of angiogenesis. (g) Intramuscular (i.m.) or intravenous (i.v.) delivery of AdsKitL (single dose of 108 p.f.u.) into Mmp9−/− mice accelerated revascularization comparably in ischemic hindlimbs (upper graph). Vessel density increased in AdsKitL-treated Mmp9−/− mice compared to AdNull- or AdmKitL-treated Mmp9−/− mice (lower graph) (n = 6 per group, *P < 0.05). (h) Reversal of ischemia-induced vasculopathy was observed in Mmp9−/− mice treated with sKitL, but not with mKitL (upper panels). Intramuscular AdmKitL (108 p.f.u.) injected into ischemic hindlimb increased sKitL plasma levels in Mmp9+/+ but not in Mmp9−/− mice 3 d after ligation (n = 4 per group, *P < 0.05). AdsKitL (108 p.f.u.) increased plasma sKitL threefold in Mmp9+/+ and Mmp9−/− mice compared to AdNull (n = 4 per group, *P < 0.05).
Mobilization of hemangiocytes is impaired in Mmp9−/− mice
A plausible mechanism through which sKitL and TPO could support neovascularization is mobilization of CXCR4+VEGFR1+ hemangiocytes. As Sca1+VEGFR1+ hematopoietic cells also express functional CXCR4, impaired release of sKitL and diminished mobilization of CXCR4+VEGFR1+ cells may be responsible for the delayed revascularization seen in Mmp9−/− mice31.
To this end, after induction of hindlimb ischemia in Mmp9+/+ and Mmp9−/− mice, we determined the number of mobilized CXCR4+VEGFR1+ cells in peripheral blood every 4 d for 14 d. Mobilization of CXCR4+VEGFR1+ hemangiocytes (Fig. 3a) was impaired in Mmp9−/− mice, suggesting that impeded mobilization of these cells may result from diminished bioavailability of sKitL.
Figure 3.

Soluble hematopoietic cytokines induce release of SDF-1 from platelets and recruitment of CXCR4+VEGFR1+ cells, accelerating ischemic revascularization. (a) Mobilized CXCR4+VEGFR1+ cells isolated from peripheral blood of Mmp9+/+ and Mmp9−/− mice after induction of hindlimb ischemia were analyzed by two-color flow cytometry. Mobilization of CXCR4+VEGFR1+ cells was impaired in Mmp9−/− compared to wild-type mice (n = 4 per group, *P < 0.05). (b) Plasma SDF-1 levels were induced 2.5-fold 3 d after intravenous injection of recombinant TPO or sKitL compared to PBS-treated controls (n = 4 per group, *P < 0.05). (c) Response of plasma SDF-1 levels to hindlimb ischemia over time. In Mmp9+/+ mice, the peak plasma SDF-1 after ligation was 1.7- to 1.9-fold higher than in Mmp9−/− mice (n = 4 per group, *P < 0.05). (d) Circulating CXCR4+VEGFR1+ cells were measured 3 d after intravenous injection of recombinant sKitL (100 ng) in wild-type mice with or without CXCR4 blockade (CXCR4-specific monoclonal antibody, nonpeptide antagonist AMD3100 or peptidomimetic antagonist CTCE-0012). sKitL induced a fourfold increase in the number of mobilized CXCR4+VEGFR1+ cells compared to PBS- or IgG isotype–treated controls. Administration of sKitL with CXCR4-specific monoclonal antibody, AMD3100 or CTCE-0012 suppressed mobilization of CXCR4+VEGFR1+ cells by threefold compared to sKitL alone (n = 4 per group, *P < 0.05). (e) Wild-type mice were injected intravenously with recombinant TPO (100 ng) or a combination of TPO and CXCR4-specific monoclonal antibody (20 μg/mouse, clone 2B11). After 3 d, CXCR4-specific monoclonal antibody decreased TPO-induced mobilization of CXCR4+VEGFR1+ cells by 83% (n = 4 per group, *P < 0.05). (f) Wild-type mice were injected intravenously with recombinant EPO (100 units) or GM-CSF (100 ng) with or without administration of CXCR4-specific monoclonal antibody (20 μg; clone 2B11) along with PBS- and IgG-treated mice as controls. Neutralizing CXCR4-specific monoclonal antibody completely blocked EPO- and GM-CSF–induced mobilization of CXCR4+VEGFR1+ cells as compared to cytokine-treated groups (n = 4 per group, *P < 0.05). (g) TPO or sKitL conferred a 2.5-fold higher mobilization activity of CXCR4+VEGFR1+ cells than either EPO or GM-CSF (n = 4 per group; P < 0.05). (h) Platelets from wild-type mice were isolated from peripheral blood, stimulated with thrombin or cytokines as indicated, and SDF-1 levels in platelet releasate were measured by ELISA. TPO and sKitL induced a similar dose-dependent release of SDF-1 from platelets, whereas GM-CSF had a minimal effect. Data represent SDF-1 detected in 1 ml platelet releasate obtained from 1 × 107 platelets (n = 3 per group, *P < 0.05).
Elevation of SDF-1 supports mobilization of hemangiocytes
As SDF-1 supports mobilization of hematopoietic cells, we speculated that sKitL- and TPO-mediated intravascular release of SDF-1 may induce mobilization of CXCR4+VEGFR1+ hemangiocytes. Intravenous injection of recombinant sKitL or TPO increased plasma SDF-1 levels (Fig. 3b). We then evaluated whether there was impaired release of SDF-1 in Mmp9−/− mice after hindlimb ischemia. Although plasma SDF-1 levels continued to increase in wild-type mice during the first 48 h after ligation, there was impaired elevation of plasma SDF-1 levels in Mmp9−/− mice (Fig. 3c). These data suggest that sKitL and TPO may support neovascularization by SDF-1–mediated mobilization of CXCR4+ VEGFR1+ hemangiocytes.
