

SUMMARY
Intrinsic innate immune mechanisms are the first line of defense against pathogens, and exist to control infection autonomously in infected cells. Here we show that autophagy, an intrinsic mechanism that can degrade cytoplasmic components, plays a direct anti-viral role against the mammalian viral pathogen Vesicular Stomatitis Virus (VSV) in the model organism Drosophila. We found that the surface glycoprotein, VSVG, is likely the pathogen-associated molecular pattern (PAMP) that initiates this cell-autonomous response. Once activated, autophagy decreases viral replication, and repression of autophagy leads to increased viral replication and pathogenesis in cells and animals. Lastly, we show that the antiviral response is controlled by the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway which normally regulates autophagy in response to nutrient availability. Altogether these data uncover an intrinsic antiviral program that that links viral recognition to the evolutionarily conserved nutrient signaling and autophagy pathways.
INTRODUCTION
Detection and clearance of viruses by the innate immune system relies on several distinct and essential pathways that are evolutionarily conserved (Janeway and Medzhitov, 2002). Extracellular virions are recognized via Toll-like receptors present on the cell surface and within endolysosomal compartments. These receptors were first identified in Drosophila and are now recognized as the canonical pathogen recognition system in all metazoans (Uematsu and Akira, 2006). Cytoplasmic pattern recognition receptors, originally identified in plants (Meylan et al., 2006), also act as intracellular pathogen detectors. Yet another intracellular pattern recognition system, responsible for the degradation of viral dsRNA genomes or replication intermediates, is the RNA interference (RNAi) pathway (Li and Ding, 2005). This ancient mechanism of viral RNA degradation plays important roles in anti-viral immunity in plants as well as metazoans including Drosophila (Cherry and Silverman, 2006). Recently, autophagy, another ancient and conserved intracellular pathway, has also been implicated in innate immunity and pathogen destruction (Gutierrez et al., 2004; Kirkegaard et al., 2004; Levine and Deretic, 2007; Liu et al., 2005; Nakagawa et al., 2004; Rich et al., 2003).
Autophagy was first genetically characterized in yeast as a response to starvation and is the process by which cells degrade cytoplasmic components including organelles for recycling (Klionsky et al., 2003; Klionsky and Ohsumi, 1999; Kuma et al., 2004; Mizushima et al., 2004). There are various types of autophagy, including micro- and macroautophagy, as well as chaperone-mediated autophagy, and they differ in their mechanisms and functions (Mizushima et al., 2008). Macroautophagy, which will be referred to as autophagy throughout this article, occurs through the de novo formation of double-membraned vesicles called autophagosomes that envelop cytoplasmic regions, which mature and grow, and subsequently fuse with the lysosome for degradation of the internalized cytoplasmic contents. Autophagy is involved in a plethora of processes, including the removal of damaged organelles and intracellular protein aggregates, turnover of long-lived proteins, and cell death (Mizushima et al., 2008). Autophagy is required both for normal development, and survival from nutrient depravation. For these processes, it has been shown that autophagy is controlled by the PI3K/Akt signaling pathway. Activitation of the signaling pathway inhibits autophagy, whereas the loss of signaling through this cascade relieves the negative repression of TOR (Target of Rapamycin) kinase on Atg1, an essential upstream component of the autophagy pathway (Stephan and Herman, 2006). Therefore, there is a direct link between nutrient availability and levels of autophagy.
In mammalian systems, autophagy was recently shown to play a role in innate immune defense against intracellular pathogens(Levine and Deretic, 2007). Studies on bacterial pathogens have revealed that some bacteria are cleared from the cytoplasm by autophagy (Andrade et al., 2006; Gutierrez et al., 2004; Ling et al., 2006; Nakagawa et al., 2004; Ogawa et al., 2005; Singh et al., 2006). However, it has yet to be established whether autophagy is induced by bacterial infection or if it plays a role at the organismal level. There have also been studies implicating autophagy in antiviral defense. For example, many viruses can be observed inside of autophagic compartments including Herpes Simplex virus Type-1 (HSV-1), and Sindbis virus (Seay et al., 2005; Talloczy et al., 2006). Moreover, data suggest that beclin 1 (the human homolog of Atg6), perhaps via autophagy, restricts viral encephalitis induced by both HSV-1 and Sindbis (Liang et al., 1998; Orvedahl et al., 2007). In plants, autophagy prevents the spread of cell death during the hypersensitive response which restricts viral replication (Liu et al., 2005). However, it has not been established that autophagy itself is sufficient to control the replication of these viruses, or if there are other facets of activated pathways such as cell death that are impacting viral growth. Importantly, it has not been demonstrated genetically that autophagy restricts viral replication in animals. Moreover, which viral component induces autophagy and which signaling pathways are responsible are currently unknown.
Given the potentially critical role of autophagy in defense from infection, and the fact that there is little known about intrinsic anti-viral mechanisms, we set out to determine whether autophagy can directly control viral replication in the genetically tractable organism, Drosophila melanogaster. Drosophila has no acquired immune system, relying only on innate systems to combat pathogens. This simplified immune system allows us to test the role of innate factors in isolation. Here we show that autophagy controls Vesicular Stomatits Virus (VSV) replication in both cultured Drosophila cells and adult flies. VSV infection via the surface glycoprotein is sensed by flies, and this in turn activates autophagy, independent of viral replication. Increased autophagic activity is mediated by repression of the nutrient signaling cascade, which alleviates repression of autophagy activating the antiviral program. This pathway is critical to control of viruses because of the fact that loss of autophagy converts a non-pathogenic VSV infection into a lethal one.
RESULTS
VSV infection of Drosophila cells is controlled by autophagy
We first characterized infection of Drosophila cells with VSV. VSV is the prototypical member of the non-segmented negative sense RNA virus family, and is transmitted to cattle by insects (Letchworth et al., 1999; Palese et al., 1996). This virus infects and spreads between arthropods and mammals, yet is controlled by the immune system of each, resulting in little mortality in either host. To study mechanisms of defense against this virus, we challenged Drosophila S2 cells with VSV that expresses a GFP reporter (VSV-GFP) upon successful replication (Ramsburg et al., 2005).
We found that VSV-GFP replicates in Drosophila cells in a dose-dependent manner. New viral antigens were detected by 20 h post-infection as measured by GFP production as well as the production of the viral glycoprotein VSV-G (Figure 1A) (Cherry et al., 2005). We also monitored viral replication using immunoblot analysis to detect the production of viral antigens. Whole cell lysates from cells infected with increasing multiplicities of infection (MOI) were examined using an antibody against GFP or VSV-M (Figure 1B). Thus, we can monitor a dose-dependent increase in viral infection using both assays.
Figure 1. VSV infects Drosophila cells.
