

Abstract
Objective:
To use a case-control study to assess and compare patterns of gray matter loss across groups of subjects with different mutations in the microtubule-associated protein tau (MAPT) gene.
Methods:
We identified all subjects from Mayo Clinic, Rochester, Minnesota, that screened positive for mutations in MAPT and had a head MRI (n = 22). Voxel-based morphometry was used to assess patterns of gray matter atrophy in groups of subjects with the IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M mutations compared with age- and sex-matched controls.
Results:
All MAPT groups showed gray matter loss in the anterior temporal lobes, with varying degrees of involvement of the frontal and parietal lobes. Within the temporal lobe, the subjects with IVS10+16, IVS10+3, N279K, and S305N mutations (mutations that influence the alternative splicing of tau pre–messenger RNA) all showed gray matter loss focused on the medial temporal lobes. In contrast to these groups, the subjects with P301L or V337M mutations (mutations that affect the structure of the tau protein) both showed gray matter loss focused on the lateral temporal lobes, with a relative sparing of the medial temporal lobe.
Conclusion:
There seem to be differences in patterns of temporal lobe atrophy across the MAPT mutations, which may aid in the differentiation of the different mutation carriers. Furthermore, there seems to be a possible association between mutation function and pattern of temporal lobe atrophy.
GLOSSARY
- ADPR
= Alzheimer’s Disease Patient Registry;
- ADRC
= Alzheimer’s Disease Research Center;
- AVLT
= Auditory Verbal Learning Test;
- BNT
= Boston Naming Test;
- bvFTD
= behavioral variant frontotemporal dementia;
- bvFTD + P
= behavioral variant frontotemporal dementia with parkinsonism;
- CDR-SB
= Clinical Dementia Rating Scale sum of boxes;
- DD
= disease duration;
- FDR
= false discovery rate;
- FTLD
= frontotemporal lobar degeneration;
- MAPT
= microtubule-associated protein tau;
- MNI
= Montreal Neurological Institute;
- mRNA
= messenger RNA;
- NA
= not applicable;
- PPA
= primary progressive aphasia;
- STMS
= Short Test of Mental Status;
- VBM
= voxel-based morphometry.
Frontotemporal lobar degeneration (FTLD) is a heterogenous progressive disorder characterized by behavioral and language abnormalities.1,2 Approximately 40% of subjects have a positive family history with an autosomal dominant pattern of inheritance.3,4 A large proportion of these familial cases have been found to have a mutation in the microtubule-associated protein tau (MAPT) gene located on chromosome 17q21.5 Mutations in MAPT are associated with deposits of hyperphosphorylated tau in the form of intraneuronal neurofibrillary tangles, Pick-like bodies, and glial inclusions in the frontal and temporal cortices of the brain. To date, 44 different pathogenic mutations in MAPT have been identified6 (http://www.molgen.ua.ac.be), including missense mutations, silent mutations, in-frame codon deletions, and intronic mutations.7 The most common of these mutations are the C to T substitution corresponding to P301L in exon 105 and IVS10+16C>T (commonly referred to as IVS10+16) in intron 107. Even though the 44 mutations are all present in the same gene, there is considerable clinical and pathologic heterogeneity across the different mutations.8 We have demonstrated using a group-level analysis that subjects with mutations in MAPT have atrophy particularly involving the anteromedial temporal lobes9; however, it is unclear whether variation exists in the patterns of atrophy across the different MAPT mutations. The aim of this study was to assess different MAPT mutations to determine whether the patterns of atrophy vary across mutations. This knowledge would provide valuable information about the disease course in these patients and may help to identify and even predict different mutations.
METHODS
Subjects.
We identified all subjects seen at Mayo Clinic, Rochester, Minnesota, that had screened positive for mutations in MAPT, were symptomatic, and had a volumetric head MRI scan (n = 22). These included 4 subjects with the P301L mutation in exon 10 (c.1907C>T; p.Pro301Leu) from 2 families, 4 subjects with a mutation at position +16 in intron 10 (referred to as IVS10+16) (c.1920+16C>T; IVS10+16C>T) from 1 family, 3 subjects with a mutation at position +3 in intron 10 (referred to as IVS10+3) (c.1920+3G>A; IVS10+3G>A) from 1 family, 3 subjects with the N279K mutation (c.1842T>G; p.Asn279Lys) from 1 family, 3 subjects with the V337M (c.2014G>A; Val337Met) mutation from 1 family, 2 subjects with the S305N mutation (c.1919G>A; p.Ser305Asn) from 1 family,10 1 subject with the G389R mutation (c.2170G>A; p.Gly389Arg), 1 subject with the R406W (c.2221C>T; p.Arg406Trp) mutation, and 1 subject with a mutation at position −10 in intron 9 (referred to as IVS9-10) (c.1827-10G>T; IVS9-10G>T).11 These cases had been prospectively studied in our Alzheimer’s Disease Research Center (ADRC) or Alzheimer’s Disease Patient Registry (ADPR) between 1991 and 2008. The historic records of all cases were reviewed by an expert in neurodegenerative diseases (K.A.J.) for the abstraction of data, including sex, age at onset, illness duration, Short Test of Mental Status (STMS) score,12 Clinical Dementia Rating Scale sum of boxes (CDR-SB),13 Boston Naming Test (BNT),14 and Auditory Verbal Learning Test (AVLT)15 30-minute delayed recall. Clinical diagnoses were made according to clinical criteria for behavioral variant frontotemporal dementia (bvFTD)2 and primary progressive aphasia (PPA).16 Subject demographics are shown in table 1. Five of these subjects underwent autopsy with pathologic findings of widespread tau deposition in neurons and glia.
