

Abstract
Background:
Mutations of voltage-gated sodium channel αII gene, SCN2A, have been described in a wide spectrum of epilepsies. While inherited SCN2A mutations have been identified in multiple mild epilepsy cases, a de novo SCN2A-R102X mutation, which we previously reported in a patient with sporadic intractable childhood localization-related epilepsy, remains unique. To validate the involvement of de novo SCN2A mutations in the etiology of intractable epilepsies, we sought to identify additional instances.
Methods:
We performed mutational analyses on SCN2A in 116 patients with severe myoclonic epilepsy in infancy, infantile spasms, and other types of intractable childhood partial and generalized epilepsies and did whole-cell patch-clamp recordings on Nav1.2 channels containing identified mutations.
Results:
We discovered 2 additional de novo SCN2A mutations. One mutation, SCN2A-E1211K, was identified in a patient with sporadic infantile spasms. SCN2A-E1211K produced channels with altered electrophysiologic properties compatible with both augmented (a ∼18-mV hyperpolarizing shift in the voltage dependence of activation) and reduced (a ∼22-mV hyperpolarizing shift in the voltage dependence of steady-state inactivation and a slowed recovery from inactivation) channel activities. The other de novo mutation, SCN2A-I1473M, was identified in a patient with sporadic neonatal epileptic encephalopathy. SCN2A-I1473M caused a ∼14-mV hyperpolarizing shift in the voltage dependence of activation.
Conclusions:
The identified de novo mutations SCN2A-E1211K, -I1473M, and -R102X indicate that SCN2A is an etiologic candidate underlying a variety of intractable childhood epilepsies. The phenotypic variations among patients might be due to the different electrophysiologic properties of mutant channels.
GLOSSARY
- BFNIS
= benign familial neonatal-infantile seizures;
- EMA
= epilepsy with myoclonic absence;
- FLE
= frontal lobe epilepsy;
- GEFS+
= generalized epilepsy with febrile seizures plus;
- IS
= infantile spasms;
- OLE
= occipital lobe epilepsy;
- PE
= partial epilepsy;
- SMEB
= borderline severe myoclonic epilepsy in infancy;
- SMEI
= severe myoclonic epilepsy in infancy;
- VGSC
= voltage-gated sodium channel;
- WT
= wild-type.
Voltage-gated sodium channels (VGSCs) are essential for the action potential generation and propagation in brain, muscle, and heart. VGSCs consist of 1 α pore-forming main subunit and 1 or 2 β accessory subunits that modulate the voltage dependence and cellular localization of the α subunit. Four α subunits (αI–III and VI) and 4 β subunits (βI–IV) are highly expressed in human brain. Mutations in the brain-type VGSC αI, αII, and βI genes have been described in multiple epileptic disorders.1,2
Mutations in VGSC αI gene, SCN1A, encoding Nav1.1, have been reported in generalized epilepsy with febrile seizures plus (GEFS+: OMIM no. 604233), severe myoclonic epilepsy in infancy (SMEI or Dravet syndrome; OMIM no. 607208), and borderline SMEI (SMEB), including intractable childhood epilepsy with generalized tonic-clonic seizures.3–8 GEFS+ is a dominantly inherited epilepsy characterized by febrile seizures in early childhood that progress to afebrile seizures in late childhood and is responsive to antiepileptic drugs.9 By contrast, SMEI is a severe and intractable infantile epilepsy. Typically, the first seizure in SMEI is most often a unilateral or generalized tonic-clonic or clonic seizure often associated with fever, which is progressively followed by additional generalized and partial seizures, ataxia, and mental decline.10 Most SMEI cases occurred sporadically and are associated with de novo SCN1A mutations. SCN1A mutations have also been documented in a broad spectrum of early childhood intractable epilepsies including infantile spasms, myoclonic-astatic epilepsy, Lennox-Gastaut syndrome, and other forms of early childhood focal and generalized epilepsies. While SCN1A is the major and thus far the best characterized responsible gene for cryptogenic intractable childhood epilepsies, other candidate genes underlying SCN1A-negative cases remain largely unknown.11,12
We previously discovered a mutation of VGSC αII gene SCN2A, encoding Nav1.2, in a family with atypical GEFS+.13 Other groups subsequently reported several other SCN2A mutations in families with benign familial neonatal-infantile seizures (BFNIS; OMIM no. 607745), which is as mild as GEFS+.14–17 In addition to these inherited SCN2A mutations in mild epilepsies, we previously described a de novo nonsense mutation, SCN2A-R102X, in a patient with sporadic intractable childhood localization-related epilepsy associated with severe mental decline.18 To date, SCN2A-R102X mutation still remains as a single case with sporadic intractable epilepsy. We hypothesized that other de novo SCN2A mutations occur, with altered electrophysiologic properties, and their existence would validate their pathophysiologic role in intractable epilepsies.
