

Abstract
Whole cell patch-clamp recordings were performed in brain slices to investigate mechanisms regulating the excitability of paraventricular nucleus (PVN) neurones that project directly to the rostral ventrolateral medulla (RVLM) (PVN–RVLM neurones) of rats. In voltage-clamp recordings, step depolarization elicited a calcium-dependent outward tail current that reversed near EK. The current was nearly abolished by apamin and by UCL1684, suggesting mediation by small-conductance Ca2+-activated K+ (SK) channels. In current-clamp recordings, depolarizing step current injections evoked action potentials that underwent spike-frequency adaptation (SFA). SK channel blockade with apamin or UCL1684 increased the spike frequency without changing the rate of SFA. Upon termination of step current injection, a prominent medium after-hyperpolarization potential (mAHP) was observed. SK channel blockade abolished the mAHP and revealed an after-depolarization potential (ADP). In response to ramp current injections, the rate of sub-threshold depolarization was increased during SK channel blockade, indicating that depolarizing input resistance was increased. Miniature EPSC frequency, amplitude, and decay kinetics were unaltered by bath application of apamin, suggesting that SK channel blockade likely increased excitability by a postsynaptic action. We conclude that although SK channels play little role in generating SFA in PVN–RVLM neurones, their activation nevertheless does dampen excitability. The mechanism appears to involve activation of a mAHP that opposes a prominent ADP that would otherwise facilitate firing.
A widely distributed CNS network generates and regulates rhythmic activity in peripheral sympathetic nerve activity (SNA) that is essential for maintaining arterial blood pressure and cardiovascular homeostasis (Guyenet, 2006). Among neurones comprising this network, presympathetic neurones in the hypothalamic paraventricular nucleus (PVN) have been the focus of considerable interest now for many years (Porter & Brody, 1985; Lovick & Coote, 1988; Stern 2001; Allen 2002; Akine et al. 2003; Toney et al. 2003; Li & Pan, 2007). The picture that has emerged indicates that discharge of these neurones is normally suppressed by strong GABA-mediated synaptic inhibition (Martin et al. 1991; Chen et al. 2003; Li et al. 2006). Consistent with this, electrophysiological studies demonstrate that most presympathetic PVN neurones are either quiescent or exhibit slow (1–2 spikes s−1) irregular discharge in vivo (Lovick & Coote, 1988; Chen & Toney, 2000, 2003). Interestingly, interruption of PVN GABAergic inhibition increases SNA and transforms SNA burst discharge into a low frequency, high amplitude pattern (Kenney et al. 2001). The latter suggests that ongoing synaptic inhibition may prevent burst firing of presympathetic PVN neurones.
In spite of having little tonic activity under normal conditions (Lovick & Coote, 1988; Chen & Toney, 2000, 2003) and contributing only modestly to the maintenance of ongoing SNA (Allen, 2002), presympathetic PVN neurones are nevertheless considered essential for increasing SNA under a variety of circumstances; including during physiological perturbations (e.g. dehydration, hypoxaemia, etc.) (Stocker et al. 2005; Reddy et al. 2007; Cruz et al. 2008) and during more chronic disease conditions (e.g. heart failure, hypertension, cardiac ischaemia, etc.) (Haywood et al. 2001; Li & Pan, 2006; Patel, 2008). Haywood and colleagues, for example, reported that GABAergic inhibition of the PVN is reduced in rats with renal-wrap hypertension in which elevated blood pressure is maintained, at least in part, by elevated SNA (Haywood et al. 2001). Using the same hypertensive model, the Stern laboratory recently reported that the intrinsic excitability of presympathetic PVN neurones is increased in association with a reduction of the transient A-type potassium current (Sonner et al. 2008). Thus, persistently elevated discharge of PVN neurones appears to be achieved both by changing their synaptic input (i.e. decreased inhibition and/or increased excitation) and by modifying their intrinsic membrane properties.