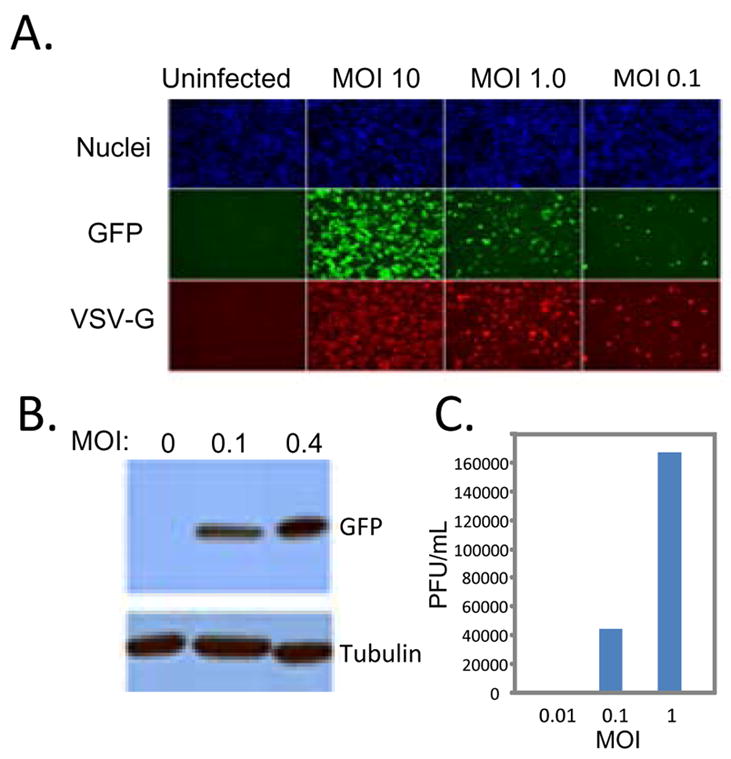
A. Schneider cells were infected with VSV-GFP at the indicated multiplicity of infection (MOI) for the 20 hours. Cells were processed for immunofluorescence and imaged using an automated microscope (ImagXpress Micro). Infected cells express GFP and the viral glycoprotein, VSV-G and are counterstained with Hoechst 33342 to observe nuclei. B. Viral antigen production at the indicated MOI at 24 hours post infection was measured by immunoblot against the virally produced antigen GFP or the cellular control tubulin. These data show a representative experiment; similar findings were made in at least three repetitions. C. Viral titers from cells infected with the indicated MOI of VSV at 24 hours post infection. These data show a representative experiment; similar findings were made in at least two repetitions.
We also determined whether VSV productively infects Drosophila cells. To this end, we monitored the production of new viral progeny. Cells were infected with increasing amounts of virus, and 24 hours post infection, the amount of virus produced was measured by plaque assay on mammalian cells demonstrating a full replication cycle in Drosophila cells (Figure 1C). Altogether, these studies show that VSV productively infects Drosophila cells, generating virus that is infectious to mammalian cells.
Drosophila has homologues of 11 yeast autophagy-related genes, and many of these have been confirmed to be required for autophagy in flies during development or starvation (Scott et al., 2004). To test whether loss of the autophagy pathway affects viral replication in tissue culture cells, dsRNAs were generated against three of these highly conserved genes previously shown to be involved in autophagy in flies (Atg5, Atg8a, and Atg18) (Scott et al., 2004). Three different genes were tested, in order to rule out off-target effects of the dsRNAs, as well as to confirm that effects observed are due to the autophagy pathway rather than an unrelated pathway or process affected by any individual RNAi reagent. To deplete the proteins we treated cells with dsRNA or a control dsRNA for three days then challenged the cells with VSV-GFP. We used automated imaging and analysis to monitor infection. Using this assay, we observed a significant increase in the percentage of infected cells by knocking down each autophagy-related gene (Figure 2A). Semi-quantitative RT-PCR and RT-qPCR was used to demonstrate that treatment with dsRNAs leads to a depletion of the cognate mRNA (Supplementary Figure 1). In addition, immunoblot analysis demonstrated that treatment of cells with dsRNA against Atg8a led to a complete loss of Atg8a protein (Figure 3E). The increase in viral replication measure by fluorescence was verified biochemically: cells were depleted for Atg5, Atg8a or Atg18, infected with VSV, and probed by immunoblot for GFP (Figure 2B). We observed an increase in the amount of GFP in the autophagy-depleted cells.
Figure 2. Autophagy is antiviral in Drosophila cells.
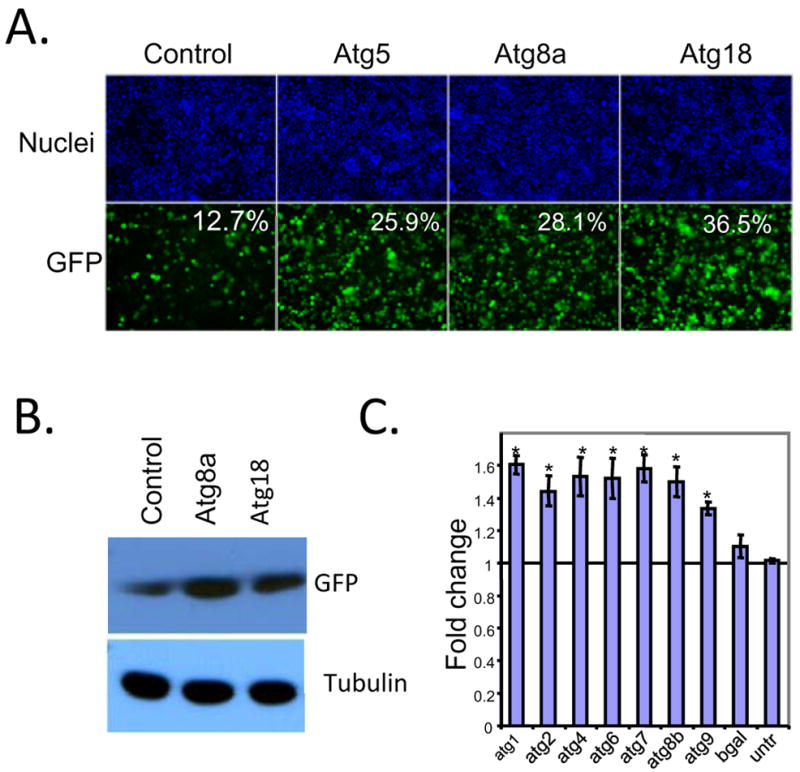
A. S2 cells were pre-treated with dsRNA against the autophagy genes Atg5, Atg8a, or Atg18 or control dsRNA (luciferase) for three days and then infected (MOI=1) for 20 hours and processed for immunofluoresence and imaged as above. Infected cells express GFP and the percent infection is calculated using automated image analysis (MetaXpress) from 3 wells, 3 sites per well. Loss of autophagy genes significantly increases VSV infection (p<0.05). B. Depletion of autophagy genes also increases the production of viral antigens by immunoblot. Cells pretreated with the indicated dsRNAs were infected (MOI=0.1) and processed for Western blot at 24 hours post infection. Viral antigens were measured by anti-GFP and normalized to the control protein tubulin. These data show representative experiments; similar findings were made in at least three repetitions. C. Depletion of additional autophagy genes by RNAi leads to an increase in the percent infection as compared to control dsRNA. Data is shown for four independent experiments as the fold effect compared to control dsRNA and the standard error is shown (MOI=0.1–0.25, 24 hours post infection); * p<0.05 student’s ttest.
Figure 3. VSV infection activates autophagy independent of replication.