METHODS
Patients.
Ethics Committees in Akita University School of Medicine, the Shizuoka Institute of Epilepsy and Neurological Disorders, Kanagawa Children’s Medical Center, and RIKEN Institute approved this study. Each adult participant or, where necessary, responsible guardians of adult subjects, as well as the parents or legal guardians of subjects who were minors at the time the study began, signed an informed consent form as approved by the Ethics Committees.
Proband 1.
The proband (figure 1A; II-1) is a 22-year-old man. There are no close relatives with a history of epilepsy or febrile convulsion or other neuropsychiatric disorders. He was born by cesarean section at 41st week of gestation with mild asphyxia due to meconium aspiration. Birth weight was 3,000 g. He showed marked developmental delay and severe intellectual disability: he stood alone at age 7 years and spoke only 1 word. He started to have daily seizures at age 11 months with series of spasms (face frowning, body stiffening, then raising both upper limbs for 1–2 seconds), and was diagnosed with infantile spasms. At age 11 months and age 2 years, he received adrenocorticotropic hormone with cessation of seizures, although transiently. At age 2–3 years, infantile spasms evolved to tonic seizures, with extension of both upper arms occasionally accompanied by short clonic movements. Seizures were refractory to various conventional medications including valproate, phenobarbital, phenytoin, and clobazam. The seizure duration was for 10–20 seconds and the frequency of seizure attacks was 1 to 10 times per day, both during sleep and awake, especially frequently when falling asleep and during afternoon nap. After age 10 years, seizures were often triggered by fever above 37.5°C. No myoclonic seizures or absence seizures were observed. At age 17 years, he was brought to a hospital because of status epilepticus, which caused respiratory arrest necessitating tracheotomy. Thereafter, he became quadriplegic and speechless. At present, he has daily tonic seizures rarely followed by clonic movement and occasional status epilepticus. The recent EEG recording showed the background activity lacking alpha waves but containing abundant slow waves. There was no photo-paroxysmal response. Right hemisphere dominant diffuse sharp waves or polyspikes were seen. Ictal EEG showed diffuse recruiting fast spike activity preceded by diffuse flattening. MRI showed mild cerebral atrophy with wider lateral ventricle of the left side. SPECT showed no remarkable findings.

Figure 1 Novel SCN2A nucleotide changes leading to amino acid substitutions identified in patients with intractable childhood epilepsies
(A) Pedigree for proband 1 (II-1) with a history of infantile spasms that evolved to symptomatic generalized epilepsy who showed the nucleotide change c.3631C>T (E1211K). The putative haplotypes determined by analyzing microsatellite markers, namely, D2S151, D2S2330, and D2S335, flanking the SCN2A locus are shown. Markers are given in order, from the p telomere to the q telomere. + = Wild-type allele; m = mutated allele; filled square = infantile spasms. (B) Pedigree for proband 2 (II-1) with neonatal epileptic encephalopathy who showed the nucleotide change c.4419A>G (I1473M). The putative haplotypes determined by analyzing microsatellite markers flanking the SCN2A locus are shown. Genetic recombination occurred in the proband’s or her brother’s maternal chromosome within the region between D2S151 and D2S2330. + = Wild-type allele, m = mutated allele; hatched circle = neonatal epileptic encephalopathy; slash = deceased. (C) Pedigree for proband 3 (II-2) with SMEB who showed the nucleotide change c.1724C>T (A575V). The nucleotide change was also detected in her asymptomatic father. Genomic DNA of her sister was not available for the analysis. + = Wild-type allele; m = mutated allele; hatched circle = SMEB; symbol with a dot = asymptomatic mutation carrier.
Proband 2.