To test this hypothesis, we blocked CXCR4-mediated signaling to challenge the ability of sKitL (Fig. 3d), TPO (Fig. 3e) or GM-CSF (Fig. 3f) to induce mobilization of CXCR4+VEGFR1+ hemangiocytes. In wild-type mice, elevation of plasma sKitL (Fig. 3d,g) or TPO levels (Fig. 3e,g) induced a four- or fivefold increase in the mobilization of CXCR4+VEGFR1+ cells, respectively. EPO or GM-CSF were less effective and induced a 2.5- or 1.8-fold increase in the number of mobilized CXCR4+VEGFR1+ cells, respectively (Fig. 3f,g). Inhibition of CXCR4 signaling by a neutralizing monoclonal antibody (clone 2B11)19,32 effectively blocked mobilization of CXCR4+VEGFR1+ cells mediated by sKitL (Fig. 3d), TPO (Fig. 3e), EPO and GM-CSF (Fig. 3f). In addition, a peptidomimetic antagonist to CXCR4 (CTCE-0012)33,34 also inhibited the mobilization of CXCR4+VEGFR1+ cells (Fig. 3d). Notably, chronic daily treatment with AMD3100, a nonpeptide bicyclam antagonist of CXCR4, was as effective as CTCE-0012 and neutralizing monoclonal antibody in inhibiting sKitL-induced mobilization of CXCR4+ VEGFR1+ cells (Fig. 3d). These data indicate that cytokine-induced mobilization of hemangiocytes is mediated in part through elevation of plasma SDF-1 levels and activation of the CXCR4 signaling pathway.
Cytokines induce release of SDF-1 from platelets
Hematopoietic and endothelial cells produce and store SDF-1 (ref. 35), suggesting that cytokines may stimulate release of SDF-1 from the hematopoietic compartment. Platelets store various angiogenic factors, including VEGF-A36 and angiopoietins37. They also co-express receptors for TPO and sKitL38. Therefore, we speculated that soluble cytokines may induce release of SDF-1 from circulating platelets. Unstimulated platelets did not release SDF-1 (Fig. 3h). But TPO- or sKitL-induced activation of platelets freshly isolated from wild-type mice resulted in a substantial increase of SDF-1 in the releasate (Fig. 3h). GM-CSF did not induce release of SDF-1 by platelets, consistent with the lack of GM-CSF receptor expression on platelets29. These findings suggest that the extent of mobilization of CXCR4+VEGFR1+ cells is dictated in part by the capacity of hematopoietic cytokines to release SDF-1 from platelets (Fig. 3h).
Because proteases such as elastases have been thought to be involved in the release of SDF-1, we speculated that MMP9 may also mediate the release of SDF-1 from platelets. TPO and sKitL did not cause release of SDF-1 from platelets isolated from MMP9-deficient mice (Supplementary Fig. 3 online). These data suggest that TPO and sKitL, but not GM-CSF, increase plasma levels of SDF-1 in part through MMP9-mediated deployment from circulating platelet stores.
Elevation of SDF-1 is impaired in thrombocytopenic mice
If TPO-induced release of SDF-1 from platelets supports recruitment of CXCR4+VEGFR1+ cells, then plasma elevation of SDF-1 should be impaired in thrombocytopenic Mpl−/− or Thpo−/− mice. In wild-type mice, SDF-1 was upregulated after hindlimb ischemia (Fig. 4a), which led to increased mobilization of CXCR4+VEGFR1+ hemangiocytes (22% of circulating mononuclear cells; Fig 4b). In contrast, the SDF-1 response in Mpl−/− and Thpo−/− mice was blunted (Fig. 4a), resulting in defective mobilization of Sca1+ and CXCR4+VEGFR1+ hemangiocytes (5.7% of circulating mononuclear cells; Fig. 4b). Thus, attenuated levels of SDF-1 in Thpo−/− mice may be attributed to low platelet levels. These results suggest that TPO, by increasing platelet levels and enhancing release of SDF-1, contributes to the mobilization of CXCR4+VEGFR1+ hemangiocytes and thereby accelerates revascularization.
Figure 4.

SDF-1 reverses the neoangiogenesis defects in Mmp9−/− and Thpo−/− mice in a dose-dependent manner. (a) Hindlimb ischemia after femoral artery ligation was performed in Mpl−/−, Thpo−/− or wild-type mice. Plasma levels of SDF-1 were measured at indicated time points by ELISA. Plasma SDF-1 levels were persistently higher (3.2-fold at 48 h) in wild-type mice as compared to Mpl−/− or Thpo−/− mice during the first 72 h after femoral artery ligation (n = 6 per group, *P < 0.05). (b) The number of circulating CXCR4+VEGFR1+ cells in wild-type mice was 5.5-fold and 5-fold higher than that of Mpl−/− mice 3 and 8 d after femoral artery ligation, respectively (n = 4 per group, *P < 0.05). (c) Administration of AdSDF-1 (low dose, 108 p.f.u. intravenously) to Thpo−/− mice after femoral artery ligation restored perfusion in the ischemic hindlimb by twofold as compared to the AdNull-treated controls. A higher dose (5 × 108 p.f.u. intravenously) improved ischemic perfusion by 2.8-fold compared to AdNull-treated controls (n = 4 per group, *P < 0.05). (d) There was increased vessel density (CD31+ vessels/gastrocnemius or adductor muscle fiber, n = 4 per group) and less muscle necrosis in SDF-1–treated Thpo−/− mice compared to AdNull-treated controls (n = 4 per group, data represent average from five separate high-power fields (HPFs)). Original magnification, ×400. (e) AdSDF-1 injection (low dose, 108 p.f.u. intravenously) in Mmp9−/− mice restored perfusion in the ischemic hindlimb by 1.95-fold compared to AdNull-treated controls. A higher dose (5 × 108 p.f.u. intravenously) improved ischemic perfusion further by 2.5-fold compared to AdNull-treated controls (n = 4 per group, *P < 0.05).
Magnitude of SDF-1 elevation determines revascularization
If the release of SDF-1 is impaired in Thpo−/− and Mmp9−/− mice, then SDF-1 should restore angiogenesis in these mice. Elevation of plasma SDF-1 levels restored revascularization in Thpo−/− (Fig. 4c,d) and Mmp9−/− (Fig. 4e) mice in a dose-dependent manner. Increasing the dose of AdSDF-1 augmented the rate and extent of improvement in the revascularization of ischemic hindlimbs (Fig. 4c,e). Histological examination showed an increase in the density of CD31+ vessels and an absence of necrotic areas in both the gastrocnemius and adductor muscles in Thpo−/− (Fig. 4d) and Mmp9−/− mice (Supplementary Fig. 1). Collectively, these data indicate that the magnitude of SDF-1 elevation in plasma is a major determinant of the extent of revascularization in angiogenesis-defective Thpo−/− and Mmp9−/− mice.