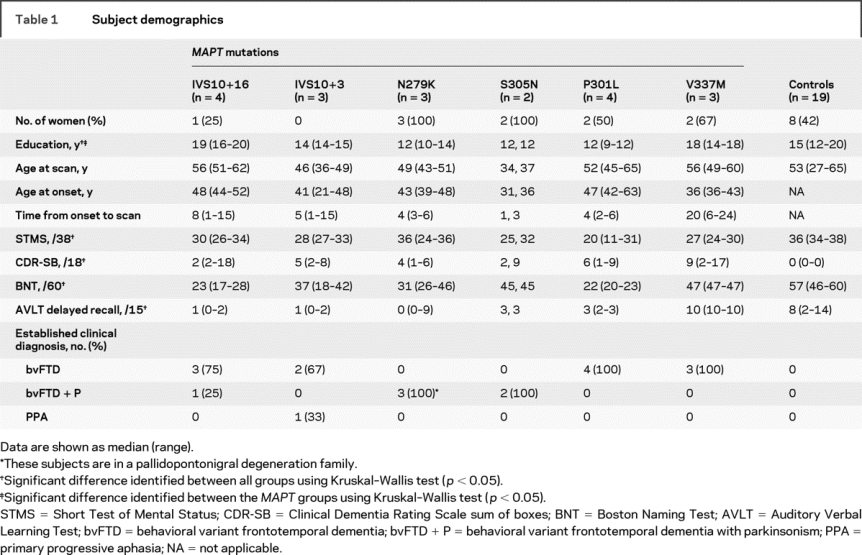
Table 1 Subject demographics
Standard protocol approvals and patient consents.
Informed consent was obtained from all subjects for participation in the studies, which were approved by the Mayo Institutional Review Board.
Genetic analysis.
Analysis of MAPT exons 1, 7, and 9–13 was performed using primers and conditions that were previously described.5 PCR amplicons were purified using the Multiscreen system (Millipore, Billerica, MA) and then sequenced in both directions using Big Dye chemistry following the manufacturer’s protocol (Applied Biosystems, Foster City, CA). Sequence products were purified using the Montage system (Millipore) before being run on an Applied Biosystems 3730 DNA Analyzer. Sequence data were analyzed using either SeqScape (Applied Biosystems) or Sequencher software (Gene Codes, Ann Arbor, MI).
Voxel-based morphometry.
Voxel-based morphometry (VBM) was used to assess patterns of gray matter atrophy in the following groups of MAPT mutation carriers: IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M. Only single subjects were available with the G389R, R406W, and IVS9-10G>T mutations, and therefore these cases were not analyzed using VBM. Each MAPT group was compared with a group of 19 age- and sex-matched healthy control subjects. All control subjects were prospectively recruited into the ADRC or the ADPR and were identified from the ADRC/ADPR database. Control subjects were cognitively normal individuals that had been seen in internal medicine for routine physical examinations and asked to enroll in the ADRC or ADPR. All subjects were then evaluated by a neurologist to verify the normal diagnosis. Controls were identified as individuals who 1) were independently functioning community dwellers, 2) did not have active neurologic or psychiatric conditions, 3) had no cognitive symptoms, 4) had normal neurologic and neurocognitive examination results, and 5) were not taking any psychoactive medications in doses that would affect cognition.
All subjects underwent a standardized protocol head MRI scan that included a T1-weighted 3-dimensional volumetric sequence. In the majority of cases, this consisted of a spoiled gradient-echo sequence performed at 1.5 T, although a total of 6 subjects with FTLD and 4 controls had a magnetization prepared rapid acquisition gradient-echo sequence performed at 3 T. Patterns of cerebral atrophy were assessed using the automated and unbiased technique of VBM.17 An optimized method of VBM was applied using both customized templates and prior probability maps,18 implemented using SPM5 (http://www.fil.ion.ucl.ac.uk/spm). The processing steps were performed as previously described.19 Briefly, all images were normalized to a customized template and segmented by the unified segmentation procedure in SPM520 using the customized tissue probability maps into gray matter, white matter, and CSF, followed by the hidden Markov random field clean-up step. All images were modulated, and smoothed with an 8-mm full-width at half-maximum smoothing kernel.