The proband (figure 1B; II-1) was a girl with unusual neonatal epileptic encephalopathy and the only member of her family with epilepsy. Her clinical details were described previously.19 She had experienced tonic or tonic-clonic seizures from age 1 month. Other characteristic clinical features were loss of reactivity, a highly suppressed EEG with ictal burst activities, hyponatremia (120–130 mEq/L), and megalencephaly. She was partially rescued using lidocaine with the least effective serum concentration being 0.5 mg/L (2.1 × 10−6M). At age 7 years and 8 months, she died from unknown causes at her home.
Mutational analysis.
Mutational analyses of SCN2A in patients were performed on all of the 26 coding exons and exon-intron boundary regions as described previously.13
Haplotype analysis.
The microsatellite markers flanking the SCN2A locus, D2S151, D2S2330, and D2S335, were genotyped using ABI PRISM Linkage Mapping Set v2.5 (PE Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions.
Construction of wild-type and mutant Nav1.2 expression vectors.
The wild-type human Nav1.2 expression vector, phSCN2A_WT, has been described previously.18 The mutant vectors, phSCN2A_E1211K, phSCN2A_I1473M, and phSCN2A_A575V, were generated from phSCN2A_WT using QuikChange Mutagenesis kit (Stratagene, LA Jolla, CA) according to the manufacturer’s instructions. All constructs were verified by sequencing and confirmed not to contain any unwanted substitutions.
Cell culture and transfection.
Human embryonic kidney HEK293 cells were plated on polylysine-coated 6-well dishes and then transfected with phSCN2A_WT, phSCN2A_E1211K, phSCN2A_I1473M, or phSCN2A_A575V together with phSCN1B-IRES2-EGFP and phSCN2B-IRES2-EGFP using Lipofectamine LTX (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Patch-clamp analysis.
Patch-clamp analysis was performed as described previously.18 Using Eclipse FN1 upright microscope (Nikon, Tokyo Japan), cells were selected by their fluorescence. Currents were recorded using Axopatch 200B amplifier (Axon Instruments, Burlingame, CA). All experiments were carried out at room temperature (22°C). Recordings from cells showing peak currents less than 200 pA were excluded from the analysis in order to eliminate any potential contribution from the small (∼50 pA) Na+ currents present in a fraction of untransfected HEK293 cells. The electrophysiologists performing all experimental recordings were blinded to the HEK293 cell genotypes used. Data are given as mean ± SEM. Differences were considered significant using Student t test if p < 0.05.
RESULTS
Novel SCN2A missense mutations in patients with intractable childhood epilepsies.
We performed mutational analyses on SCN2A in 116 patients with intractable childhood epilepsies. These patients were negative for SCN1A mutations. The cohort consisted of 19, 25, 3, 3, 2, 2, 47, and 15 patients presenting SMEI, SMEB, infantile spasms, occipital lobe epilepsy, frontal lobe epilepsy, epilepsy with myoclonic absence, and unclassified partial epilepsies and unclassified generalized epilepsies. In the cohort, we found a total of 6 different nucleotide changes leading to amino acid substitutions in SCN2A (table). Among these nucleotide changes, c.3631G>A (E1211K), c.4419A>G (I1473M), c.1724C>T (A575V), and c.982T>G (F328V) were novel. E1211K, I1473M, and A575V were observed only in affected individuals but not in our pool of healthy controls (312 or 311 individuals), and therefore were subjected to further whole-cell patch-clamp recordings as described below. F328V was found in patients and healthy controls and therefore conjectured to be nonpathogenic. The other 2 nucleotide changes, c.56G>A (R19K) and c.1571G>A (R524Q), were previously reported to produce unremarkable changes of the electrophysiologic properties of rat Nav1.2 and therefore assigned as nonpathogenic variants.13
Table Nucleotide changes leading to amino acid substitutions in SCN2A
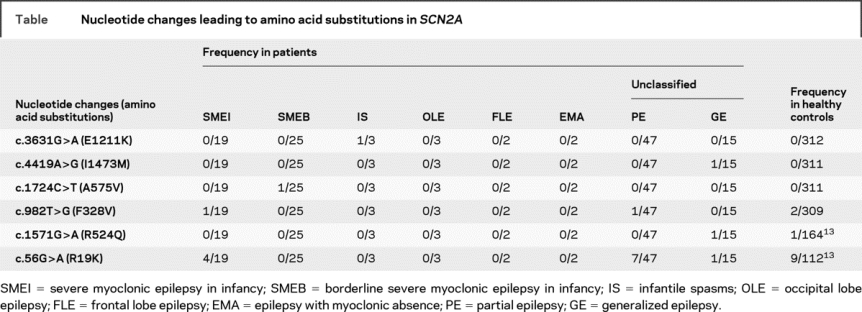
E1211K and I1473M are de novo mutations whereas A575V is inherited.