VEGF-A mobilizes hemangiocytes via CXCR4 activation
TPO induces expression of VEGF-A through stimulation of hypoxia inducing factor 1 α (HIF-1α)39. To determine whether TPO directly induces angiogenesis independently of VEGF-A through plasma elevation of SDF-1 levels under normoxic conditions, we injected low-dose recombinant TPO (rTPO) subcutaneously into the ear pinna of wild-type mice. TPO induced sprouting of neoangiogenic vasculature that was comparable to that induced by VEGF-A (Fig. 5a). In contrast to VEGF-A, rTPO caused less tissue edema (Fig. 5a). In vivo Matrigel plug implants loaded with rTPO also showed formation of vascular channels with profuse branching, similar to that of implants loaded with VEGF-A (Fig. 5b). Administration of rTPO to the ear pinna induced a 2- and 2.4-fold increase in plasma TPO and SDF-1 levels, respectively (Fig. 5c). Treatment with rTPO induced a similar neovascular pattern as did treatment with VEGF-A, suggesting that TPO may exert its proangiogenic effect through upregulation of VEGF-A and induced release of SDF-1.
Figure 5.
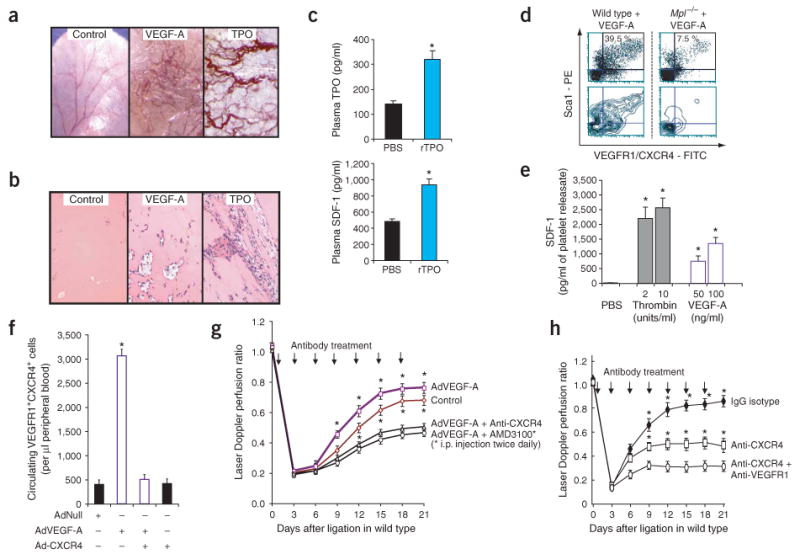
Inhibition of CXCR4 and, to a lesser degree, of VEGFR1 blocks VEGF-A–induced mobilization of hemangiocytes and ischemic revascularization. (a) In the mouse ear pinna angiogenesis model, subcutaneous recombinant TPO (200 ng every 3 d) induced substantial formation and sprouting of neovessels similar to subcutaneous treatment with VEGF-A but with less edema formation (original magnification, ×10). PBS-injected ear pinna was used as a control. (b) Matrigel plugs with recombinant TPO showed substantial formation and branching of vascular channels comparable to VEGF-A (original magnification, ×400). Matrigel plugs loaded with PBS were used as controls. (c) Subcutaneous injection of low-dose recombinant TPO into ear pinna led to a twofold increase in plasma TPO and SDF-1 levels compared to PBS-treated controls (n = 4 per group, *P < 0.05). (d) Three days after intravenous AdVEGF-A (single dose of 108 p.f.u.), a fivefold decrease in circulating CXCR4+VEGFR1+ cells also expressing Sca1 in Mpl−/− versus wild-type mice was detected by flow cytometry. Upper, cytometric distribution of Sca1+VEGFR1+CXCR4+ cells. Lower, corresponding contour mapping of fluorescent signaling intensity. (e) Thrombin and VEGF-A induced dose-dependent release of SDF-1 from wild-type platelets in vitro. Data represent amounts of SDF-1 in 1 ml of platelet releasate obtained from 1 × 107 platelets (n = 3 per group, *P < 0.05). (f) Monoclonal antibody to CXCR4 (clone 2B11, 20 μg intravenously) blocked VEGF-A–induced mobilization of CXCR4+VEGFR1+ cells by 80% in wild-type compared to AdNull-treated mice (n = 4 per group, *P < 0.05). (g) Wild-type C57Bl/6 mice after femoral artery ligation were treated intravenously with AdVEGF-A (single dose of 108 p.f.u.) alone or in combination with either CXCR4-specific monoclonal antibody (clone 2B11, 20 μg intravenously every 3 d) or AMD3100 (1.25 mg/kg intraperitoneally twice daily). CXCR4 blockade with either CXCR4-specific monoclonal antibody (2B11) or AMD3100 aborted enhanced vascular recovery conferred by VEGF-A. Concurrent treatment of AdVEGF-A with either 2B11 or AMD3100 reduced perfusion restoration by 36% and 42.3%, respectively, compared to treatment with AdVEGF-A alone (n = 4 per group, *P < 0.05). Control mice were treated with IgG and AdNull. (h) CXCR4-specific monoclonal antibody (20 μg intravenously every 3 d) reduced the restoration of perfusion by 50% in wild-type mice (n = 4 per group, *P < 0.05), whereas coadministration of CXCR4-specific (20 μg intravenously every 3 d) and VEGFR1-specific (400 μg intraperitoneally every 3 d) antibodies further diminished recovery of perfusion by 63% compared to IgG-isotype controls.
VEGF-A induced the mobilization of CXCR4+VEGFR1+ hemangiocytes in wild-type mice but not in Mpl−/− mice (Fig. 5d), raising the possibility that VEGF-A may also regulate angiogenesis by inducing release of SDF-1 from platelets, which are decreased in thrombocytopenic Mpl−/− mice. Platelets have been shown to express functional VEGFR1 (ref. 40). Similar to TPO and sKitL, VEGF-A could also induce the release of SDF-1 from platelets (Fig. 5e). Moreover, treatment with neutralizing monoclonal antibody to CXCR4 was sufficient to block VEGF-A–mediated mobilization of CXCR4+VEGFR1+ cells. This result strongly suggests that VEGF-A supports mobilization of hemangiocytes through plasma elevation of SDF-1 (Fig. 5f).
VEGF-A induces revascularization through CXCR4 activation
Consistent with previously published data6, elevation of plasma VEGF-A levels can accelerate the rate and magnitude of ischemic revascularization (Fig. 5g). Injection of wild-type mice with AdVEGF-A in combination with either neutralizing monoclonal antibody to CXCR4 or chronic daily treatment with the CXCR4 antagonist AMD3100 partially blocked vascular recovery in wild-type mice or incremental revascularization afforded by the delivery of VEGF-A (Fig. 5g).