A full factorial statistical model was used to assess patterns of gray matter atrophy in each MAPT mutation group compared with the control group. Adjustment for the potential confounders of age at scan, sex, and field strength was performed by including them as covariates in the statistical model. Results were assessed after correction for multiple comparisons using the false discovery rate (FDR) at p < 0.001. Given that the results showed a different pattern of temporal lobe volume loss in the IVS10+16, IVS10+3, N279K, and S305N mutations compared with the P301L and V337M mutations, we tested this statistically by grouping the IVS10+16, IVS10+3, N279K, and S305N subjects together and comparing them with a group of subjects that consisted of the P301L and V337M subjects. As in the main analysis, age at scan, sex, and field strength were included as covariates. Time from disease onset to scan was also included as a covariate in this analysis to correct for differences in the time that each subject had the disease. This analysis was assessed at a more lenient statistical threshold of p < 0.05 (FDR corrected).
Statistics.
Statistical analyses were performed using JMP computer software (version 6.0.0; SAS Institute Inc., Cary, NC) with α set at 0.05. Sex ratios were compared across groups with the χ2 test. The Kruskal–Wallis test was used to compare continuous data across groups.
RESULTS
Table 1 shows the subject demographics. Significant differences were observed across all groups, including controls, in education, STMS, CDR-SB, BNT, and AVLT delayed recall, with the subjects with FTLD showing poorer performance on cognitive tests than controls. The only demographic variable that was significantly different across the MAPT groups was education, which may reflect educational biases across different families. The majority of all the MAPT subjects had an established clinical diagnosis of bvFTD, except for 1 subject with the IVS10+3 mutation who had a clinical diagnosis of PPA.
The 3-dimensional renderings in figure 1 show that all of the MAPT groups showed predominant gray matter loss in the temporal lobes, particularly the anterior temporal lobe, with varying degrees of frontal and parietal lobe involvement. However, figure 2 shows that the focus of loss within the temporal lobes varies across the different MAPT mutations. The subjects with IVS10+16, IVS10+3, N279K, and S305N mutations show the most severe gray matter loss in the medial temporal lobes (table 2), including the hippocampus, amygdala, parahippocampal gyrus, and fusiform gyrus. The inferior and lateral temporal lobes and temporal pole are involved although to a lesser degree. Gray matter loss was largely restricted to the temporal lobes in the IVS10+16, IVS10+3, and N279K groups, with the exception of the insula, whereas the S305N group also showed gray matter loss in the frontal and parietal lobes (figure 1). In contrast to these 4 groups, the subjects with a P301L or V337M mutation show a relative sparing of the medial temporal lobes, with greater loss observed in more inferior and lateral temporal regions (figure 2 and table 2). Both the P301L and V337M groups also showed gray matter loss in the frontal lobes and basal ganglia, with some minor involvement of the parietal lobe. Gray matter loss across all 6 groups was bilateral.

Figure 1 Gray matter loss in subjects with IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M mutations
Results are shown on 3-dimensional renderings of the brain and show regions of gray matter loss in each group compared with controls. CDR-SB = median Clinical Dementia Rating Scale sum of boxes; DD = median disease duration (time from onset to scan).