Evidence indicating that PVN neurones are important for setting the level of SNA prompted interest in mechanisms that regulate their excitability and discharge. Many studies have focused on PVN neurones that project directly to the rostral ventrolateral medulla (RVLM), as the latter region is the major source of excitatory synaptic drive to spinal sympathetic preganglionic neurones. PVN–RVLM neurones make close synaptic appositions with sympathoexcitatory RVLM neurones and are mostly glutamatergic (Stocker et al. 2006), though they may co-express a number of neuropeptides. In keeping with this, studies performed in intact rats indicate that PVN stimulation increases the discharge of bulbospinal RVLM neurones by activating ionotropic glutamate receptors and vasopressin V1 receptors (Yang & Coote, 1998; Yang et al. 2001).
In brain slice studies, presympathetic PVN neurones exhibit complex patterns of discharge. Application of l-glutamate, for example, evokes high frequency trains of action potentials followed by significant postexcitatory depression (Pakhomov et al. 2006). A similar pattern of discharge has been observed among PVN–RVLM neurones in response to the neuropeptide angiotensin II (Cato & Toney, 2003) and in response to depolarizing current injections, which also elicit high frequency discharge accompanied by significant spike-frequency adaptation (SFA) and a prominent after-hyperpolarization potential (AHP) (Tasker & Dudek, 1991; Stern, 2001; Sonner & Stern, 2007). Based on literature evidence that these activity patterns often result from activation of one or more Ca2+-sensitive K+ conductances subsequent to an increase in intracellular calcium (i.e. through activation of voltage-dependent Ca2+ channels) (Vogalis et al. 2003; Stocker, 2004; Villalobos et al. 2004; Teruyama & Armstrong, 2007, but see Teagarden et al. 2008), we hypothesized that small-conductance Ca2+-activated K+ (SK) channels suppress excitability of PVN–RVLM neurones and contribute to the patterning of their discharge. To test this hypothesis, the present study was conducted in vitro using hypothalamic slices. Whole cell patch-clamp recordings were performed in PVN–RVLM neurones identified by retrograde axonal tracing.
Methods
Animals
Male Sprague–Dawley rats (n= 25, 400–500 g) were purchased from Charles River Laboratories (Wilmington, MA, USA) and were individually housed in a temperature controlled room (22–23°C) with a 14 h–10 h light–dark cycle. Rats had free access to standard chow and tap water. All experimental and surgical procedures were approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio and were performed in strict adherence to procedures set for the NIH Guide to the Care and Use of Laboratory Animals.
Retrograde labelling of PVN–RVLM neurones
PVN neurones were retrogradely labelled from the ipsilateral RVLM as previously described (Cato & Toney, 2005). Briefly, rats were anaesthetized with isoflurane (3% in O2) and placed in a stereotaxic frame, and a small burr hole was made to expose the cerebellum. A glass micropipette was lowered into the pressor region of RVLM (co-ordinates: −12.7 mm caudal to bregma, 1.8 mm lateral to midline and 8.9 mm below the skull) and rhodamine-containing microspheres were microinjected in a volume of 50 nl. Each animal received a daily injection of penicillin G (30 000 units (100 g bw)−1, s.c.) for 3 days after surgery. The location of the tracer was verified histologically (Cato & Toney, 2005; Stocker et al. 2006).
Electrophysiology
Five to seven days after tracer injections into RVLM, rats were anaesthetized with isoflurane (3% in O2) and decapitated. Brains were removed and placed in ice-cold cutting solution containing (in mm): sucrose, 206; KCl, 2; MgSO4, 2; NaH2PO4, 1.25; NaHCO3, 26; CaCl2, 1; MgCl2, 1; d-glucose, 10; ascorbic acid, 0.4. Osmolarity and pH were adjusted to 290–295 mosmol l−1 and 7.4, respectively. Solution pH and 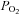 were maintained by equilibration with a 95% O2–5% CO2 gas mixture. Coronal slices through the PVN were cut to a thickness of 300 μm on a vibrating microtome (Leica VT 1000S; Leica, Nussloch, Germany). Slices were incubated at room temperature (24–26°C) for at least 2 h in continuously gassed artificial cerebrospinal fluid (ACSF) containing (in mm): NaCl, 125; KCl, 2; MgSO4, 2; NaH2PO4, 1.25; NaHCO3, 26; CaCl2, 2; d-glucose, 10; ascorbic acid, 0.4 (osmolarity 295–300 mosmol l−1; pH 7.4). Slices were transferred to a glass-bottomed recording chamber and viewed through an upright microscope (E600FN, Nikon) equipped with DIC optics, epi-fluorescence, an infrared (IR) filter and an IR-sensitive video camera (C2400, Hamamatsu, Bridgewater, NJ, USA). Appropriate filter was used to visualize neurones retrogradely labelled with rhodamine.