Nevertheless, blocking CXCR4 alone did not completely abrogate the proangiogenic activity of VEGF-A. Moreover, inhibition of CXCR4 in conjunction with VEGFR1 blockade was necessary to effectively impede revascularization of ischemic hindlimbs in wild-type mice (Fig. 5h). Therefore, whereas CXCR4 activation has a crucial role in mediating VEGF-A–independent mobilization and recruitment of CXCR4+VEGFR1+ hemangiocytes, the engagement of VEGFR1 is important for the proangiogenic function of these cells.
VEGF-A supports the proangiogenic activity of hemangiocytes
Once recruited to the neoangiogenic niche, CXCR4+VEGFR1+ cells may contribute to revascularization either by releasing angiogenic factors or by stabilizing neovessels by localizing to the perivascular sites. Placental growth factor (PlGF), which signals exclusively through VEGFR1, induced marrow-derived CXCR4+VEGFR1+ hemangiocytes to form spindle-shaped cells (Supplementary Fig. 3). These cells expressed markers that are characteristic of hematopoietic progenitor cells, including Sca1, c-Kit, Tie-2, and of myeloid lineage, such as CD11b and PECAM (Supplementary Table 1 online and data not shown). They did not express markers characteristic of smooth muscle cells (that is, smooth muscle α-actin) or endothelial cells (VE-cadherin, von Willebrand factor or E-selectin). Stimulation of VEGFR1+CXCR4+ hemangiocytes with PlGF resulted in the release of angiopoietin-2 (Ang-2) (Supplementary Fig. 3 and Supplementary Methods online), suggesting that these cells could functionally contribute to neovascularization by elaborating angiogenic factors.
SDF-1 retains hemangiocytes within the ischemic niche
VEGF-A recruits endothelial progenitors through upregulating the expression of SDF-1 (ref. 41), suggesting that expression of CXCR4 may be essential for the retention of hemangiocytes within the ischemic zone. Therefore, we set out to assess the capacity of CXCR4+ cells to rescue the revascularization defect in Mmp9−/− mice. We isolated and expanded CXCR4− and CXCR4+ cells from E13.5 fetal livers of Cxcr4−/− and Cxcr4+/+ embryos, respectively. Subsequently, we injected 5 × 105 PKH26-labeled CXCR4− or CXCR4+ cells separately into the ischemic limbs of Mmp9−/− mice. Compared to CXCR4+ cells, local delivery of CXCR4− cells was less effective in restoring revascularization of ischemic hindlimbs in Mmp9−/− mice (Fig. 6a). Moreover, five times more CXCR4+ cells were retained in ischemic hindlimbs as compared to CXCR4− cells 15 d after ligation and implantation (Fig. 6b,c). These data indicate that CXCR4 activation is important for the retention of hemangiocytes within the neovascular niche.
Figure 6.
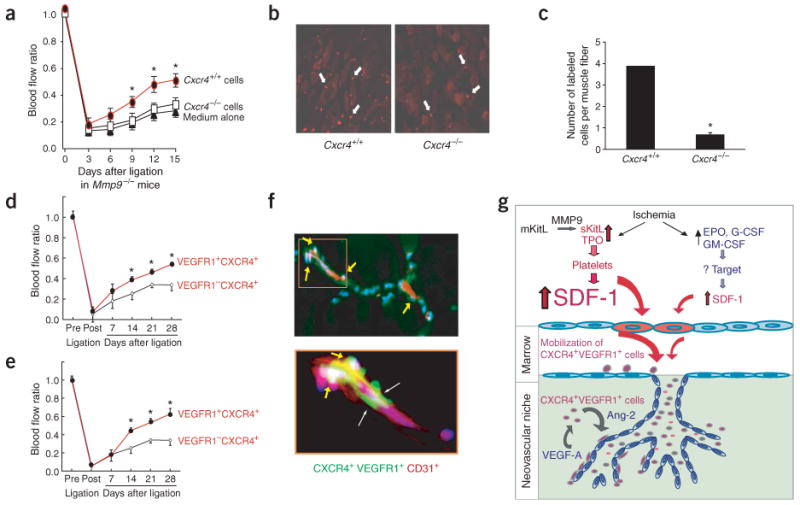
Retention and incorporation of CXCR4+VEGFR1+ cells, but not CXCR4− or VEGFR1− cells, restore functional neoangiogenesis in Mmp9−/− mice. (a) Hematopoietic cells from liver of Cxcr4+/+ or Cxcr4−/− embryos (E13.5) were expanded ex vivo with KitL, fluorescently labeled with PKH26 and subsequently transplanted locally into the ischemic hindlimb of Mmp9−/− mice 24 h after femoral artery ligation. Implantation of Cxcr4+/+ cells (5 × 105, filled circles), but not Cxcr4−/− cells (5 × 105, open squares) or vehicle medium (filled triangles) in ischemic hindlimb of Mmp9−/− mice conferred a 33% increase in perfusion recovery (n = 3 per group, *P < 0.05). (b) PKH26 fluorescently labeled Cxcr4+/+ and Cxcr4−/− cells (white arrows) localized within ischemic limbs 15 d after femoral artery ligation. Original magnification, ×200. (c) The retention of hematopoietic Cxcr4+/+ cells within the ischemic hindlimb in Mmp9−/− mice was fourfold higher than that of Cxcr4−/− cells (n = 3 per group, *P < 0.05). (d,e) Delivery of CXCR4+VEGFR1+ cells (3 × 105), but not CXCR4+VEGFR1− cells (3 × 105), either intramuscularly (d) or intravenously (e) into ischemic hindlimb, significantly improved the rate and extent of revascularization in Mmp9−/− mice (n = 6 per group; P < 0.05). (f) Donor-derived CXCR4+VEGFR1+ cells (green, labeled with PKH2GL) were coimmunostained with CD31 (PECAM-1)-PE (red) and counterstained with DAPI (blue). CXCR4+VEGFR1+ cells were predominantly localized to the perivascular and interstitial region on the abluminal aspect of the collateral CD31+ neovessels 7 d after transplantation (f, arrows in upper panels; original magnification, ×100). The same pattern was observed in all tested ischemic hindlimbs (n = 6 per group). Higher magnification of the indicated area in the upper panel is shown in the lower panel (original magnification, ×400). (g) Schematic model: SDF1-CXCR4 as the molecular hub for hematopoietic cytokine-induced neovascularization.