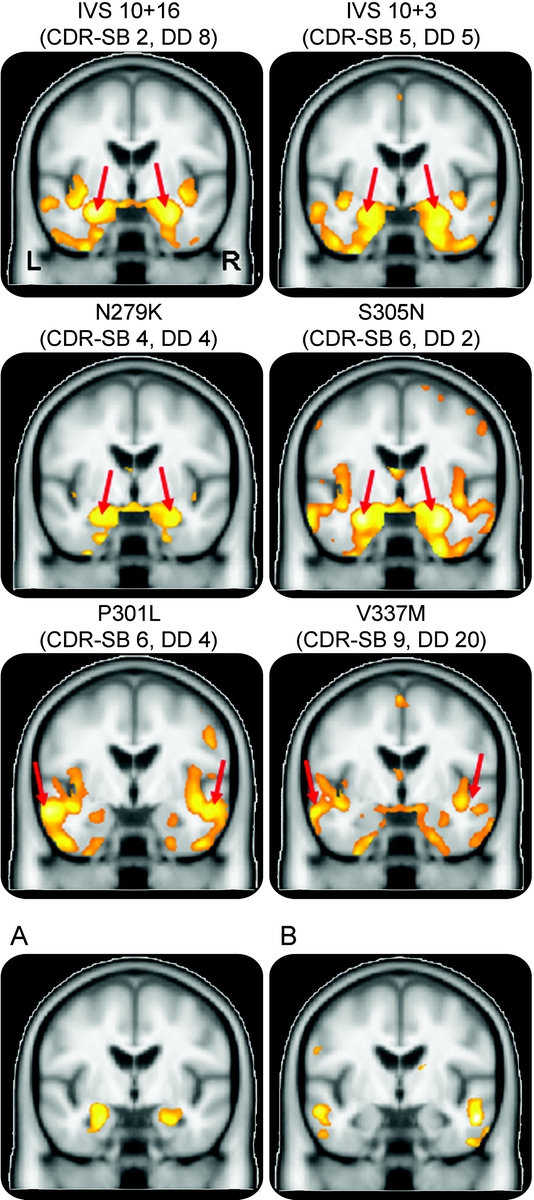
Figure 2 Patterns of temporal lobe gray matter loss in subjects with IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M mutations
Coronal slices through the temporal lobes showing gray matter loss in each group compared with controls. The IVS10+16, IVS10+3, N279K, and S305N groups show the most significant loss in the medial temporal lobes (red arrows), whereas the P301L and V337M groups show the most significant loss in the lateral temporal lobes (red arrows). Bottom coronal slices show results of direct comparisons between the “medial temporal” subjects (IVS10+16, IVS10+3, N279K, and S305N) and the “lateral temporal” subjects (P301L and V337M). (A) Regions that show greater loss in the “medial temporal” group than the “lateral temporal” group; (B) regions that show greater loss in the “lateral temporal” group than the “medial temporal” group. CDR-SB = median Clinical Dementia Rating Scale sum of boxes; DD = median disease duration (time from onset to scan).
Table 2 Location of the voxel showing the most significant gray matter loss on VBM in each of the MAPT mutations compared with controls
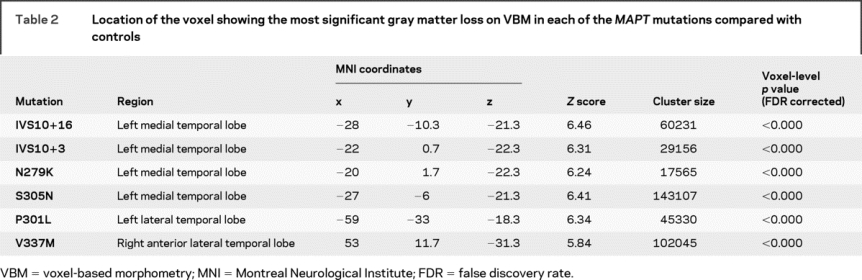
A direct comparison between MAPT mutation groups correcting for differences in time from onset to scan demonstrated that the IVS10+16, IVS10+3, N279K, and S305N subjects have significantly greater gray matter loss in the hippocampus and amygdala than the P301L and V337M subjects (figure 2 and table e-1 on the Neurology® Web site at www.neurology.org). Conversely, the P301L and V337M subjects showed greater gray matter loss in the middle and inferior lateral temporal gyri and in the frontal lobes than the other MAPT mutation subjects (figure 2 and table e-1).
Example MRI scans from patients with each of these MAPT mutations illustrating the differences in temporal lobe involvement are shown in figure 3.
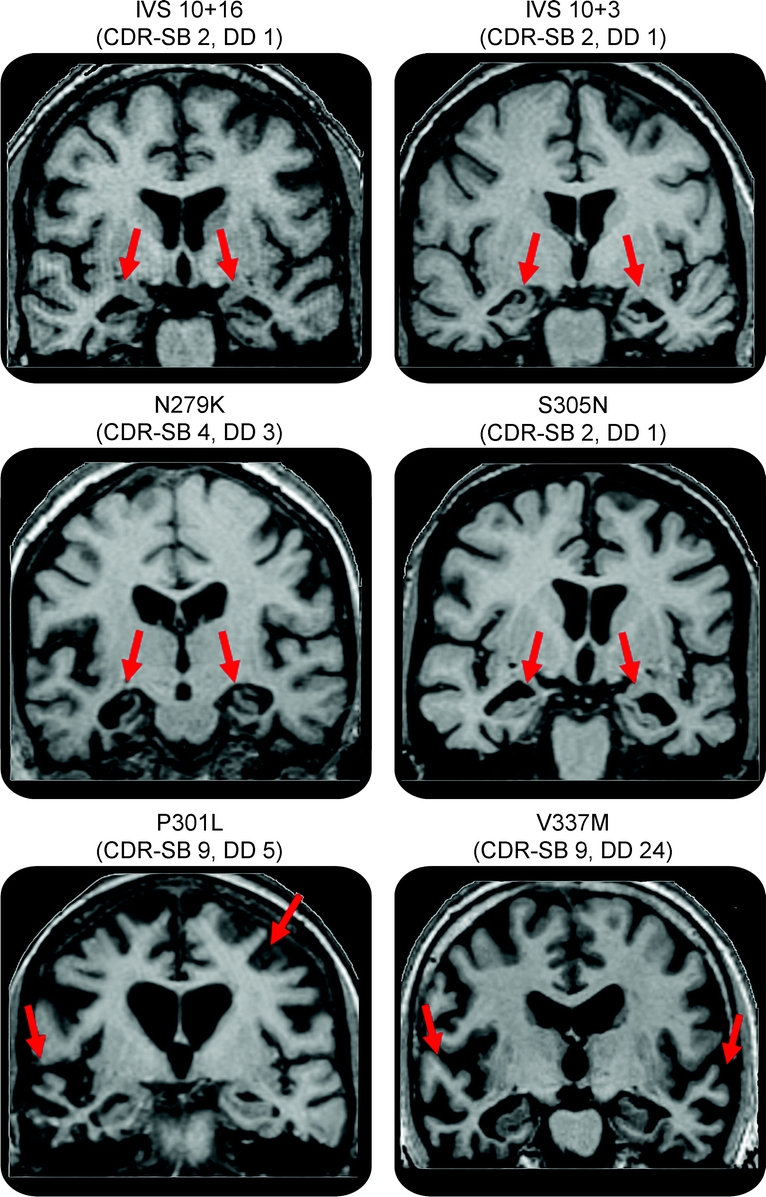
Figure 3 MRI scans from 6 subjects each with a different MAPT mutation
The IVS10+16, IVS10+3, N279K, and S305N subjects show atrophy in the medial temporal lobes with relative sparing of the lateral temporal lobes. The P301L and V337M subjects show severe atrophy of the lateral temporal lobes with relative sparing of the medial temporal lobe. CDR-SB = Clinical Dementia Rating Scale sum of boxes for each subject; DD = disease duration for each subject (time from onset to scan).