A. VSV infection induces autophagosome formation as measured by electron microscopy. Representative images are shown for cells that are either uninfected or infected at an MOI=5, 20 hours post infection. B. Higher magnification images of autophagosomes in VSV-infected cells. C. Active and UV-inactivated virus induce autophagic vesicles. Cells were infected with UV-inactivated VSV-GFP as above. Autophagic bodies were counted for at least 20 cells from each treatment and presented as a notched box plot. The horizontal darkline represents the median for each category. The box represents the interquartile range and the whiskers encompass the most extreme data values. The notches represent a confidence interval for the median and nonoverlapping notches indicate different medians at the 5% significance level. There is a significant difference between uninfected and infected or UV-virus infected (* Wilcoxon test, p-value = 9.0e-5 and p-value =3.7e-3), respectively. However, there is no significant difference in the number of autophagosomes per cell between infected and UV-virus infected (Wilcoxon test, p-value = 0.22). D. The induction of autophagic vesicles by VSV is Atg5-dependent. S2 cells were pre-treated with either control (LacZ) or Atg5 dsRNA for 3 days and infected with VSV-GFP at an MOI=5 for 20 hours. Autophagic bodies were counted for 35 cells from each treatment and presented as a notched box plot. The horizontal black line represents the median for each category. Whiskers include the most extreme data value. Nonoverlapping notches indicate different medians at the 5% significance level. There is a significant difference between control and Atg5-depleted cells (* Wilcoxon test, p-value = 1.2e-07). These findings were observed in at least two independent experiments. E. Immunoblot analysis of cells treated with control dsRNA or dsRNA against Atg8 (Atg8a and Atg8b). Atg8 is observed in resting cells (Atg8-I, ~16kD). F. A smaller form of Atg8 is induced in cells that are serum starved, or infected with VSV, or UV inactivated VSV (Atg8-II, ~14kD). These data show representative experiments; similar findings were made in at least three repetitions.
To further validate that the autophagy pathway as a whole is required for the phenotype, we tested additional conserved autophagy genes including Atg1, Atg2, Atg4, Atg6, Atg7, Atg8b, or Atg9. Cells were treated with the indicated dsRNA for three days and then infected with VSV. Using microscopy and automated image analyses, we quantified the percentage of infected cells across four independent experiments and normalized the infection to untreated cells. When compared to the non-targeting control dsRNA (b-gal) we observed a significant increase in VSV infection upon depletion of autophagy genes (Figure 2C). We also found a similar increase in viral titers produced in cells depleted for autophagy genes (Supplementary Figure 2). Altogether, these data demonstrate that autophagy controls viral replication, consistent with its playing an antiviral role in cultured cells.
VSV infection activates autophagy in cultured cells
The finding that autophagy inhibits VSV replication suggests that autophagy might be induced by VSV infection of cells. Autophagy is routinely monitored by three assays: electron microscopy, fluorescent GFP-LC3 punctae or as lipidated LC3 by immunoblot (Klionsky et al., 2008). We first monitored autophagy by electron microscopy. This approach allows identification of intracellular organelles and structures, and to show that virally-induced compartments are of autophagic origin. Double-membrane vesicles that contain cytoplasmic components or degrading organelles such as mitochondria are not known to arise by any other mechanism. Therefore, we performed electron microscopy analysis on both VSV-infected and uninfected S2 cells. Upon quantification of autophagic bodies, we found a >2.5-fold increase in the number of autophagosomes in the infected cells (Figure 3A, C). Higher magnification images show double membrane vesicles with cytoplasmic contents or membranous contents (degrading mitochondria) indicative of autophagic compartments (Figure 3B).
To verify that the VSV-induced vesicular structures we observed were indeed autophagic in origin, we tested whether Atg5 depletion by RNAi could prevent their induction. Indeed we found that loss of Atg5 inhibited the induction of these autophagic compartments as measured by electron microscopy (Figure 3D). Altogether, these data suggest that viral infection induces autophagy, which in turn attenuates viral replication.
Given that VSV infection activates autophagy, there were two possible viral inducers: incoming virions and viral replication products. To test whether virus replication is required, we challenged cells with either replication competent or UV-inactivated virus, and measured the effect on autophagy using electron microscopy. UV-inactivated virus binds cells, undergoes endocytosis, and fuses within the endolysosomal compartment (Dubovi and Youngner, 1976). The core, released into the cytoplasm, is replication incompetent. We verified that the UV-inactivated virus was indeed replication defective by fluorescence microscopy (Supplementary Figure 3). Using this assay, we found that both replication-competent and UV-inactivated VSV induce autophagic vesicles (Figure 3C). Indeed, quantification reveals that UV-inactivated virus induces the formation of autophagic bodies to a similar extent as replication competent virus. This suggests that the incoming virus presents the cells with a pre-existing molecule (perhaps a PAMP) that is sufficient to activate the antiviral program.
During autophagy cytosolic LC3 (LC3-I) is conjugated on its carboxyl terminus with phosphatidylethanolamine forming lipidated LC3 (LC3-II) which localizes to the autophagic membrane. The amount of LC3-II correlates with the number of autophagosomes and can be monitored by immunoblot (Klionsky et al., 2008). In Drosophila there are two LC3 homologs that are 95% similar, Atg8a and Atg8b. We developed an antibody that recognized the C-terminus of both homologs as treatment of cells with dsRNA against Atg8 (dsRNA has overlap with both targets) completely abolished the signal (Figure 3E). In addition, we observed an increase in the production of Atg8-II upon starvation validating our antibody (Figure 3F). Next, we tested whether infection with wild type or UV-inactivated virus induced the production of Atg8-II. Indeed, we observed significant induction of Atg8-II upon treatment with both replication competent and incompetent virus (Figure 3F).
To further study the role of autophagy in VSV infection we took advantage of an assay that depends on the translocation of the autophagosome protein LC3 (Atg8) from the cytosol (diffuse distribution) to newly formed autophagosomes, which appear as bright cytoplasmic punctae (Kabeya et al., 2000). GFP-LC3 is an autophagosome-specific membrane marker detected in puctae upon autophagosomes induction in both mammalian and Drosophila systems (Rusten et al., 2004; Scott et al., 2004; Yano et al., 2008). This assay allows us to monitor the induction of autophagy cell-by-cell. We expressed this GFP tagged LC3 (GFP-LC3) in cultured cells and found that there is diffuse cytoplasmic staining (Figure 4A). However, upon infection, with either wild type or UV-inactivated virus we observed a significant increase in the percentage of punctae-containing cells. This is quantified in Figure 4B. Moreover, we found that there was also an increase in the number of punctae per cell upon infection with either wild type or UV-inactivated VSV (Figure 4C). Lastly, we tested whether the activation of autophagy was cell-autonomous. To this end, cells expressing GFP-LC3 were uninfected, infected, or infected at a multiplicity of infection such that 30% of the cells were VSV-M+. Next, we quantified the percentage of cells with punctae that were either VSV+ or VSV−. We found that only the infected cells had an increase in GFP-LC3 punctae (Figure 4D). Together, the combination of electron microscopy, the induction of Atg8-II and GFP-LC3 punctae formation we show that VSV infection, in the absence of viral replication, led to a robust, cell-autonomous increase in autophagy in Drosophila cells.
Figure 4. VSV infection induces autophagy in Drosophila cells.