E1211K was found in the proband 1 with infantile spasms that evolved to severe symptomatic generalized epilepsy of a tonic seizure phenotype (figure 1A and figure e-1A [on the Neurology® Web site at www.neurology.org]; see Methods for the patient’s clinical details). E1211K was not detected in his asymptomatic parents and therefore was defined as a de novo mutation in the proband. The putative haplotypes determined by analyzing the microsatellite markers flanking the SCN2A locus support their genetic kinship.
I1473M was found in the proband 2 with sporadic neonatal epileptic encephalopathy, which was categorized as unclassified generalized epilepsy in the table (figure 1B and figure e-1B). The clinical features were similar but distinct from those of early infantile epileptic encephalopathy with suppression burst (also known as Ohtahara syndrome).19 I1473M was a de novo mutation in the proband because it was not detected in her asymptomatic parents. The putative haplotypes determined by analyzing the microsatellite markers flanking the SCN2A locus support their genetic kinship.
A575V was found in the proband 3 with SMEB (figure 1C and figure e-1C; see e-Methods for the patient’s clinical details). A575V was also found in her asymptomatic father.
E1211 and I1473 are evolutionarily conserved among vertebrate and invertebrate VGSC α subunits.
The glutamate residue of SCN2A-E1211K putatively located on transmembrane segment 1 (S1) of domain III (DIII) was highly conserved among vertebrate and invertebrate VGSC α subunits and human voltage-gated calcium channel α subunits (figure 2). Similarly, the isoleucine residue of SCN2A-I1473M putatively located on S6 of DIII was perfectly conserved through vertebrate and invertebrate VGSC α subunits. These conserved amino acids implicated a pathogenic potential for E1211K and I1473M.

Figure 2 SCN2A nucleotide changes leading to amino acid substitutions detected in intractable childhood epilepsies
An asterisk indicates a nonsense mutation, SCN2A-R102X, described before.18 SCN2A-A575V is assigned to the intracellular linker between domain I (DI) and DII. The alanine residue A575 (highlighted in black) is conserved among human, mouse, and rat Nav1.2 but not in other types of mammalian VGSC α subunits. SCN2A-E1211K is localized to transmembrane segment 1 (S1) of DIII. The glutamate residue E1211 (highlighted in black) is significantly conserved through vertebrate and invertebrate VGSC α subunits and human calcium channel α subunits. I1473M is localized to S6 of DIII. The isoleucine residue I1473 (highlighted in black) is perfectly conserved through vertebrate and invertebrate sodium channel α subunits. Filled square = de novo nonsense mutation; filled circle = de novo missense mutation; open circle = possible nonpathogenic variant. Sources of amino acid sequences are as follows (notations refer to accession numbers): Human Nav1.2, NP_001035232; mouse Nav1.2, NP_001092768; rat Nav1.2, NP_036779; human Nav1.1, NP_008851; human Nav1.3, NP_008853; human Nav1.4, NP_000325; human Nav1.5, NP_932173; human Nav1.6, NP_055006; human Nav1.7, NP_002968; human Nav1.8, NP_006505; human Nav1.9, NP_054858; cockroach sodium channels, AAC47483 and AAK01090; human Cav1.1, NP_000060; human Cav1.2, NP_000719; human Cav1.3, NP_000711; human Cav1.4, NP_005174; human Cav2.1, NP_000059; human Cav2.2, NP_000709; human Cav2.3, NP_000712.
The alanine residue of SCN2A-A575V putatively located at the intracellular linker connecting DI and DII was conserved in human, mice, and rat Nav1.2, but not in other human VGSC α subunits (figure 2).
E1211K and I1473M mutant channels exhibit significantly altered electrophysiologic properties.