were maintained by equilibration with a 95% O2–5% CO2 gas mixture. Coronal slices through the PVN were cut to a thickness of 300 μm on a vibrating microtome (Leica VT 1000S; Leica, Nussloch, Germany). Slices were incubated at room temperature (24–26°C) for at least 2 h in continuously gassed artificial cerebrospinal fluid (ACSF) containing (in mm): NaCl, 125; KCl, 2; MgSO4, 2; NaH2PO4, 1.25; NaHCO3, 26; CaCl2, 2; d-glucose, 10; ascorbic acid, 0.4 (osmolarity 295–300 mosmol l−1; pH 7.4). Slices were transferred to a glass-bottomed recording chamber and viewed through an upright microscope (E600FN, Nikon) equipped with DIC optics, epi-fluorescence, an infrared (IR) filter and an IR-sensitive video camera (C2400, Hamamatsu, Bridgewater, NJ, USA). Appropriate filter was used to visualize neurones retrogradely labelled with rhodamine.
Patch electrodes were pulled (Flaming/Brown P-97, Sutter Instrument, Novato, CA, USA) from borosilicate glass capillaries and polished to a tip resistance of 4–5 MΩ. Electrodes were filled with a solution containing (in mm): potassium gluconate, 135; Hepes, 10; EGTA, 0.1; MgCl2, 1.0; NaCl, 1.0; Na2ATP, 2.0; Na2GTP, 0.5 (osmolarity 280–285 mosmol l−1; pH 7.3). Note that a relatively low concentration of EGTA (0.1 mm) was used to allow intracellular free Ca2+ to accumulate and activate SK channels during membrane potential depolarization (Brenner et al. 2005). Whole-cell current- and voltage-clamp recordings were made using an Axopatch 200B amplifier and pCLAMP software (v10.0, Axon Instruments, Union City, CA, USA). The calculated liquid junction potential was −15 mV. Signals were filtered at 1 kHz, digitized at 10 kHz (Digidata 1322A, Axon Instruments), and saved on a computer for offline analysis. After achieving a gigaohm seal and the whole-cell configuration, cell capacitance, access resistance and resting membrane potential (Vm) were recorded until stable. Series resistance (16 ± 1 MΩ) was compensated in voltage-clamp recordings. Cells that met the following criteria were included in the analysis: action potential amplitude ≥50 mV from threshold to the peak, input resistance (Rinput) larger than 0.5 GΩ when hyperpolarizing current injections of −20 pA were delivered from a holding potential of −80 mV, resting Vm negative to −50 mV, and less than 20% change in series resistance during the recording.
Identification of SK current
To study SK current, voltage-clamp recordings were performed with intracellular solution containing the cAMP analogue 8-(4-chlorophenylthio)-3′,5′-cyclic adenosine monophosphate (8CPT-cAMP, 50 μm) to block the slow after-hyperpolarization (sAHP) current (IsAHP) (Stocker et al. 1999). Membrane potential was clamped at −60 mV and stepped to +10 mV for 100 ms. Upon returning Vm to −60 mV, an outward tail current was recorded. After control discharge responses were obtained, apamin (100 nm) or UCL1684 (100 nm) was bath applied to selectively block SK channels. A similar approach was used to assess the Ca2+ dependence of outward tail current. Stable tail currents were recorded under control conditions and slices were then perfused with Ca2+ free ACSF containing 2 mm MgCl2. Tail current recorded during each treatment was subtracted from the corresponding control tail current to isolate SK-mediated current. Henceforth this current is referred to as ImAHP. Recordings were performed in the presence of 1.0 μm tetrodotoxin (TTX) and 1 mm tetraethylammonium (TEA). Decay kinetics of the SK mediated ImAHP were analysed by fitting the subtracted current with a one-phase exponential.