VSV induces autophagosomes as measured by LC3 re-localization. A. Drosophila cells were transfected with a GFP-LC3 reporter (green). Cells were either uninfected or infected with wild type VSV or UV inactivated VSV for 24 hours, fixed, probed with anti-VSVM (Red) and with Hoechst 33342 (blue). A representative image is shown. Arrows indicate GFP-LC3+ puncta. B. The percentage of cells with punctae were counted for five experiments and fold change was graphed for cells that were uninfected (blue) infected with wild type VSV (red) or UV inactivated VSV (green); error bars indicate standard deviation; * p<0.01 student’s ttest. C. There is an increase in the number of punctae per cell upon infection with either VSV or UV inactivated VSV graphed as the number of punctae per cell normalized to 100% for each treatment (n>150 for each condition). D. The induction of GFP-LC3 punctae is cell-autonomous. The percentage of cells with punctae were counted for three experiments and fold change was graphed for cells that were either uninfected (blue), infected (red), or uninfected in a well where 30% of the cells were infected (orange). Only the infected cells have an increase in the GFP-LC3 punctae; error bars indicate standard deviation; * p<0.01 student’s ttest. E. There is no increase in the percentage of cells with punctae in cells that were transfected with viral RNA (red) compared to controls (blue). The percentage of cells with punctae was counted for three experiments and fold change was; error bars indicate standard deviation. F. There is no increase in the percentage of cells with punctae in cells that were transfected with viral RNP (red) compared to controls (blue). The percentage of cells with punctae was counted for three experiments and fold change was graphed; error bars indicate standard deviation. G. There is an increase in the percentage of cells with punctae in cells that were infected with VSV G+ VPs compared to blebs isolated from untransfected control cells. The average of three experiments is shown; error bars indicate standard deviation; * p<0.01 student’s ttest.
The viral glycoprotein, VSV G, induces autophagy
The VSV virion is largely composed of the viral RNA genome, the proteins that are bound to the viral RNA making up the RNA-nucleoprotein complex or RNP, matrix proteins associated with the nucleocapsid and the membrane-bound surface envelope spike glycoprotein which extends from the viral membrane. We set out to determine which component of the virion is the pathogen associated molecular pattern or PAMP that is recognized by Drosophila cells, leading to the induction of antiviral autophagy. To this end, we purified virions from which we isolated the RNP and the viral RNA (Gupta et al., 1995). Because VSV is a negative strand RNA virus, the naked nucleic acid is not capable of initiating an infection in the absence of the viral proteins that make up the RNP (N, P, and L). We verified that the purified RNA was non-functional and that the RNP was functional by co-transfection with a reporter to mark the transfected cells. As expected, we found that 100% of the cells transfected with the RNP but none of the cells transfected with viral RNA produced VSV-M as measured by immunofluorescence (Supplementary Figure 4A). Next, we tested whether cells transfected with the viral RNA or RNP induced autophagy as measured by GFP-LC3 punctae formation, and found that neither PAMP was able to induce autophagy using this assay (Figure 4E, F).
Next, we set out to test whether the viral glycoprotein VSV G could induce autophagy. To purify VSV G containing vesicular particles (VPs) devoid of any other viral component we either mock transfected or transfected 293T cells with VSVG. VSV G alone is sufficient to induce blebbing and the formation of VPs that are infectious and have no other viral component (Abe et al., 1998; Rolls et al., 1994). We purified the VPs and found that only the VPs from the VSV G transfected cells carry VSV G by immunoblot (data not shown), and used these VPs to infect Drosophila cells expressing GFP-LC3. We found that Drosophila cells treated with the VSV G VPs but not the preparation from mock-transfected cells were positive for VSV G by immunofluorescence (Supplementary Figure 4B). Next, we challenged cells expressing GFP-LC3 with the purified blebs and found that treatment of cells with VSV G+ VPs were sufficient to induce LC3-GFP+ punctae (Figure 4G). Taken together, our data suggest that VSV G is the PAMP that is recognized by Drosophila cells.
VSV infection induces autophagy both in primary cells and in adult flies
The above findings indicate a critical role for autophagy in cultured cells. We extended our findings to primary cells by taking advantage of transgenic animals that express GFP-LC3 in larval hemocytes. Hemocytes are the Drosophila phagocytic macrophage-like cell type from which S2 cells are derived. We used the Gal4/UAS system, in which transgenes under the control of Gal4 transcription factor binding sites (UAS sites) can be induced by Gal4 expression (Brand and Perrimon, 1993). This allows us to drive expression of the autophagosome marker GFP-LC3 using a specific hemocyte promoter, hemolectin. Third instar larval hemocytes expressing GFP-LC3 (hemolectin-Gal4, UAS-GFP-LC3) were isolated and challenged with VSV ex vivo. In uninfected primary hemocytes, there is diffuse, low level GFP-LC3 expression (Figure 5A). In contrast, by 2 hours post infection, the hemocytes show bright GFP positive punctae indicative of autophagosome induction (Figure 5A) and quantified in Figure 5B. This time point is prior to the initiation of viral replication, further supporting the hypothesis that a preformed component of the virus, rather than replicating virus, is recognized by cells to induce autophagy.
Figure 5. VSV infection induces autophagy in primary cells and in adult flies.

A. Primary hemocytes expressing GFP-LC3 (Hml-gal4>UAS-eGFP-huLC3, green) were either uninfected or infected ex vivo for two hours, then fixed and stained with Hoescht 33342 to visualize nuclei (blue) by fluorescence microscopy. A representative image is shown. B. Percentage of cells with punctae were counted (n>40 for each condition). Error bars indicate range of two independent experiments. Increased punctae formation is observed in the infected cells; * p<.001, chi-square test in each experiment. C. Adult wild type flies were infected with VSV-GFP for three days. The flies were monitored for infection (GFP+) and autophagy (Lysotracker+) and counterstained with Hoescht 33342 to observe the nuclei. There is increased Lysotracker staining in the infected cells in vivo in the animal. White arrows indicate that cells that are Lysotracker+ are also GFP+. Blue arrows (merged image) show that the GFP− cells throughout the tissue are not Lysotracker+. These data show representative experiments; similar findings were made in at least three repetitions.
To further demonstrate that infection induces autophagy in vivo, we infected adult flies with VSV-GFP and monitored autophagy using Lysotracker staining, a marker for acidified compartments that has been extensively used in flies (Bilen and Bonini, 2007; Chen et al., 2008; Juhasz and Neufeld, 2006). Using this assay, we found that VSV infection (GFP expression) leads to an increase in autophagy (Lysotracker+) in vivo (Figure 5C). Moreover, this induction is again cell-autonomous; only GFP+ cells were Lysotracker+. These data show that both Drosophila cells and animals respond to VSV infection by inducing an autophagic program.