DISCUSSION
This study used the automated technique of VBM to assess patterns of gray matter atrophy in groups of subjects with different MAPT mutations. Remarkably consistent patterns of gray matter atrophy were identified across subjects with the IVS10+16, IVS10+3, N279K, and S305N mutations. All 4 of these groups showed gray matter loss predominantly in the anterior temporal lobes, and particularly involving the medial temporal lobe structures. While subjects with P301L and V337M mutations also showed the most severe loss in the anterior temporal lobes, the loss was focused more on the lateral temporal cortex with relative sparing of the medial temporal lobe. These patterns of atrophy may help differentiate subjects with the P301L or V337M mutations from subjects with the IVS10+16, IVS10+3, N279K, or S305N mutations.
Medial temporal lobe atrophy therefore seems to be a striking feature of the IVS10+16, IVS10+3, N279K, and S305N mutations. Previous studies of individual mutations have found similar results, although no studies have compared multiple mutations. Severe medial temporal lobe atrophy has been observed in a couple of VBM studies that assessed subjects with the IVS10+1621 and IVS10+322 mutations, and medial temporal lobe atrophy and 18-F-fluoro-deoxyglucose PET hypometabolism have been observed in individual cases of N279K mutation carriers.23 Predominant temporal lobe atrophy has also been reported in subjects with the S305N mutation.24,25 In contrast to the findings in the IVS10+16, IVS10+3, N279K, and S305N mutations, subjects with the P301L and V337M mutations showed gray matter loss predominantly in the lateral temporal lobes. In fact, subjects with the P301L or V337M mutations, when grouped together, showed significantly more involvement of the lateral temporal lobes than the subjects with other MAPT mutations on direct comparison. A comparison in the opposite direction showed that the subjects with mutations in IVS10+16, IVS10+3, N279K, or S305N showed greater medial temporal lobe atrophy than the P301L or V337M subjects. These results are in keeping with the fact that performance on the test of episodic memory was better on average in the P301L and V337M mutation carriers (average score of 5 vs 2).