CXCR4+VEGFR1+ cells reverse angiogenic defect in Mmp9−/− mice
To determine whether physical delivery and localization of CXCR4+VEGFR1+ cells to the ischemic niche may reverse the revascularization defect in Mmp9−/− mice, we isolated purified marrow-derived CXCR4+VEGFR1+ and CXCR4+VEGFR1− mononuclear cells from wild-type mice and injected them either intravenously into the tail vein or intramuscularly in the vicinity of the ligated femoral artery in Mmp9−/− mice (Fig. 6d,e). Mmp9−/− mice transplanted with VEGFR1+ but not VEGFR1− cells showed an increase in blood flow recovery (Fig. 6d,e). Although transplantation of VEGFR1+ cells did not completely restore revascularization of ischemic hindlimbs, it functionally rescued Mmp9−/− mice from ischemic necrosis and autoamputaion (data not shown). At day 7 after transplantation, donor-derived VEGFR1+ cells were localized to the periendothelial region at the abluminal side of the neovessels (Fig. 6f). Therefore, in addition to promoting the mobilization of CXCR4+ hemangiocytes from the marrow, SDF-1 also supports retention and localization of these cells within the neovascular niche, whereas VEGFR1 modulates the angiogenic performance of CXCR4+ hemangiocytes (Fig. 6g).
Discussion
Here, we set forth a new concept by which ischemia induces plasma elevation of stem and progenitor cell-active cytokines, including sKitL and TPO, and, to a lesser extent, progenitor-active cytokines, such as GM-CSF and EPO. TPO and sKitL induced the release of SDF-1 from platelets, thereby increasing systemic plasma levels of SDF-1. This resulted in a substantial mobilization of CXCR4+VEGFR1+ cells, accelerating revascularization of the ischemic limbs. We have termed this unique class of CXCR4+VEGFR1+ cells ‘hemangiocytes,’ which represent a heterogeneous population of VEGF-responsive nonendothelial, proangiogenic hematopoietic progenitors consisting of immature and differentiated myelomonocytic cells. Although endothelial progenitors, smooth muscle cells and hemangiocytes share some phenotypic and functional characteristics, hemangiocytes can be distinguished from their vascular-associated counterparts based on lack of expression of VE-cadherin, von Willebrand factor, E-selectin and smooth muscle α-actin. Hemangiocytes also express progenitor markers, including Sca1 and Tie2, and induce neovascularization by releasing angiogenic factors as well as by physically supporting the assembly of endothelial cells1,41. Although recruited hemangiocytes localize primarily to the perivascular region3,5–10, vascular cells differentiating from endothelial progenitors are believed to integrate into the lumen of neovessels1,5,6,9,21,23,41. Together, our data support the proposition that the SDF-1–CXCR4 signaling pathway is the main molecular hub that mediates recruitment of CXCR4+VEGFR1+ hemangiocytes and thereby accelerates ischemic revascularization.
The magnitude of mobilization and incorporation of CXCR4+VEGFR1+ hemangiocytes and the extent of functional revascularization correlated with the absolute plasma SDF-1 levels. Relative to GM-CSF and EPO, which had only minor effects on the mobilization of CXCR4+VEGFR1+ cells, TPO and sKitL increased SDF-1 levels by fourfold, promoting a proportional increase in the number of circulating CXCR4+VEGFR1+ cells and rapid revascularization of ischemic limbs. Intravascular deployment of SDF-1 was achieved through direct sKitL- and TPO-mediated release of SDF-1 from platelets. GM-CSF did not induce release of SDF-1 from platelets. These results were validated in mouse genetic models, in which we showed that the revascularization defect in sKitL-deficient Mmp9−/− as well as thrombocytopenic Thpo−/− and Mpl−/− mice was also associated with diminished plasma elevation of SDF-1 and impaired mobilization of CXCR4+VEGFR1+ cells. Intravenous or local delivery of CXCR4+ VEGFR1+ cells, but not of CXCR4− or VEGFR1− cells, into the ischemic limbs of Mmp9−/− mice restored revascularization and rescued ischemic limbs from necrosis. Inhibition of CXCR4 blocked cytokine-mediated mobilization of CXCR4+VEGFR1+ cells and revascularization of ischemic limbs in wild-type mice, whereas elevation of plasma SDF-1 levels restored revascularization in Thpo−/− and Mmp9−/− mice in a dose-dependent manner. These data suggest that the SDF-1–CXCR4 signaling pathway primarily supports cytokine-mediated angiogenesis through mobilization of CXCR4+VEGFR1+ cells, whereas VEGFR1 modulates the proangiogenic activity of these cells within the ischemic niche.
SDF-1, acting through CXCR4, is the principal chemokine that modulates trafficking of hematopoietic stem and progenitor cells42,43. The precise role of CXCR4 in mobilization of hematopoietic cells during neo-angiogenic processes has not been well defined. Consistent with previously published data, we showed that SDF-1 and its peptidomimetic analogs promote mobilization of marrow-derived hematopoietic cells to the circulation20,30,34,44–46. Paradoxically, other studies have suggested that inhibition of CXCR4 by proteases and the nonpeptide antagonist AMD3100 (refs. 47,48) may also promote mobilization of hematopoietic cells. In those studies, however, the effect of AMD3100 on mobilization of hematopoietic cells was examined only in the first few days of treatment. Here, we showed that chronic inhibition of CXCR4 beyond 3 d, by sustained daily treatment with AMD3100, diminished mobilization of CXCR4+ cells and impaired revascularization. Neutralizing monoclonal antibody to CXCR4 and a peptidomimetic CXCR4 antagonist, CTCE-0012, also blocked mobilization, providing complementary evidence to support the notion that chronic inhibition of CXCR4 impedes, rather than induces, mobilization of CXCR4+VEGFR1+ cells and thereby impairs neovascularization. Whether the strain of mice also influences the extent of SDF-1–mediated mobilization of hemangiocytes is unknown and remains to be determined.
As compared to the lineage-specific cytokines, such as GM-CSF and EPO, cytokines with activity for stem and progenitor cells, including TPO, sKitL and VEGF-A, induced a larger degree of plasma elevation of SDF-1, enhancing mobilization of CXCR4+VEGFR1+ hemangiocytes. Thus, TPO and sKitL were more effective in accelerating ischemic revascularization. A plausible explanation for this differential effect is the fact that receptors for TPO, sKitL and VEGF-A (c-Mpl, c-Kit and VEGFR1, respectively) are expressed not only on stem and progenitor cells but also on platelets38. Therefore, as compared to the lineage-specific cytokines, TPO, sKitL and VEGF-A can induce release of SDF-1 from a broader spectrum of hematopoietic cells, particularly from platelets. This possibility was supported by the finding that in thrombocytopenic TPO- and c-Mpl–deficient mice, there was a profound defect in the plasma elevation of SDF-1 after induction of hindlimb ischemia.