To investigate the functional consequences of SCN2A-E1211K, I1473M, and A575V, we examined the electrophysiologic properties of the human wild-type (WT) and mutant (E1211K, I1473M, and A575V) Nav1.2 expressed heterologously in HEK293 cells in the presence of human VGSC βI and βII subunits using whole-cell patch-clamp recordings. Western blot analysis showed no differences in protein expression levels among WT, E1211K, I1473M, and A575V (figure e-2 and e-Methods).
Half-activation potentials in E1211K and I1473M were significantly shifted to the hyperpolarized direction relative to WT (p = 0.001 and 0.004); in contrast, A575V displayed values similar to WT (figure 3, A and B). Average half activation potentials calculated from the pooled data were −17.1 ± 3.8 mV (n = 12) for WT, −35.2 ± 3.5 mV (n = 12) for E1211K, −31.3 ± 3.0 mV (n = 15) for I1473M, and −19.9 ± 2.9 mV (n = 8) for A575V.

Figure 3 Voltage-gated Na+ currents recorded from HEK293 cells expressing human wild-type and mutant (E1211K, I1473M, and A575V) Nav1.2 channels
(A) Peak sodium conductance-voltage relationships for wild-type (WT; closed circle), E1211K (closed triangle), I1473M (open triangle), and A575V (open circle). Na+ currents were evoked by 10 msec depolarizations to various test potentials (−80 mV to 20 mV) from a holding potential of −120 mV. Sodium conductance (gNa) was calculated according to the equation gNa = INa/(Vg − Vr), where INa is the peak amplitude of the Na+ current, Vg is the test potential, and Vr is the reversal potential for Na+. To compare voltage dependence of activation, data were fitted by the least-squares fit of the data to a Boltzmann function, according to the equation gNa/gNamax = 1 − 1/{1 − exp[(Vh − V1/2)/k]}, where gNamax is the maximum conductance, Vh is the potential of individual step pulses, V1/2 is the potential at which gNa is one-half maximal, and k is the slope factor. The data points represent the average of gNa/gNamax. Note that E1211K and I1473M opened at significantly lower voltages. (B) Half activation potentials of Nav1.2 channels. Half activation potentials were calculated for individual cells and averaged. (C) Steady-state voltage dependence of inactivation for WT (closed circle), E1211K (closed triangle), I1473M (open triangle), and A575V (open circle). Cells were pre-pulsed for 2 seconds at various holding potentials (from −120 mV to 10 mV in 10-mV increments), and then Na+ current was evoked by a step depolarization to 0 mV. The peak amplitudes of the Na+ currents measured at individual test potentials were normalized to the peak amplitude of the Na+ current measured at a holding potential of −120 mV. Data were fitted by the least-squares fit of the data to a Boltzmann function, according to the equation I/Imax = 1/{1 + exp[(Vh − V1/2)/k]}, where Imax is the magnitude of the peak Na+ current observed at a holding potential of −120 mV, Vh is the holding potential, V1/2 is the potential at which the Na+ current is one-half maximal, and k is the slope factor. The data points represent the average of I/Imax. Note that the curve for E1211K is significantly shifted to the hyperpolarized direction. (D) Half inactivation potentials of Nav1.2 channels. Half inactivation potentials were calculated for individual cells and averaged. Values represent means ± SEM, *p < 0.05, **p < 0.01.
Next, we examined the steady-state voltage dependence of inactivation. Half-inactivation potentials in E1211K displayed a significant shift to the hyperpolarized direction compared to WT (p = 0.0001) (figure 3, C and D). Average half inactivation potentials calculated from the pooled data were −64.8 ± 4.7 mV (n = 11) for WT, −86.9 ± 2.2 mV (n = 12) for E1211K, −59.5 ± 1.9 mV (n = 13) for I1473M, and −70.1 ± 2.6 mV (n = 7) for A575V.
We then investigated the time course of recovery from the inactivated state using a double pulse protocol. E1211K showed a pronounced delay of recovery from inactivation compared to WT (p = 0.02), whereas I1473M and A575V exhibited time courses similar to WT (figure 4A). Average of Arec calculated from the pooled data were 0.58 ± 0.12 (n = 11) for WT, 0.30 ± 0.03 (n = 12) for E1211K, 0.78 ± 0.12 (n = 15) for I1473M, and 0.53 ± 0.09 (n = 8) for A575V (figure 4B).