Testing neuronal excitability
Neuronal excitability was studied in current-clamp mode by adjusting Vm to −80 mV by injecting negative current through the patch electrode. Injected current was then varied in steps of +25 pA, each for a duration of 800 ms. To study the mAHP and ADP, recordings were made from a potential of −60 mV, and +150 pA current injections (500 ms) were made followed by a return of Vm to −60 mV for 5 s. To determine the spike threshold (Vt) and depolarizing Rinput below Vt, ramp current injections (0.2 pA ms−1, 1000 ms) were made from a potential of −80 mV. All current-clamp recordings were performed without TTX, TEA or 8CPT-cAMP.
Recording synaptic activity
To mimic conditions used in excitability testing, miniature excitatory postsynaptic currents (mEPSCs) were recorded in voltage-clamp mode at a holding potential of −80 mV. Recordings were performed in the presence of 1 μm TTX, 100 μm picrotoxin (a GABA-A receptor antagonist), and 2 mm MgCl2 to block action potential dependent synaptic activity, GABAergic mIPSC activity, and NMDA receptor-mediated mEPSC activity. Effects of SK channel blockade on the frequency, amplitude and decay time constant of remaining AMPA receptor mediated mEPSC activity was determined by comparing events recorded under control conditions with those recorded from the same neurones during bath application of apamin (100 nm). Detection of events was accomplished by setting a threshold below the noise level, which was determined by bath application of 20 μm CNQX to block non-NMDA receptors.
Chemicals
All chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) with the exception of tetrodotoxin (Tocris Bioscience, UK) and tetraethylammonium (Fluka BioChemika, Switzerland).
Statistical analysis
Summary data are reported as means ±s.e.m. Depending on the experiment, group means were compared using either Student's t-test for unpaired data, or a one-way or two-way ANOVA. When differences were found, Bonferroni's post hoc test was used for multiple pair-wise comparisons (GraphPad Prism, v5.0; GraphPad Software Inc., La Jolla, CA, USA). Mini-Analysis software (Synaptosoft, Leonia, NJ, USA) was used to analyse mEPSC activity. Cumulative probabilities of mEPSC inter-event intervals and amplitudes were compared using Kolomogorov–Smirnov tests. At least 200 mEPSCs were used for each analysis. Differences between means were considered significant at P < 0.05.
Results
Whole cell patch-clamp recording was performed on 45 retrogradely labelled PVN–RVLM neurones (n= 25 rats). Retrograde tracer was confined to the RVLM, which is located ventral to the nucleus ambiguus and 200–500 μm posterior to the caudal pole of the facial nucleus. Under conditions used in these experiments, recorded neurones lacked spontaneous discharge before and during SK channel blockade. This was the case except for two cells that had irregular spontaneous activity at relatively depolarized resting Vm (−48 mV). Data from these cells were excluded from analysis.
Identification of SK channel-mediated ImAHP in PVN–RVLM neurons
In voltage-clamp recordings, SK current was isolated in a group of PVN–RVLM neurones that had a mean resting Vm of −58 ± 2 mV and a mean cell capacitance of 27 ± 4 pF (n= 22). To isolate SK current, Vm was stepped from a holding potential of −60 mV to +10 mV for 100 ms. Upon returning Vm to −60 mV, an outward tail current was observed. Bath application of the SK channel-selective blockers apamin and UCL1684 was used to isolate the SK current (ImAHP) by subtraction. An apamin-sensitive ImAHP was recorded in 4 of 4 neurones tested. Figure 1A shows a representative trace of isolated apamin-sensitive ImAHP (Fig. 1A, inset). An UCL1684-sensitive ImAHP was detected in another four cells. Figure 1B shows a representative trace of isolated UCL1684-sensitive ImAHP (Fig. 1B, inset). As with SK channel blockade, perfusion with a Ca2+-free bath solution (Fig. 1C, inset) effectively eliminated the tail current. Neither the amplitude (Fig. 1E) nor the decay time constant (Fig. 1F) of ImAHP was significantly different between cells treated with apamin and UCL1684 and Ca2+-free extracellular solution. To test whether current rundown affected the amplitude of ImAHP, time control experiments were performed (n= 5). Figure 1D shows representative traces of outward tail currents recorded immediately before and 15 min after switching to a second bath that also contained standard ACSF. Note that neither the amplitude (65.7 ± 18 versus 67.3 ± 21 pA) nor the decay time constant (285 ± 9 versus 318 ± 17 ms) changed significantly as a function of time.