Platelets have been shown to carry a myriad of proangiogenic factors, including VEGF-A and angiopoietins36,37. Platelets can also release antiangiogenic factors, such as thrombospondins and platelet factor 4. But what determines the platelet activity as a proangiogenic modulator? It is conceivable that the release of SDF-1 is selectively modulated by stimulation with different cytokines such as TPO, sKitL and VEGF-A. These cytokines may selectively induce release of SDF-1 from platelets, but not release of other antiangiogenic factors, thereby facilitating mobilization of CXCR4+VEGFR1+ hemangiocytes and accelerating revascularization.
In vitro studies have shown that hematopoietic cytokines can promote the survival and motility of primary cultured endothelial cells13,14,16,17, raising the possibility that SDF-1 may directly induce angiogenesis by recruiting preexisting vascular cells within the ischemic niche. Implantation of CXCR4− and VEGFR1− cells, however, did not restore neovascularization of the ischemic limbs in Mmp9−/− mice. These data suggest that the proangiogenic effect of hematopoietic cytokines is mediated in part through SDF-1–induced mobilization and incorporation of CXCR4+VEGFR1+ cells, and not entirely through local recruitment of preexisting CXCR4+ or VEGFR1+ neovessels. In support of this conclusion, we showed that mKitL induced neovascularization in wild-type, but not in Mmp9−/− mice. These results suggest that release of sKitL to the circulation is essential to restore neovascularization through a systemic effect by mobilizing hemangiocytes. In addition, plasma elevation of SDF-1 or TPO, or systemic delivery of CXCR4+VEGFR1+ hemangiocytes, was as effective as their local delivery to support revascularization of the ischemic limbs. Therefore, hematopoietic cytokines exert their proangiogenic effects systemically through mobilization and recruitment of hemangiocytes to the neoangiogenic niche.
Subsets of hematopoietic cells may contribute to the revascularization of ischemic limbs or tumor angiogenesis3,5–10,49. We have now shown that CXCR4+VEGFR1+ hemangiocytes can reverse the revascularization defect in Mmp9−/− mice. The magnitude of plasma elevation of SDF-1 correlated with the number of mobilized CXCR4+VEGFR1+ cells and the rate of improvement in ischemic revascularization. These findings suggest that the cellular stoichiometry of recruited CXCR4+VEGFR1+ cells is dictated by the level of cytokine-mediated release of SDF-1 and is one of the crucial determinants for functional neovascularization. Therefore, delivery of hematopoietic cytokines, SDF-1 or hemangiocytes can be considered as a plausible therapeutic strategy for accelerating ischemic revascularization.
Expression of CXCR4 on marrow-derived cells has been shown to mediate oncogene addiction in breast tumors. Elevation of plasma SDF-1 levels by tumor fibroblasts supported recruitment of marrow-derived Sca1+CD31+ cells, thereby accelerating tumor angiogenesis and growth50. Thus, inhibition of CXCR4 may provide an effective means to block tumor growth, whereas administration of SDF-1 could support revascularization of ischemic tissues. Furthermore, SDF-1 levels in plasma and platelets may serve as surrogate biomarkers to assess neoangiogenic activity during ischemic revascularization and tumor angiogenesis.
Methods
Mice, adenoviral vectors, recombinant cytokines and antibodies
The derivations of TPO-deficient27, c-Mpl–deficient28, MMP9-deficient3, and G-CSF- and GM-CSF–deficient mice29 have been previously described. We purchased strain-matched wild-type (C57BL/6) mice from the Jackson Laboratory. All mice were age-(6–8 weeks), weight-(20–25 grams) and sex-matched. The experiments were performed with the authorization of the institutional review board and the Animal Care and Use Committee of the Weill Cornell Medical College. Mice received adenoviral vectors (Ad5-derived, E1A−E3−E4+)26 expressing SDF-1 (AdSDF-1), TPO (AdTPO), VEGF-A (AdVEGF-A), EPO (AdEPO), GM-CSF (AdGM-CSF), mKitL (AdmKitL) and sKitL (AdsKitL) or empty vector (AdNull) in 100 μl PBS, administered by a single tail-vein or intramuscular injection on day 1 after femoral artery ligation.
We purchased mouse recombinant TPO, GM-CSF, G-CSF, sKitL, VEGF-A, and SDF-1 from Peprotech and EPO from Amgen. To inhibit CXCR4 in vivo for the mobilization of hematopoietic cells, we injected mice intravenously with 20 μg azide-free, endotoxin-free monoclonal antibody to CXCR4 (clone 2B11, BD Biosciences)32 every 3 d for a total of seven injections. Similarly, we intraperitoneally administered neutralizing monoclonal antibody against VEGFR1 (clone MF-1, ImClone Systems) at a dose of 400 μg concurrently with CXCR4-specific monoclonal antibody for the blocking studies. Control mice received 100 μl IgG-isotype through tail vein injection or intraperitoneally on the same schedule as CXCR4-specific or VEGFR1-specific monoclonal antibody, respectively. We dissolved CTCE-0012 (Chemokine Therapeutic Corp.), which is a peptidomimetic CXCR4 antagonist33,34, at 4 μg in 100 μl PBS and injected it intravenously through tail vein every 6 h. We administered AMD3100 (Sigma Chemical Co.) as a twice-daily intraperitoneal injection at a dose of 1.25 mg/kg.
SDF-1 release from platelets
We obtained platelet-rich plasma from 750 μl mouse blood that we collected from the retro-orbital plexus and mixed it with 450 μl physiologic mouse saline (165 mM NaCl) and 50 μl 3.2% sodium citrate as an anticoagulant. We centrifuged the diluted blood mixture at 1,000 r.p.m. at 24–27 °C for 5 min and harvested the supernatant as platelet-rich plasma (PRP). The platelet count in PRP was determined by an automated Advia-120 Hematology System. We collected platelets by centrifugation of PRP at 2,500 r.p.m. for 5 min at 24–27 °C, and subsequently subjected them to activation by thrombin (Sigma) and various cytokines. The levels of SDF-1 were measured by Quantikine ELISAs (R&D) and expressed as pg/ml of platelet releasate, (which contained 1 × 107 platelets/ml releasate).