Figure 4 Recovery from inactivated state for human wild-type and mutant (E1211K, I1473M, and A575V) Nav1.2 channels
(A) Recovery from inactivated state for WT (closed circle), E1211K (closed triangle), I1473M (open triangle), and A575V (open circle). Two depolarizing pulses (step to 10 mV, 10 msec in duration) with various interpulse intervals (from 0.5 msec to 50 msec) were successively applied to activate Na+ currents. The peak amplitude of Na+ currents evoked by the second pulse (I2) was normalized to the peak amplitude of Na+ currents evoked by the first pulse (I1) and I2/I1 is expressed as recovery ratios. Data were fitted by the equation I2/I1 = 1 − exp(−Arect), where Arec is the factor to determine the speed of recovery. The data points represent the average of I2/I1. Note that the recovery for E1211K was significantly prolonged. (B) Arec of Nav1.2 channels. Arec was calculated for individual cells and averaged. Values represent means ± SEM, *p < 0.05, **p < 0.01.
Taken altogether, E1211K and I1473M markedly altered the voltage-dependence of Nav1.2, indicating high pathogenic potentials of E1211K and I1473M. In contrast, A575V caused no significant effects on the voltage dependence of Nav1.2, suggesting low pathogenicity of A575V.
DISCUSSION
We identified 2 plausible pathogenic de novo SCN2A mutations, E1211K and I1473M, in 2 sporadic intractable childhood epilepsy cases. Together with the de novo R102X,18 which we previously discovered in a patient with sporadic intractable childhood epilepsy, these mutations indicate that SCN2A is a plausible etiologic candidate gene underlying intractable childhood epilepsies.
One de novo mutation, E1211K, was identified in a patient with sporadic infantile spasms that progressed into severe symptomatic generalized epilepsy. Although the patient was born with mild asphyxia, we surmise that it would be insufficient to cause the sporadic infantile spasms and favor the notion that the de novo E1211K is the most possible primary cause. A respiratory arrest caused by a status epilepticus at age 17 years plausibly accounts for the rapid deterioration of his physical and mental condition.
The electrophysiologic analyses revealed that E1211K significantly altered the functional properties of Nav1.2 channel. E1211K caused a large hyperpolarizing shift of the voltage dependence of activation, affecting a reduction in the threshold of depolarization required for activation. E1211K also caused the left shift of the voltage dependence of steady-state inactivation, suggesting that, at the physiologic resting membrane potential (−70 to −60 mV), more than 85% of Nav1.2 carrying E1211K would be in the inactivated state in comparison to ∼50% wild-type Nav1.2. Furthermore, E1211K delayed the recovery from inactivated state possibly leading to a reduction in channel availability during repetitive firings. Taken altogether, E1211K produced mutant channels with mixed electrophysiologic properties indicating both augmented and reduced channel activities. Overall, the effects of E1211K on the excitability of neurons are currently difficult to predict. The consequences on the ultimate neuronal excitability might depend on other cellular conditions and the localization in specific neuronal types.
The other de novo mutation, I1473M, was identified in a patient with unusual neonatal epileptic encephalopathy.19 Nav1.2 channel carrying I1473M showed a significant shift of the voltage dependence of activation to the hyperpolarized direction, suggesting that this mutation may increase the channel activity and thereby may cause hyperexcitation of neurons leading to epileptic seizures. Epileptic seizures responded to lidocaine, which might possibly reduce VGSC activities by affecting not only the mutant Nav1.2 channel but also wild-type Nav1.2 and other brain-type VGSCs.
We also identified an inherited mutation, A575V, in a patient with SMEB, which was not observed in our 311 healthy control individuals. Given that A575V was found in her asymptomatic father and that the functional effect of A575V on human Nav1.2 was insignificant, A575V is most likely a rare nonpathogenic variant. However, it could be also possible that A575V contributes to the susceptibility to develop epileptic seizures when combined with additional genetic or environmental modifiers, present in the patient but not in the father.