The severity and distribution of gray matter loss varied across the different MAPT mutations. This may in part be due to variability in the time from disease onset to the time of scan across the groups. For example, the V337M group showed a widespread pattern of gray matter loss and had an average time from onset to scan of 20 years, which is longer than any of the other MAPT mutation groups, suggesting that they may be further along in their disease course than the other groups. However, the P301L subjects also showed widespread loss including the frontal lobes but had a relatively short time from onset to scan of only 4 years, which suggests either that frontal lobe loss is an early feature of P301L mutations or P301L mutations may have a more rapidly progressive disease, and therefore atrophy has spread further through the brain in the same time. Previous studies have typically reported both temporal and frontal atrophy in P301L patients,8,26–28 and others have suggested that P301L subjects are rapidly progressive.8 We also identified severe involvement of the basal ganglia in both the P301L and V337M subjects. Basal ganglia involvement has been previously observed in single P301L cases.27,29 Similarly, the S305N subjects showed gray matter loss in the frontal and parietal lobes but had a short time from onset to scan, although this group only consisted of 2 subjects. Subjects with P301L, V337M, and S305N also performed more poorly on tests of dementia severity (CDR-SB and STMS) than subjects with the other mutations. Therefore, although disease duration could account for the widespread pattern of atrophy observed in the V337M group and even possibly in the P301L group, it seems that S305N may simply be a more severe phenotype given that the disease duration was short (1–3 years). Further investigation with larger numbers of subjects will be needed to determine whether the involvement of the frontal and parietal lobes are mutation specific or consequences of variability in disease duration. Nevertheless, the P301L and V337M groups showed a fundamentally different anatomic pattern of temporal lobe involvement compared with the other mutations which survived in the analysis that corrected for time from onset to scan and therefore could not be a product of simple differences in disease severity. These patterns of atrophy may therefore be useful in distinguishing subjects with the P301L and V337M mutations from those with the IVS10+16, IVS10+3, N279K, or S305N mutations.
The IVS10+16, IVS10+3, N279K, and S305N mutations are all predicted to cause disease by influencing the alternative splicing of tau pre–messenger RNA (mRNA).7,30 They all increase the splicing of exon 10, thus changing the ratio between 3R and 4R tau isoforms and resulting in an increase in 4R tau. Mutation S305N located at the splice site of exon 10 and intronic mutations, IVS10+16 and IVS10+3, are predicted to directly affect a stem-loop RNA structure spanning the splice donor site of intron 10,5,31 whereas mutation N279K has been proposed to increase the inclusion of exon 10 by strengthening a cis-acting polypurine element. The P301L and V337M mutations are also located in exon 10, but in contrast to the other mutations, they do not affect splicing of exon 10 but instead affect the structure and functional properties of the tau protein.7,30 Tau proteins that contain the P301L and V337M mutations are more favorable substrates for phosphorylation32 and hence lead to the aggregation of tau.7,30,33 This dichotomy in function across the mutations seems to correlate with the dichotomy observed in the patterns of temporal lobe atrophy, suggesting a possible relationship between the effect of the mutation on tau and the resultant patterns of atrophy in MAPT mutation carriers. However, how these different disease mechanisms may influence these anatomic changes is unclear, and further investigation will be needed to investigate whether this relationship generalizes to different mutations.
Although the numbers of subjects in our MAPT groups were small, we have found evidence that the P301L and V337M mutations have different patterns of atrophy to the IVS10+16, IVS10+3, N279K, and S305N mutations. A feature that is consistent across all these mutations was the fact that all showed the greatest degree of loss in the anterior temporal lobes, which explains the poor performance on tests of memory and naming observed in all the MAPT mutation groups. We have previously shown that this feature helps to differentiate subjects with mutations in MAPT from those with mutations in progranulin.9 Indeed, as we have previously published, some of these patients were given an initial diagnosis of mild cognitive impairment or Alzheimer disease9 reflecting the memory impairment. The predominance of IVS10+16, IVS10+3, N279K, and S305N mutation carriers in our previous study explains why we found significant anteromedial temporal lobe atrophy in our group of MAPT mutation carriers.9 Our results also suggest that there may be a possible association between mutation function and atrophy, with mutations that influence splicing of tau pre-mRNA showing medial temporal volume loss and mutations affecting protein structure showing lateral temporal loss. Future studies will need to expand on these results, particularly with a view to understanding the relationship among disease mechanisms, pathology, and brain atrophy in FTLD.
AUTHOR CONTRIBUTIONS
Statistical analysis was performed by Keith A. Josephs.
ACKNOWLEDGMENT
The authors thank Dr. Dennis W. Dickson from the Department of Neuroscience and Neuropathology, Mayo Clinic Jacksonville, FL, and Dr. Joseph E. Parisi from the Department of Laboratory Medicine and Pathology, Mayo Clinic Rochester, MN, for conducting pathologic analyses.