Antibodies for flow cytometry
We labeled mouse circulating mononuclear cells with the following monoclonal antibodies: PE-conjugated Sca1-specific (E13.161.7 and D7), PE-conjugated CD11b-specific (clone M1/70), and PE-conjugated CXCR4-specific (clone 2B11) from BD PharMingen; and FITC-conjugated VEGFR1-specific (clone MF-1, ImClone Systems) antibodies.
Identification of CD31+ vessels
We deparaffinzied 5-μm sections of formaldehyde-fixed, paraffin-embedded muscles (gastrocnemius and adductor) and incubated them with primary antibody to CD31 (PECAM-1; 1:300; BD PharMingen) overnight at 4 °C. We then incubated sections with a rat-specific rabbit IgG (1:200) and the Vectastain Elite ABC reagents per the manufacturer's recommendation. Immunohistochemical signals were developed with 3-3′ diaminobenzidine substrate and counterstained with hematoxylin.
Hindlimb ischemia model
For the hindlimb ischemia model, mice underwent ligation and segmental resection of left femoral vessel as previously described6. To induce hindlimb ischemia, we made an incision in the skin at the left inguinal area and carried out extraperitoneal dissection laterally along the tissue planes to expose the femoral vessels. We then ligated the femoral vessels both proximally and distally using 6-0 silk sutures and resected the ligated vessels between the ligatures without damaging the nervus femoralis. We then closed the left groin incision using 4-0 polysorb sutures.
We performed the sham operation by incision and suturing of the skin alone without ligation or severing of the femoral vessels. We measured the rate of blood flow every 3 d after the ligation by means of a laser-Doppler perfusion scanner (PeriScan System, Perimed) in both the ischemic and nonischemic hindlimbs of the same mouse. We then calculated the ratio of relative blood flow by dividing the laser Doppler-derived perfusion of the ischemic hindlimb by that of the nonischemic hindlimb.
Ear pinna angiogenesis model and Matrigel plug assay
We established a mouse syngeneic tumor transplant model using LLC, B16 melanoma, EL2 sarcoma or B6RV2 lymphoma by injecting respective tumors subcutaneously into the right flank of Thpo−/−, Mpl−/− or C57Bl/6J (wild type) mice at a dosage of 2 × 106 cells per inoculation. We measured tumors twice weekly with calipers and calculated tumor volumes as (w1 × w2 × w3), where w1 and w2 represent the largest and smallest tumor diameter, respectively; w3 represents the thickness of the tumor. For the metastasis study, we transplated LLC cells as described above. Mice were subsequently killed and the organs removed, weighed and examined to measure the number of metastatic nodules.
In the murine ear pinna angiogenesis model, we investigated the angiogenic response to recombinant TPO and VEGF-A. We injected mice (n = 3–5) subcutaneously with either recombinant murine VEGF-A (50 ng every 3 d), recombinant TPO (200 ng every 3 d) or PBS (negative control) in a volume of 50 μl at the base of the ear pinna. Mice were killed 14 d after injection and the ear pinna examined under dissecting microscope for vascularity.
For the in vivo Matrigel assay, we anesthetized mice and injected them subcutaneously at abdominal midline with 0.5 ml growth factor–depleted Matrigel (BD Biosciences) supplemented with 100 ng mouse recombinant TPO or 10 μg VEGF-A. We injected control mice with Matrigel without adding exogenous growth factors. We removed Matrigel plugs 21 d after implantation, photographed them and prepared them for histological examination.
Isolation and transplantation of CXCR4+VEGFR1+ cells
We isolated marrow-derived CXCR4+VEGFR1+ or CXCR4+VEGFR1− cells from 8-week-old mice using biotin-conjugated monoclonal mouse-specific rat antibodies to CXCR4 (2B11) and VEGFR1 (MF-1), followed by avidin-Microbeads isolation by MACS (Miltenyi Biosystems). We labeled CXCR4+VEGFR1+ cells with green fluorescence membrane linker (PKH2-GL, Sigma Chemical Co) and used CXCR4+VEGFR1− cells as controls. We resuspended 300,000 cells (either CXCR4+VEGFR1+ or CXCR4+VEGFR1−) in 100 μl serum-free X-VIVO 20 medium (Cambrex Biosciences), then injected them either intramuscularly into four separate zones within the underlying quadriceps muscle in the ischemic limb area or intravenously through the tail vein with the use of a 27.5-gauge needle (n = 6 for each implantation group).
Ex vivo expansion and transplantation of fetal liver-derived Cxcr4−/− hematopoietic cells
Pregnant mice from matings between Cxcr4+/− mice were killed at gestation age of embryonic day (E)13.5 and liver from each fetus was harvested. We extracted DNA from each fetus and determined the genotype by RT-PCR to distinguish between Cxcr4+/+ and Cxcr4−/− genotypes as per Jackson Laboratory Protocols. We expanded hematopoietic cells ex vivo with 100 ng/ml sKitL. After 48 h of incubation, we harvested the nonadherent cell population and labeled the cells with red fluorescent membrane linker PKH26 (Sigma). We injected a total of 5 × 105 cells of either Cxcr4+/+ or Cxcr4−/− genotype intramuscularly into four separate zones within the underlying quadriceps muscle in the ischemic thigh area 24 h after femoral artery ligation in Mmp9−/− mice (n = 3).
Supplementary Material
Acknowledgments
We wish to acknowledge the technical assistance of P. Lau, M. Choy, G. Kanhai, R. Tejada, G. Lam and A. Intrator. We would also like to thank F. de Sauvage from Genentech for providing the Thpo−/− and Mpl−/− mice. S.R. is an investigator of Howard Hughes Medical Institute and is supported by the American Cancer Society, the Lymphoma and Leukemia Society and the National Institutes of Health (RO1-HL075234, HL59312, HL66592 and HL67839). D.K.J. is supported by the Hermione Foundation. D.L. is supported by the Doris Duke Charitable Foundation, the Children's Blood Foundation and a grant from the National Cancer Institute.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
Competing Interests Statement: The authors declare competing financial interests (see the Nature Medicine website for details).