Autophagy plays an antiviral role in adult flies
To determine whether autophagy also plays an innate anti-viral role in the adult organism in vivo, we characterized viral infection of adult flies by challenging them with VSV and monitored infection. We found that VSV infection is non-pathogenic in adult flies as measured by mortality post-infection (Figure 6A). We next set out to determine if depletion of autophagy genes alters the susceptibility of flies to infection. Mutant flies were generated in vivo by driving expression of transgenes bearing long hairpin double-stranded RNA constructs targeting specific autophagy genes in vivo, leading to loss-of-function phenotypes. We crossed control and transgenic flies with hairpin transgenes against Atg18 (UAS-Atg18 IR), previously shown to be required for starvation-induced autophagy in flies, to actin-GAL4 resulting in high levels of ubiquitous transgene expression (Scott et al., 2004). We validated that Atg18 is indeed depleted in these flies by semi-quantitative RT-PCR and RT-qPCR (Supplementary Figure 1). Whereas UAS-Atg18 IR singly transgenic flies, and flies expressing a control dsRNA (Supplementary Figure 5) were resistant to VSV-induced lethality, actin-GAL4/UAS-Atg18 IR doubly transgenic flies that were depleted for Atg18 became sensitive to infection and succumbed to the virus. A representative experiment is shown in Figure 6A). Flies depleted for Atg18 had a normal lifespan, and were not more sensitive to infection with Drosophila C virus (Supplementary Figure 5). Together, this shows that the altered pathology of the autophagy mutant is specific and that this pathway is protective against VSV infection.
Figure 6. Autophagy is antiviral in adult flies.

A. Adult flies expressing ubiquitous and high level dsRNA against Atg18 (Actin-Gal4; UAS-Atg18IR) or sibling controls (+; UAS-Atg18IR) were challenged with 104 pfu VSV and morbitity was monitored as a function of time post-infection. Log rank test reveals that loss of Atg18 significantly increases susceptibility (p<0.001). A representative experiment is shown; similar findings were made in at least three experiments. B. Adult flies expressing ubiquitous and high level dsRNA against Atg18 (Actin-Gal4; UAS-Atg18IR) or their sibling controls (+; UAS-Atg18IR) were challenged with VSV and monitored over time for viral replication as measured by immunoblot against virally produced GFP and normalized to a cellular control protein. * indicates a non-specific band. C. Adult flies expressing ubiquitous and high level dsRNA against Atg18 (Actin-Gal4; UAS-Atg18IR, red bars) or their sibling controls (+; UAS-Atg18IR, blue bars) were challenged with VSV and monitored over time for viral replication as measured by viral titers. Plaque assays reveal a significant increase in viral titers in Atg18-depleted flies post infection. D. Adult flies expressing ubiquitous and high level dsRNA against Atg18 (Actin-Gal4; UAS-Atg18IR) or their sibling controls (+; UAS-Atg18IR) were challenged with VSV and monitored over time for viral replication as measured by Northern blot. There is a significant increase in the levels of GFP RNA in the Atg18-depleted animals. E. Adult flies expressing ubiquitous and high level dsRNA against Atg7 (Actin-Gal4; UAS-Atg7IR) or their respective sibling controls (+; UAS-Atg7IR) were challenged with VSV and monitored over time for viral replication as measured by Northern blot. There is a significant increase in the levels of GFP RNA in the Atg7-depleted animals. F. Adult flies expressing ubiquitous and high level dsRNA against Atg12 (Actin-Gal4; UAS-Atg12IR) or their respective sibling controls (+; UAS-Atg12IR) were challenged with VSV and monitored over time for viral replication as measured by Northern blot. There is a significant increase in the levels of GFP RNA in the Atg12-depleted animals.
We also found that the increased mortality correlates with increased viral replication in flies. At all time points post-infection there was a marked increase in the levels of VSV antigen production, as measured by immunoblot (Figure 6B). In addition, we measured the levels of virus production by plaque assays and found that there was an increase in viral titers in the Atg18-depleted flies at early time-points post infection, prior to lethality (Figure 6C). Taken together, our data suggest that loss of autophagy in adult flies leads to increased viral replication leading to mortality from an otherwise non-lethal infection.
To verify that our data for Atg18 is due to an effect on autophagy and not another role for Atg18, we tested whether depletion of additional autophagy genes, Atg7 or Atg12, previously shown to be required for starvation-induced autophagy in flies, also affects viral replication {Scott, 2004 #13}. We found that there was a significant increase in the amount of VSV RNA replication in animals depleted for Atg18 (Figure 6D), Atg7 (Figure 6E), or Atg12 (Figure 6F), as measured by Northern blot. We also monitored viral replication by Western blot analysis of the virus-encoded GFP, and again observed a significant increase in viral growth in flies depleted for the autophagy genes Atg7 and Atg12 (Supplementary Figure 6). We also validated that RNAi itself does not impact viral replication by expressing a control dsRNA against the beta-galactosidase gene, and found that there was no effect on viral growth (data not shown). Next, we determined whether loss of Atg7 also had an effect on survival post challenge with VSV. Indeed, we found that whereas UAS-Atg7 IR singly transgenic flies were resistant to VSV-induced lethality, actin-GAL4/UAS-Atg7 IR doubly transgenic flies were more sensitive to infection although it takes longer to succumb to the infection (Supplementary Figure 5).
Lastly, we tested whether loss of Atg18 had an effect on hemocyte numbers or functionality. Hemocytes are macrophage-like cells which control infection of intracellular pathogens (Lemaitre and Hoffmann, 2007). To monitor this phagocytic cell type we injected flies that were either wild type or depleted for Atg18 with fluorescent beads. Fluorescent beads are efficiently phagocytosed by hemocytes allowing for the number and activity of this cell type to be monitored (Elrod-Erickson et al., 2000). The abodomens of these flies were imaged and we found that there was no difference in the number of hemocytes or the level of phagocytic activity in the hemocytes of adult flies depleted for Atg18 (Supplementary Figure 7). Taken together, we demonstrate that autophagy is induced by infection in primary cells, inhibits VSV replication in animals, and prevents viral pathogenesis.
Nutrient signaling controls autophagy during viral infection in animals
Given that autophagy is induced by infection, we were interested in determining which signaling pathway might control this antiviral response. The PI3K/Akt pathway has been previously shown to regulate autophagy during development and starvation. Therefore, we tested whether this pathway also senses VSV infection in vivo. In resting cells autophagy is repressed, at least in part, by the PI3K signaling pathway. Akt, a key kinase in this pathway, has been demonstrated to control autophagy via activation of the TOR (Target of Rapamycin) kinase (Lum et al., 2005). TOR is a global cell regulator that responds to nutritional conditions and represses autophagy via Atg1 (Lee et al., 2007b; Scott et al., 2007). Inhibiting this pathway either by decreased inputs from extra-cellular ligands or by over-expression of negative regulators leads to decreased signaling and increased autophagy (Rusten et al., 2004; Scott et al., 2004).
To modulate this pathway, and test whether the PI3K/Akt pathway controls VSV-induced autophagy, we took advantage of a well-studied negative regulator, PTEN. PTEN is a phosphatase that blocks PI3K signaling by dephosphorylating PI(3,4,5)P (Wullschleger et al., 2006). Over-expression of this factor leads to an increase in autophagy in the larval fat body (Arico et al., 2001; Scott et al., 2004). We over-expressed this gene in transgenic animals using a heat shock-Gal4 driver. Under these conditions there was no significant effect on fly viability (data not shown). We tested whether PTEN also controls autophagy in the adult fat body using Lysotracker, a standard assay in Drosophila (Bilen and Bonini, 2007; Chen et al., 2008; Juhasz and Neufeld, 2006). Using this assay, we found that over-expression of PTEN induces autophagy in fat bodies of well fed adults (Supplementary Figure 8).