DISCLOSURE
Dr. Whitwell and Mr. Senjem report no disclosures. Dr. Jack serves as a consultant for Elan Corporation; and receives research support from Pfizer, Inc., the NIH [R01-AG11378 (PI), P50-AG16574 (Coinvestigator), and U01 AG024904-01 (Coinvestigator)], and the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Foundation. Dr. Boeve has served as a consultant to GE Healthcare; and receives research support from Myriad Genetics Inc., the Alzheimer’s Association (PI), and the NIH as a Coinvestigator [P50 AG16574, UO1 AG06786, and RO1 AG15866]. Mr. Baker holds US Patent 12/302,691 [Detecting and Treating Dementia (2008)] and US Patent 12/413,869 [Methods and Materials for Detecting and Treating Dementia (2009)]. Dr. Ivnik serves on the editorial boards of The Clinical Neuropsychologist and Aging, Neuropsychology, and Cognition; and receives research support from the NIA as Coinvestigator [AG 06786 and AG 16574]. Dr. Knopman serves as an Associate Editor for Neurology; has served on a data safety monitoring board for Sanofi-Aventis Pharmaceuticals; is an investigator in a clinical trial sponsored by Elan Pharmaceuticals and Forest Pharmaceuticals; and receives research support from the NIH [R01-AG023195 (PI), R01-AG11378 (Coinvestigator), P50-AG16574 (Coinvestigator), U01 AG06786 (Coinvestigator), and R01 HL70825 (Coinvestigator)]. Dr. Wszolek serves as Co–Editor-in Chief of Parkinsonism and Related Disorders, as Co–Editor-in- Chief of the Polish Edition of Neurology, as Regional Editor of the European Journal of Neurology, and on the editorial boards of Neurologia i Neurochirurgia Polska, Advances in Rehabilitation, the Medical Journal of the Rzeszow University, and Clinical and Experimental Medical Letters; holds and has contractual rights for receipt of future royalty payments from the following patents: Norwegian Patent NO20045612A0, Polynucleotide (Filed: 12/23/2004, Pub: 01/15/2007); Norwegian Patent NO20052535A0, Polynucleotide (Filed: 05/27/2005, Pub: 05/27/2005); Norwegian Patent NO0323175B1, Framgangsmate for a pavise en mutasjon som forarsaker arvelig parkinsonisme (Filed: 05/27/2005, Pub: 01/15/2007); Australian Patent AU5319787AA, A novel polynucleotide involved in heritable Parkinson’s disease (Filed: 12/19/2005, Pub: 06/29/2006); World Patent WO06068492A1, A novel polynucleotide involved in heritable Parkinson’s disease (Filed: 12/19/2005, Pub: 6/29/2006); US Patent US2008/0009454 A1, Polynucleotide (Filed: 12/19/2005, Pub: 01/10/2008); Canadian Patent CA2606672AA, A novel polynucleotide involved in heritable Parkinson’s disease (Filed: 12/19/2005, Pub: 06/29/2006); Canadian Patent CA2606672AA, A novel polynucleotide involved in heritable Parkinson’s disease (Filed: 12/19/2005, Pub: 06/29/2006); European Patent EP1838871A1, A novel polynucleotide involved in heritable Parkinson’s disease (Filed: 12/19/2005, Pub: 10/03/2007); receives royalties from publishing Parkinsonism and Related Disorders (Elsevier, 2007, 2008, 2009), the Polish Edition of Neurology (Medycyna Praktyczna, 2007, 2008, 2009) and the European Journal of Neurology (Wiley-Blackwell, 2007, 2008, 2009); receives research support from Allergan, Inc., the Pacific Alzheimer Research Foundation (Canada) [C06-01], the Morris K. Udall Parkinson’s Disease Research Center of Excellence (NIH/NINDS P50 NS40256), Mayo Clinic Florida, Research Committee CR program, the NIH [NIA P01AG017216-1 (Coinvestigator), NIA R01AG015866-1 (Coinvestigator), and NINDS P50NS 40256 (Coinvestigator)], and the CIHR [P 121849 (Coinvestigator)]. Dr. Petersen serves on scientific advisory boards for Elan Pharmaceuticals, Wyeth Pharmaceuticals, and GE Healthcare; receives royalties from publishing Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH [U01-AG06786 (PI), P50-AG16574 (PI), R01-AG11378 (Coinvestigator), and U01-24904 (Coinvestigator)]. Dr. Rademakers has received honoraria for non–industry-sponsored activities; holds US Patent 12/302,691 [Detecting and Treating Dementia (2008)] and US Patent 12/413,869 [Methods and Materials for Detecting and Treating Dementia (2009)]; and receives research support from the Pacific Alzheimer Research Foundation (Canada) [C06-01], the Association for Frontotemporal Dementia (PI), and the NIH [P50-AG16574 (PI on Project 2)]. Dr. Josephs is funded by the NIH Roadmap Multidisciplinary Clinical Research Career Development Award Grant (K12/NICHD)-HD49078 (PI) and the Morris K. Udall Parkinson’s Disease Research Center of Excellence (NIH/NINDS P50 NS40256).
Supplementary Material
Address correspondence and reprint requests to Dr. Keith A. Josephs, Department of Neurology, Mayo Clinic, 200 First St. SW, Rochester, MN 55905 josephs.keith@mayo.edu
Supplemental data at www.neurology.org
Support for several investigators was provided by the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program of the Mayo Foundation and the NIH Construction Grant NIH C06 RR018898 (Coinvestigator).