This and our previous studies have provided a total of 3 de novo SCN2A mutations: E1211K, I1473M, and R102X.18 Notably, these mutations associated with distinct epileptic phenotypes and affected channel properties of Nav1.2 differentially. There are 2 possible explanations for the phenotypic varieties. First, phenotypic variability could be due to the individual mutation or its location in the Nav1.2 protein, which could differentially affect protein stability, subcellular trafficking, or channel functions. Alternatively, SCN2A mutation mosaicism might be related to phenotypic variations. When de novo mutations occur during early development, the patients could be somatic mosaics for their mutations, and therefore brain regions in those patients could be affected differentially, leading to phenotypic variability. In support of this idea, SCN1A mutation mosaicism occurs in some mildly affected or asymptomatic patients’ parents in several familial SMEI cases.20–23
Of SCN2A mutations identified in BFNIS patients so far,14–17 4 were studied using patch-clamp recordings and had Nav1.2 channels with altered channel properties predicting a gain of function24,25 or with reduced cell surface expression implicating a loss of function.26 Although there was a discrepancy among the results, the amounts of changes in half activation and half inactivation potentials by the BFNIS mutations were within ±6 mV. By contrast, the amounts of changes in half activation and half inactivation potentials in SCN2A-E1211K were ∼−18-mV and ∼−22-mV, and that of the half activation potential in SCN2A-I1473M was ∼−14-mV. Thus, both E1211K and I1473M altered the channel properties of Nav1.2 to a greater extent than the BFNIS mutations, suggesting a mechanism for more severe epileptic phenotypes. In order to confirm these correlations, the effects of the mutations on the functions of the neurons responsible for seizure development, which are currently unknown, should be examined further.
SCN1A mutant mice with spontaneous epileptic seizures have reduced SCN1A expression.27,28 Further, SCN1A is prominently expressed in the parvalbumin-positive inhibitory interneurons and we proposed that SCN1A mutations might cause functional defects in the parvalbumin-positive interneurons, which then fail to suppress overexcitation of neural circuits and result in epileptic seizures.28 In contrast to the case for SCN1A mutations, the molecular mechanisms underlying seizures caused by SCN2A mutations remain largely unknown. Since SCN2A is highly expressed in both principal neurons and interneurons in rat hippocampus,29,30 SCN2A mutations might alter the global function of the implicated neurons. Although loss or reduced SCN2A expression did not generate spontaneous epileptic seizures in mice,31 our present study and others’ provide solid genetic evidence implicating SCN2A in the etiology of human epilepsies.14–18 Further studies using mouse models with the SCN2A mutations identified in human epilepsies may help to understand the impact of such mutations on the epileptic brain.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. M. Kaneda.
ACKNOWLEDGMENT
The authors thank all the patients and families for their contributions and cooperation. They also thank Drs. Oyama and Nukina (RIKEN Brain Science Institute) for rabbit anti-VGSC βI antibody, Drs. Kaneko (Hirosaki University) and Hirose (Fukuoka University) for providing materials, Ms. Shibazaki for laboratory support, and Research Resource Center of RIKEN Brain Science Institute for DNA sequencing and genotyping analyses.
Supplementary Material
Address correspondence and reprint requests to Dr. Kazuhiro Yamakawa, Laboratory for Neurogenetics, RIKEN Brain Science Institute, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan yamakawa@brain.riken.jp
Supplemental data at www.neurology.org
Supported by grants from the RIKEN Brain Science Institute, the Ministry of Health, Labour and Welfare of Japan, the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the NIH (GM-49711).
Disclosure: The authors report no disclosures.
Received February 25, 2009. Accepted in final form June 18, 2009.