We then challenged flies over-expressing PTEN, and thus undergoing constitutive autophagy, with VSV and monitored viral replication. We observed a decrease in viral protein production compared to controls by immunoblot analysis at all time points tested (Figure 7A). Therefore, we conclude that inhibition of the PI3K/Akt pathway leads to a significant decrease in VSV replication. To further dissect the role of PI3K signaling in the control of VSV infection, we tested whether inhibition of the pathway using a different negative regulator also blocks infection. We took advantage of a deleted form of p60, the adaptor that couples the insulin receptor to the catalytic subunit of PI3K, Dp110 (Britton et al., 2002). This deletion mutant, Δp60, is a variant that lacks the Dp110-binding domain, that when over-expressed inhibits the PI3K/Akt signaling pathway similarly to the over-expressed PTEN, thereby leading to an increase in autophagy (Scott et al., 2004). Using the same heat shock/GAL4 over-expression system to drive expression of Δp60, we also observe a decrease in VSV replication as measured by immunoblot (Figure 7A). Together, these data indicate that inhibition of the PI3K/Akt pathway, which in turn activates autophagy, also inhibits VSV replication, suggesting that this pathway plays an important role in the anti-viral response.
Figure 7. The PI3K/Akt pathway controls autophagy and viral replication in adult flies.

A. Flies carrying a heat-shock inducible Gal4 were crossed to UAS-PTEN, UAS-Δp60 or control at room temperature. Progeny were collected and heat shocked at 37°C for one hour on the day of infection and every two days following challenge. Viral replication was monitored over time for viral replication by immunoblot against virally produced GFP and normalized to cellular proteins. A significant decrease in viral replication was observed post infection. B. Flies carrying a heat-shock inducible PTEN were crossed to flies heterozygous for a mutant Atg1 allele at room temperature. Progeny were collected and heat shocked at 37°C for one hour on the day of infection and every two days following challenge. Viral replication was monitored over time for viral replication by immunoblot against virally produced GFP and normalized to a cellular control protein. A significant increase in viral replication was observed in the heterozygous animals post infection. C. Flies carrying a heat-shock inducible Gal4 were crossed to UAS-Akt IR, at room temperature. Progeny were collected and heat shocked at 37°C for one hour on the day of infection and every two days following challenge. Viral replication was monitored over time for viral replication by immunoblot against virally produced GFP and normalized to cellular proteins. A significant decrease in viral replication was observed post infection. D. Flies were injected with insulin in the presence or absence of VSV. Phospho-Akt and Total-Akt were monitored by immunoblot. A significant decrease in Phospho-Akt was observed post VSV infection. These data show representative experiments; similar findings were made in at least three repetitions.
To verify that the increase in VSV replication observed upon inhibition of the PI3K/Akt pathway is indeed due to activation of autophagy and not another pathway regulated by this signaling cascade, we performed epistasis analysis. Atg1, a conserved serine/threonine-specific protein kinase, is a crucial component of the autophagy machinery and is a key regulator of autophagy. All known pathways of regulation of autophagy appear to converge upon Atg1. Loss of Atg1 blocks autophagy, whereas overexpression leads to an increase in autophagy in flies (Scott et al., 2007; Scott et al., 2004). Therefore, it is a limiting component of this pathway. First, we tested whether loss of one copy of Atg1 had an effect on infection and found that this had no affect on VSV levels (Supplementary Figure 9). Next, we performed the epistasis analysis in a sensitized background, by crossing flies carrying a heat shock promoter driving expression of PTEN (Heat shock-PTEN) to flies that were heterozygous for Atg1. This allowed us to compare flies that over-expressed PTEN, and had two copies of Atg1, to flies that were heterozygous for Atg1. We expected that the decreased viral replication mediated by over-expression of PTEN would be attenuated in animals that had reduced Atg1 levels. We challenged these siblings and found that removing one copy of Atg1 relieves the PTEN-mediated repression of VSV replication (Figure 7B). This experiment demonstrates that the effect of PTEN on VSV is mediated by an increase in autophagy.
The previous experiments relied on over-expression phenotypes therefore; we tested whether we observed a similar dependence on nutrient signaling using a loss-of-function strategy. Loss of Akt signaling is predicted to activate autophagy and inhibit VSV replication. To this end, we tested whether loss of Akt by RNAi, using a previously validated transgene, would result in decreased viral replication (Wang et al., 2008). Using the same heat shock/GAL4 over-expression protocol used above, we drove expression of an Akt dsRNA (UAS-Akt IR) and found that depletion of Akt by RNAi led to a decrease in VSV replication as measured by immunoblot (Figure 7C).
Together, these data suggest that infection by VSV is sensed by flies, leading to a decrease in PI3K/Akt signaling, which increases autophagy and inhibits viral replication. However, these data do not show whether infection itself modulates this signaling pathway. A prediction is that VSV infection is sensed by the fly, leading to the repression of PI3K/Akt signaling to activate the anti-viral autophagy program. We set out to test this hypothesis by monitoring signaling during infection. One method to monitor signaling through this pathway is to measure the levels of phosphorylation of Akt by immunoblot. To monitor Akt activity upon infection in vivo in adult flies, we tested whether we could detect phospho-Akt in uninfected adult flies, but were unable to detect basal levels (data not shown). To increase the levels of active Akt in flies, we induced the pathway by injecting insulin into the flies. Under these conditions, we observed activation of the signaling pathway as determined by phospho-Akt (Figure 7D). Next we challenged the flies with VSV in the presence of insulin and compared the levels of phospho-Akt to flies that were insulin-treated but uninfected. We observed a significant decrease in phospho-Akt levels in VSV infected animals (Figure 7D). In contrast, levels of total Akt protein remained constant. This demonstrates that VSV infection leads to an inhibition of Akt signaling in animals.
DISCUSSION
While autophagy has been implicated in diverse processes, both normal and pathogenic, a role in controlling viral replication has been difficult to demonstrate directly in vivo, and is complicated by differing requirements by distinct pathogens. Using a new model system we found that autophagy plays an important anti-viral role against VSV in tissue culture cells as well as in adult flies. By modulating the pathway in vivo, either inhibiting it using RNAi against components of the autophagic machinery, or activating it by repressing the function of upstream members of the PI3K/Akt signaling pathway, we demonstrate a reciprocal effect on viral replication: activation of autophagy inhibits, and inhibition of autophagy exacerbates infection. Moreover, we showed that infection is sensed by flies and leads to inhibition of the Akt signaling pathway. This in turn leads to activation of the anti-viral autophagic program, thereby protecting the animal from an otherwise lethal virus infection.