References
- 1.Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–712. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- 2.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–490. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takakura N, et al. A role for hematopoietic stem cells in promoting angiogenesis. Cell. 2000;102:199–209. doi: 10.1016/s0092-8674(00)00025-8. [DOI] [PubMed] [Google Scholar]
- 5.Aicher A, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 6.Luttun A, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002;8:831–840. doi: 10.1038/nm731. [DOI] [PubMed] [Google Scholar]
- 7.Cursiefen C, et al. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest. 2004;113:1040–1050. doi: 10.1172/JCI20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang L, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 9.Lyden D, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 10.Conejo-Garcia JR, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–958. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 11.De Palma M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi T, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 13.Heeschen C, et al. Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood. 2003;102:1340–1346. doi: 10.1182/blood-2003-01-0223. [DOI] [PubMed] [Google Scholar]
- 14.Kertesz N, Wu J, Chen TH, Sucov HM, Wu H. The role of erythropoietin in regulating angiogenesis. Dev Biol. 2004;276:101–110. doi: 10.1016/j.ydbio.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 15.Henke M, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet. 2003;362:1255–1260. doi: 10.1016/S0140-6736(03)14567-9. [DOI] [PubMed] [Google Scholar]
- 16.Matsui J, Wakabayashi T, Asada M, Yoshimatsu K, Okada M. Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J Biol Chem. 2004;279:18600–18607. doi: 10.1074/jbc.M311643200. [DOI] [PubMed] [Google Scholar]
- 17.Brizzi MF, et al. Thrombopoietin stimulates endothelial cell motility and neoangiogenesis by a platelet-activating factor-dependent mechanism. Circ Res. 1999;84:785–796. doi: 10.1161/01.res.84.7.785. [DOI] [PubMed] [Google Scholar]
- 18.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 19.Avecilla ST, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004;10:64–71. doi: 10.1038/nm973. [DOI] [PubMed] [Google Scholar]
- 20.Petit I, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 21.Butler JM, et al. SDF-1 is both necessary and sufficient to promote proliferative retinopathy. J Clin Invest. 2005;115:86–93. doi: 10.1172/JCI22869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walter DH, et al. Impaired CXCR4 signaling contributes to the reduced neovascularization capacity of endothelial progenitor cells from patients with coronary artery disease. Circ Res. 2005;97:1142–1151. doi: 10.1161/01.RES.0000193596.94936.2c. [DOI] [PubMed] [Google Scholar]
- 23.Ceradini DJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 24.De Falco E, et al. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood. 2004;104:3472–3482. doi: 10.1182/blood-2003-12-4423. [DOI] [PubMed] [Google Scholar]
- 25.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hattori K, et al. Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med. 2002;8:841–849. doi: 10.1038/nm740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunting S, et al. Normal platelets and megakaryocytes are produced in vivo in the absence of thrombopoietin. Blood. 1997;90:3423–3429. [PubMed] [Google Scholar]
- 28.Gurney AL, Carver-Moore K, de Sauvage FJ, Moore MW. Thrombocytopenia in c-mpl-deficient mice. Science. 1994;265:1445–1447. doi: 10.1126/science.8073287. [DOI] [PubMed] [Google Scholar]
- 29.Basu S, et al. “Emergency” granulopoiesis in G-CSF-deficient mice in response to Candida albicans infection. Blood. 2000;95:3725–3733. [PubMed] [Google Scholar]
- 30.Heissig B, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires mmp-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson C, Sung HJ, Lessner SM, Fini ME, Galis ZS. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues: potential role in capillary branching. Circ Res. 2004;94:262–268. doi: 10.1161/01.RES.0000111527.42357.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forster R, et al. Intracellular and surface expression of the HIV-1 coreceptor CXCR4/fusin on various leukocyte subsets: rapid internalization and recycling upon activation. J Immunol. 1998;160:1522–1531. [PubMed] [Google Scholar]
- 33.Crump MP, et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16:6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez LE, et al. Increased plasma levels of stromal-derived factor-1 (SDF-1/CXCL12) enhance human thrombopoiesis and mobilize human colony-forming cells (CFC) in NOD/SCID mice. Exp Hematol. 2004;32:300–307. doi: 10.1016/j.exphem.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Dar A, et al. Chemokine receptor CXCR4-dependent internalization and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nat Immunol. 2005;6:1038–1046. doi: 10.1038/ni1251. [DOI] [PubMed] [Google Scholar]
- 36.Mohle R, Green D, Moore MA, Nachman RL, Rafii S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci USA. 1997;94:663–668. doi: 10.1073/pnas.94.2.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li JJ, Huang YQ, Basch R, Karpatkin S. Thrombin induces the release of angiopoietin-1 from platelets. Thromb Haemost. 2001;85:204–206. [PubMed] [Google Scholar]
- 38.Avraham H, et al. Effects of the stem cell factor, c-kit ligand, on human megakaryocytic cells. Blood. 1992;79:365–371. [PubMed] [Google Scholar]
- 39.Kirito K, Kaushansky K. Thrombopoietin stimulates vascular endothelial cell growth factor (VEGF) production in hematopoietic stem cells. Cell Cycle. 2005;4:1729–1731. doi: 10.4161/cc.4.12.2197. [DOI] [PubMed] [Google Scholar]
- 40.Selheim F, Holmsen H, Vassbotn FS. Identification of functional VEGF receptors on human platelets. FEBS Lett. 2002;512:107–110. doi: 10.1016/s0014-5793(02)02232-9. [DOI] [PubMed] [Google Scholar]
- 41.Grunewald M, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–189. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 42.Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106:1901–1910. doi: 10.1182/blood-2005-04-1417. [DOI] [PubMed] [Google Scholar]
- 43.Papayannopoulou T. Current mechanistic scenarios in hematopoietic stem/progenitor cell mobilization. Blood. 2004;103:1580–1585. doi: 10.1182/blood-2003-05-1595. [DOI] [PubMed] [Google Scholar]
- 44.Hattori K, Heissig B, Rafii S. The regulation of hematopoietic stem cell and progenitor mobilization by chemokine SDF-1. Leuk Lymphoma. 2003;44:575–582. doi: 10.1080/1042819021000037985. [DOI] [PubMed] [Google Scholar]
- 45.Hattori K, et al. Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood. 2001;97:3354–3360. doi: 10.1182/blood.v97.11.3354. [DOI] [PubMed] [Google Scholar]
- 46.Pelus LM, et al. The CXCR4 agonist peptide, CTCE-0021, rapidly mobilizes polymorphonuclear neutrophils and hematopoietic progenitor cells into peripheral blood and synergizes with granulocyte colony-stimulating factor. Exp Hematol. 2005;33:295–307. doi: 10.1016/j.exphem.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 47.Broxmeyer HE, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liles WC, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102:2728–2730. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]
- 49.Losordo DW, Dimmeler S. Therapeutic angiogenesis and vasculogenesis for ischemic disease: part II: cell-based therapies. Circulation. 2004;109:2692–2697. doi: 10.1161/01.CIR.0000128596.49339.05. [DOI] [PubMed] [Google Scholar]
- 50.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.