REFERENCES
- 1.Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 2005;115:2010–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat 2005;25:535–542. [DOI] [PubMed] [Google Scholar]
- 3.Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000;24:343–345. [DOI] [PubMed] [Google Scholar]
- 4.Sugawara T, Mazaki-Miyazaki E, Ito M, et al. Nav1.1 mutations cause febrile seizures associated with afebrile partial seizures. Neurology 2001;57:703–705. [DOI] [PubMed] [Google Scholar]
- 5.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001;68:1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugawara T, Mazaki-Miyazaki E, Fukushima K, et al. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 2002;58:1122–1124. [DOI] [PubMed] [Google Scholar]
- 7.Fujiwara T, Sugawara T, Mazaki-Miyazaki E, et al. Mutations of sodium channel α subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 2003;126:531–546. [DOI] [PubMed] [Google Scholar]
- 8.Fukuma G, Oguni H, Shirasaka Y, et al. Mutations of neuronal voltage-gated Na+ channel α1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB). Epilepsia 2004;45:140–148. [DOI] [PubMed] [Google Scholar]
- 9.Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes Brain 1997;120:479–490. [DOI] [PubMed] [Google Scholar]
- 10.Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severe myoclonic epilepsy in infancy (Dravet syndrome). In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, eds. Epileptic Syndromes in Infancy, Childhood and Adolescence, 4th ed. Montrouge, France: John Libbey Eurotext; 2005:89–113. [Google Scholar]
- 11.Wallace RH, Hodgson BL, Grinton BE, et al. Sodium channel α1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology 2003;61:765–769. [DOI] [PubMed] [Google Scholar]
- 12.Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007;130:843–852. [DOI] [PubMed] [Google Scholar]
- 13.Sugawara T, Tsurubuchi Y, Agarwala KL, et al. A missense mutation of the Na+ channel αII subunit gene Nav1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci USA 2001;98:6384–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berkovic SF, Heron SE, Giordano L, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol 2004;55:550–557. [DOI] [PubMed] [Google Scholar]
- 15.Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002;360:851–852. [DOI] [PubMed] [Google Scholar]
- 16.Striano P, Bordo L, Lispi ML, et al. A novel SCN2A mutation in family with benign familial infantile seizures. Epilepsia 2006;47:218–220. [DOI] [PubMed] [Google Scholar]
- 17.Herlenius E, Heron SE, Grinton BE, et al. SCN2A mutations and benign familial neonatal-infantile seizures: the phenotypic spectrum. Epilepsia 2007;48:1138–1142. [DOI] [PubMed] [Google Scholar]
- 18.Kamiya K, Kaneda M, Sugawara T, et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci 2004;24:2690–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawaishi Y, Yano T, Enoki M, Takada G. Lidocaine-dependent early infantile status epilepticus with highly suppressed EEG. Epilepsia 2002;43:201–204. [DOI] [PubMed] [Google Scholar]
- 20.Depienne C, Arzimanoglou A, Trouillard O, et al. Parental mosaicism can cause recurrent transmission of SCN1A mutations associated with severe myoclonic epilepsy of infancy. Hum Mutat 2006;27:389. [DOI] [PubMed] [Google Scholar]
- 21.Gennaro E, Santorelli FM, Bertini E, et al. Somatic and germline mosaicisms in severe myoclonic epilepsy of infancy. Biochem Biophys Res Commun 2006;341:489–493. [DOI] [PubMed] [Google Scholar]
- 22.Marini C, Mei D, Helen Cross J, Guerrini R. Mosaic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia 2006;47:1737–1740. [DOI] [PubMed] [Google Scholar]
- 23.Morimoto M, Mazaki E, Nishimura A, et al. SCN1A mutation mosaicism in a family with severe myoclonic epilepsy in infancy. Epilepsia 2006;47:1732–1736. [DOI] [PubMed] [Google Scholar]
- 24.Scalmani P, Rusconi R, Armatura E, et al. Effects in neocortical neurons of mutations of the Nav1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci 2006;26:10100–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu R, Thomas EA, Jenkins M, et al. A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Mol Cell Neurosci 2007;35:292–301. [DOI] [PubMed] [Google Scholar]
- 26.Misra SN, Kahlig KM, George AL, Jr. Impaired NaV1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia 2008;49:1535–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006;9:1142–1149. [DOI] [PubMed] [Google Scholar]
- 28.Ogiwara I, Miyamoto H, Morita N, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 2007;27:5903–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furuyama T, Morita Y, Inagaki S, Takagi H. Distribution of I, II and III subtypes of voltage-sensitive Na+ channel mRNA in the rat brain. Brain Res Mol Brain Res 1993;17:169–173. [DOI] [PubMed] [Google Scholar]
- 30.Black JA, Yokoyama S, Higashida H, Ransom BR, Waxman SG. Sodium channel mRNAs I, II and III in the CNS: cell-specific expression Brain Res Mol Brain Res 1994;22:275–289. [DOI] [PubMed] [Google Scholar]
- 31.Planells-Cases R, Caprini M, Zhang J, et al. Neuronal death and perinatal lethality in voltage-gated sodium channel αII-deficient mice. Biophys J 2000;78:2878–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.