Furthermore, our data suggest that autophagy is directly activated by VSV infection, most likely via the surface glycoprotein VSV G, and thus activation does not require viral replication. While VSV RNA can be recognized by the cytoplasmic sensors in mammals, we show that VSV RNA is not the sensor for autophagy in flies. Moreover, this class of intracellular sensors has not yet been identified in Drosophila. Interestingly, recent work in mammalian systems indicates that VSV G can also be recognized by TLR4 to activate this class of PRRs (Georgel et al., 2007). Furthermore, it has been observed that TLR4 signaling can activate autophagy downstream of the bacterial PAMP LPS, suggesting a link between pattern recognition by TLRs and the cellular antimicrobial response (Xu et al., 2007). Drosophila has 9 TLRs raising the possibility that this VSV G dependent viral recognition pathway uses this class of pattern recognition receptors; however, other receptors may be involved. Previous studies found that in specialized antigen presenting dendritic cells which are not competent for VSV replication, autophagy is required to present VSV antigens to endosomal TLR7, suggesting that different aspects of the autophagic pathway may modulate viral replication under different conditions or in specialized cell types (Lee et al., 2007a).
Autophagy has been shown to be essential for defense against a variety of pathogens in cell culture. Our studies suggest that VSV G is recognized by a pattern recognition receptor whose activation leads to inhibition of the PI3K signaling pathway, increasing autophagy, thereby blocking viral replication. Importantly, this newly identified anti-viral pathway may play a role in the control of additional infectious agents. Moreover, it may be fruitful to explore pharmacological modulation of the pathway as a means to inhibit viral replication as there are a number of small molecule modulators that have been developed to regulate the PI3K/Akt signaling pathway.
EXPERIMENTAL PROCEDURES
Cells, antibodies, and reagents
Drosophila S2 cells were grown and maintained as described in Schneiders media supplemented with 10% FBS (JRH), penicilin/streptomycin and glutamine (Cherry and Perrimon, 2004). BHK21 cells were maintained in DMEM/10% FBS (Sigma), penicilin/streptomycin, and glutamine. Antibodies were obtained from the following sources: anti-GFP (Invitrogen; different lots have different background bands), anti-tubulin (Sigma), anti-VSVG (gift from R. Doms), anti-VSVM (gift from D. Lyles and R. Hardy), anti-phospho-Akt and anti-total Akt (Cell Signaling). Polyclonal rabbit Anti-Atg8 was generated against peptide (FDKRRAEGDKIRRKYPDR) and affinity purified by ProSci. Fluorescently labeled secondary antibodies were obtained from Jackson Immunochemicals, and other secondary antibodies from Amersham. Additional chemicals were obtained from Sigma.
Virus and Viral Components
VSV-eGFP (a kind gift from J. Rose) was grown in BHK cells as described (Ramsburg et al., 2005). VSV was UV inactivated as described (Iwasaki, 2007). Viral RNPs were purified as described (Gupta et al., 1995) and the viral RNA was purified from the RNP using Trizol following the manufacturer’s protocol. To generate VSV G VPs 293Ts were transfected with pVSV G (Invitrogen) using Fugene HD following the manufacturer’s protocol. Supernatant was clarified, ultracentrifuged through a 20% sucrose cushion, and resuspended in PBS.
RNAi and infections
dsRNAs for RNAi were generated and used for RNAi for three days as described (Cherry et al., 2005). Amplicons used are described at http://flyrnai.org. Cells were passaged into serum free media supplemented with dsRNA at 250ng/20,000 cells. One hour later, complete media was added. Three days later cells were infected with the indicated viral innoculum, and assayed at the indicated time point post infection.
Immunofluorescence
Cells were processed for immunofluorescence as previously described. Briefly, cells were fixed in 4% formaldehyde/PBS, washed in PBS/0.1% TritonX-100 (PBST) twice, and blocked in 2%BSA/PBST. Primary antibody was diluted in block and incubated overnight at 4°C. Cells were washed three times in PBST, and incubated in secondary antibody for one hour at room temperature. Cells were counterstained with Hoechst33342 (Sigma) and imaged using an automated microscope (ImageXpress Micro). Three wells per treatment, three sites per well were collected. Quantitation was performed using automated image analysis software (MetaXpress). Experiments were performed at least three times.
Drosophila S2* cells were transfected with pMT-Gal4 and UAS-GFP-LC3 using Effectene (Qiagen) following the manufacturer’s protocol. 2 days later the cells were induced with 500uM CuSO4 and 24 hours later, the cells were either infected or uninfected at an MOI=30 or transfected with Flag-RAN along with viral RNA or RNP. 20 hours later the cells were fixed, counterstained with Hoescht33342 and processed for fluorescence microscopy. >150 cells per treatment were counted for each experiment.
Hemocytes of the indicated genotype were obtained by dissection of 3rd instar larvae in complete Schneiders media. Fifteen larvae were pooled for each of two independent experiments. Cells were either uninfected, or infected with VSV (MOI=30) fixed and stained with Hoechst33342 and imaged on an inverted scope (Leica).
For lysotracker staining of adult flies their abdomens were dissected in complete media and treated with 100nM Lysotracker Red, counterstained with Hoescht33342 for 10 min at room temp, and mounted in complete media. The dissected fat bodies were imaged on an inverted scope (Leica). Three animals per genotype were imaged for each of three independent experiments.
Immunoblotting, Northern blots and Titers
Cell or flies were collected at the time points indicated. The cells or flies were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with a protease inhibitor cocktail (Boehringer). Samples were separated by 10% SDS-PAGE and blotted as previously described (Cherry and Perrimon, 2004). For Northern blot analysis, cells or flies were lysed and total RNA was extracted using Trizol (Invitrogen) according to the manufacturer. Ten micrograms of total RNA was run on a 1% gel, transferred to Hybond N+ nylon and a radiolabelled probe was generated from the indicated gene. Experiments were performed at least three times independently. For titering, cells were collected, freeze-thawed and titered on BHK21 cells. Flies were crushed in DMEM/10% FBS and titered on BHK21 cells.
Electron Microscopy
Cells were pre-treated with the indicated dsRNA for three days where indicated. Next, the cells were infected at an MOI=5 for 20 hours and fixed in 2% PFA/2% glutaraldehyde in 0.1 M Sodium cacodylate buffer, pH 7.4 for 1 hour at RT, postfixed in 2% osmium tetroxide in 0.1M sodium cacodylate and enbloc stained with 2% aqueous uranyl acetate in maleate buffer pH 5.2 for 30 minutes at RT. After dehydration in a graded ethanol series, cells were removed from the dish in propyleneoxide embedded in Epon 812. Ultrathin sections (~70 nm) were mounted on mesh copper grids, stained with 2% uranyl acetate in acetone followed by bismuth subnitrite and examined in a FEI-Tecnai T12 electron microscope and digital images were collected using Gatan CCD camera at a primary magnification of 2100X. The number of intracellular organelles was measured by randomly counting at least 20 cells per treatment per experiment. At least two independent experiments were performed for each comparison. Data was analyzed and plotted using R. Significance was assessed using a Wilcoxon test.