Disclosure: Author disclosures are provided at the end of the article.
Received February 26, 2009. Accepted in final form June 24, 2009.
REFERENCES
- 1.Josephs KA. Frontotemporal dementia and related disorders: deciphering the enigma. Ann Neurol 2008;64:4–14. [DOI] [PubMed] [Google Scholar]
- 2.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 3.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol 1999;56:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 1999;64:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 6.Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch Neurol 2008;65:460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 2004;24:277–295. [DOI] [PubMed] [Google Scholar]
- 8.van Swieten JC, Stevens M, Rosso SM, et al. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol 1999;46:617–626. [DOI] [PubMed] [Google Scholar]
- 9.Whitwell JL, Jack CR Jr, Boeve BF, et al. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 2009;72:813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boeve BF, Tremont-Lukats IW, Waclawik AJ, et al. Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation. Brain 2005;128:752–772. [DOI] [PubMed] [Google Scholar]
- 11.Malkani R, D’Souza I, Gwinn-Hardy K, et al. A MAPT mutation in a regulatory element upstream of exon 10 causes frontotemporal dementia. Neurobiol Dis 2006;22:401–403. [DOI] [PubMed] [Google Scholar]
- 12.Kokmen E, Naessens JM, Offord KP. A short test of mental status: description and preliminary results. Mayo Clin Proc 1987;62:281–288. [DOI] [PubMed] [Google Scholar]
- 13.Hughes CP, Berg L, Danziger WL, et al. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. 2nd ed. Austin, TX: Pro-ed; 2001. [Google Scholar]
- 15.Rey A. L’examen clinique en psychologie. Paris: Presses Universitaires de France; 1964. [Google Scholar]
- 16.Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol 1982;11:592–598. [DOI] [PubMed] [Google Scholar]
- 17.Ashburner J, Friston KJ. Voxel-based morphometry: the methods. Neuroimage 2000;11:805–821. [DOI] [PubMed] [Google Scholar]
- 18.Senjem ML, Gunter JL, Shiung MM, et al. Comparison of different methodological implementations of voxel-based morphometry in neurodegenerative disease. Neuroimage 2005;26:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jack CR Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain 2008;131:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashburner J, Friston KJ. Unified segmentation. Neuroimage 2005;26:839–851. [DOI] [PubMed] [Google Scholar]
- 21.Whitwell JL, Josephs KA, Rossor MN, et al. Magnetic resonance imaging signatures of tissue pathology in frontotemporal dementia. Arch Neurol 2005;62:1402–1408. [DOI] [PubMed] [Google Scholar]
- 22.Spina S, Farlow MR, Unverzagt FW, et al. The tauopathy associated with mutation +3 in intron 10 of Tau: characterization of the MSTD family. Brain 2008;131:72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arvanitakis Z, Witte RJ, Dickson DW, et al. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation). Parkinsonism Related Disord 2007;13:230–239. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi K, Hayashi M, Fukutani Y, et al. KP1 expression of ghost Pick bodies, amyloid P-positive astrocytes and selective nigral degeneration in early onset Picks disease. Clin Neuropathol 1999;18:240–249. [PubMed] [Google Scholar]
- 25.Kobayashi K, Kidani T, Ujike H, et al. Another phenotype of frontotemporal dementia and parkinsonism linked to chromosome-17 (FTDP-17) with a missense mutation of S305N closely resembling Pick’s disease. J Neurol 2003;250:990–992. [DOI] [PubMed] [Google Scholar]
- 26.Bird TD, Nochlin D, Poorkaj P, et al. A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 1999;122(pt 4):741–756. [DOI] [PubMed] [Google Scholar]
- 27.Mirra SS, Murrell JR, Gearing M, et al. Tau pathology in a family with dementia and a P301L mutation in tau. J Neuropathol Exp Neurol 1999;58:335–345. [DOI] [PubMed] [Google Scholar]
- 28.Kodama K, Okada S, Iseki E, et al. Familial frontotemporal dementia with a P301L tau mutation in Japan. J Neurol Sci 2000;176:57–64. [DOI] [PubMed] [Google Scholar]
- 29.Wszolek ZK, Pfeiffer RF, Bhatt MH, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 1992;32:312–320. [DOI] [PubMed] [Google Scholar]
- 30.Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta 2005;1739:240–250. [DOI] [PubMed] [Google Scholar]
- 31.Spillantini MG, Goedert M, Crowther RA, et al. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci USA 1997;94:4113–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alonso Adel C, Mederlyova A, Novak M, et al. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem 2004;279:34873–34881. [DOI] [PubMed] [Google Scholar]
- 33.Barghorn S, Zheng-Fischhofer Q, Ackmann M, et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 2000;39:11714–11721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.