Adult Infections
All flies were obtained from the Bloomington stock center unless stated otherwise, and were maintained on standard medium at room temperature. Flies carrying UAS-Atg12IR or UAS-Atg7IR (gift of T. Neufeld) or UAS-Atg18IR (VDRC) were crossed to Actin-GAL4/+ flies at room temperature. Flies wild type (w1118), UAS-PTEN, UAS-Δp60 (gift of M. Birnbaum) or UAS-Akt IR (VDRC) were crossed to heat-shock GAL4. Heat-shock PTEN flies were crossed to Atg1 heterozygous flies (Atg1(00305)). On the day of injection the progeny were heat shocked at 37°C for one hour and shocked every two days throughout the experiment. 4–7 day old adults of the stated genotypes were inoculated with VSV-gfp as previously described (Cherry and Perrimon, 2004). Groups of at least 20 flies were challenged for mortality studies and a log-rank test was used to determine significance. Insulin (1mg/mL) was injected in the presence or absence of VSV-gfp. Flies were processed at the indicated time point post infection. Flies carrying UAS-eGFP-huLC3 were crossed to Hemolectin-Gal4 to drive expression in hemocytes (Goto et al., 2003; Rusten et al., 2004).
Supplementary Material
Acknowledgments
We thank M. Tudor, S. Ross, N. Bonini, J. DiAngelo as well as members of the Cherry laboratory for critically reading the manuscript; P. Bates for helpful suggestions; J. Rose for VSV-gfp; T. Neufeld for Atg snapback transgenic flies; J. DiAngelo and M. Birnbaum for Akt pathway transgenic flies; The VDRC for Atg18 and Akt snapback transgenic flies; The Bloomington Stock center for other fly stocks; R. Doms for anti-VSV G antibody; D. Lyles and R. Hardy for anti-VSV M antibody; N. Shah for technical assistance and electron microscopy support. This work was supported by a training grant T32AI07324 to S.S., and NIH grants 1R01AI07451 and U54AI057168 to S.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe A, Chen ST, Miyanohara A, Friedmann T. In vitro cell-free conversion of noninfectious Moloney retrovirus particles to an infectious form by the addition of the vesicular stomatitis virus surrogate envelope G protein. J Virol. 1998;72:6356–6361. doi: 10.1128/jvi.72.8.6356-6361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116:2366–2377. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–35246. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3:1950–1964. doi: 10.1371/journal.pgen.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Britton JS, Lockwood WK, Li L, Cohen SM, Edgar BA. Drosophila’s insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell. 2002;2:239–249. doi: 10.1016/s1534-5807(02)00117-x. [DOI] [PubMed] [Google Scholar]
- Chen GC, Lee JY, Tang HW, Debnath J, Thomas SM, Settleman J. Genetic interactions between Drosophila melanogaster Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy. 2008;4:37–45. doi: 10.4161/auto.5141. [DOI] [PubMed] [Google Scholar]
- Cherry S, Doukas T, Armknecht S, Whelan S, Wang H, Sarnow P, Perrimon N. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev. 2005;19:445–452. doi: 10.1101/gad.1267905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry S, Perrimon N. Entry is a rate-limiting step for viral infection in a Drosophila melanogaster model of pathogenesis. Nat Immunol. 2004;5:81–87. doi: 10.1038/ni1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry S, Silverman N. Host-pathogen interactions in drosophila: new tricks from an old friend. Nat Immunol. 2006;7:911–917. doi: 10.1038/ni1388. [DOI] [PubMed] [Google Scholar]
- Dubovi EJ, Youngner JS. Inhibition of pseudorabies virus replication by vesicular stomatitis virus. II Activity of defective interfering particles. J Virol. 1976;18:534–541. doi: 10.1128/jvi.18.2.534-541.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod-Erickson M, Mishra S, Schneider D. Interactions between the cellular and humoral immune responses in Drosophila. Curr Biol. 2000;10:781–784. doi: 10.1016/s0960-9822(00)00569-8. [DOI] [PubMed] [Google Scholar]
- Georgel P, Jiang Z, Kunz S, Janssen E, Mols J, Hoebe K, Bahram S, Oldstone MB, Beutler B. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology. 2007;362:304–313. doi: 10.1016/j.virol.2006.12.032. [DOI] [PubMed] [Google Scholar]
- Goto A, Kadowaki T, Kitagawa Y. Drosophila hemolectin gene is expressed in embryonic and larval hemocytes and its knock down causes bleeding defects. Dev Biol. 2003;264:582–591. doi: 10.1016/j.ydbio.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Das T, Banerjee AK. Casein kinase II is the P protein phosphorylating cellular kinase associated with the ribonucleoprotein complex of purified vesicular stomatitis virus. J Gen Virol . 1995;76(Pt 2):365–372. doi: 10.1099/0022-1317-76-2-365. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Iwasaki A. Role of autophagy in innate viral recognition. Autophagy. 2007;3:354–356. doi: 10.4161/auto.4114. [DOI] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Neufeld TP. Autophagy: a forty-year search for a missing membrane source. PLoS Biol. 2006;4:e36. doi: 10.1371/journal.pbio.0040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–314. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Cregg JM, Dunn WA, Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Ohsumi Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007a;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim S, Lee J, Park J, Lee G, Kim Y, Kim JM, Chung J. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007b;8:360–365. doi: 10.1038/sj.embor.7400917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- Letchworth GJ, Rodriguez LL, Del cbarrera J. Vesicular stomatitis. Vet J. 1999;157:239–260. doi: 10.1053/tvjl.1998.0303. [DOI] [PubMed] [Google Scholar]
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HW, Ding SW. Antiviral silencing in animals. FEBS Lett. 2005;579:5965–5973. doi: 10.1016/j.febslet.2005.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–577. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Palese P, Zheng H, Engelhardt OG, Pleschka S, Garcia-Sastre A. Negative-strand RNA viruses: genetic engineering and applications. Proc Natl Acad Sci U S A. 1996;93:11354–11358. doi: 10.1073/pnas.93.21.11354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsburg E, Publicover J, Buonocore L, Poholek A, Robek M, Palin A, Rose JK. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J Virol. 2005;79:15043–15053. doi: 10.1128/JVI.79.24.15043-15053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5:455–468. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- Rolls MM, Webster P, Balba NH, Rose JK. Novel infectious particles generated by expression of the vesicular stomatitis virus glycoprotein from a self-replicating RNA. Cell. 1994;79:497–506. doi: 10.1016/0092-8674(94)90258-5. [DOI] [PubMed] [Google Scholar]
- Rusten TE, Lindmo K, Juhasz G, Sass M, Seglen PO, Brech A, Stenmark H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7:179–192. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Seay M, Dinesh-Kumar SP, Levine B. Digesting oneself and digesting microbes: autophagy as a host response to viral infection. In: Palese P, editor. Modulation of Host Gene Exprssion and Innate Immunity by Viruses. Dordrecht: Springer; 2005. pp. 245–279. [Google Scholar]
- Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- Stephan JS, Herman PK. The regulation of autophagy in eukaryotic cells: do all roads pass through atg1? Autophagy. 2006;2:146–148. doi: 10.4161/auto.2.2.2485. [DOI] [PubMed] [Google Scholar]
- Talloczy Z, Virgin HWt, Levine B. PKR-Dependent Autophagic Degradation of Herpes Simplex Virus Type 1. Autophagy. 2006;2:24–29. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- Wang B, Goode J, Best J, Meltzer J, Schilman PE, Chen J, Garza D, Thomas JB, Montminy M. The insulin-regulated CREB coactivator TORC promotes stress resistance in Drosophila. Cell Metab. 2008;7:434–444. doi: 10.1016/j.cmet.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, Takada H, Goldman WE, Fukase K, Silverman N, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.