Figure 1. Characteristics of SK channel-mediated outward current in PVN–RVLM neurones.

Representative traces show the outward tail current evoked by a 100 ms square pulse +70 mV depolarization (from −60 mV to +10 mV). Recordings were performed in the presence of bath TTX (1 μm) and TEA (1 mm), and intracellular 8CPT-cAMP (50 μm). A and B, outward tail current induced by the depolarizing pulse under control conditions (black trace) and in the presence of either apamin (A, grey trace, 100 nm) or UCL1684 (B, grey trace, 100 nm). Insets in A and B show net SK current (ImAHP) obtained by subtraction (control – blocker). C, outward tail current was absent in Ca2+-free ACSF. Inset in C shows the net SK current obtained from subtraction (control – Ca2+-free). D, outward tail current under control conditions (black) was unchanged 15 min later (grey). E, summary data showing the amplitude of subtracted SK current (ImAHP, left) and its decay time constant (right). The number of neurones recorded in each group is given inside the corresponding bar. Numbers in parentheses indicate number of animals from which data were obtained.
Reversal potential of isolated ImAHP was determined in voltage-clamp mode. Summary I-V relationships (n= 6) for subtracted ImAHP were consistently non-rectifying. Reversal potential was −101 mV after correcting for liquid junction potential (Fig. 2A). This reversal potential was fairly close to the value predicted by the Nernest equation for a K+ selective conductance, i.e. −103 mV. Figure 2B shows representative traces of apamin-sensitive ImAHP evoked by voltage steps. These properties are consistent with those of voltage-insensitive, Ca2+-activated SK channels.
Figure 2. Reversal potential of SK current in PVN–RVLM neurones.

A, mean amplitude of the subtracted SK current is plotted as a function of holding potential for PVN–RVLM neurones (n= 6) from 3 rats. Reversal potential was estimated to be −101 mV. B, representative subtracted SK current traces used to generate the I–V plot. To activate the outward current, a depolarizing pulse was delivered from a holding potential of −60 mV, followed by test potentials ranging from −110 mV to −50 mV in 10 mV increments.
SK channel blockade increases excitability of PVN–RVLM neurons
All neurones tested lacked spontaneous discharge, but depolarizing current injections consistently evoked repetitive action potential firing. Figure 3Aa shows a representative discharge response to a 200 pA depolarizing current pulse. The relationship between current injection and spike frequency (stimulus–response curve) was well fitted by a sigmoidal function. Graded current injections (25, 50, 75, 100, 125, 150, 175 and 200 pA; 800 ms) evoked graded increases in spike frequency (Fig. 3Ba).
Figure 3. SK channel blockade increases excitability of PVN–RVLM neurones.

Voltage traces show representative responses of PVN–RVLM neurones to a 200 pA depolarizing current injection (800 ms pulse) in the absence (Aa) and presence of SK channel blocker apamin (Ab, 100 nm) or UCL1684 (Ac, 100 nm). SFA is seen in Aa–c. Ba, the relationship between depolarizing current injected and spike frequency in the absence (Control, filled squares) and presence of apamin (filled rhombus) or UCL1684 (filled inverted triangles). Graded current injections evoked graded increases in spike frequency. The maximum spike frequency was achieved at ∼150 pA of depolarizing current. Apamin and UCL1684 each significantly increased the maximum spike frequency. Responses between 0 and 125 pA were nearly linear. Apamin and UCL1684 also increased the slope of the linear phase of the stimulus–response relation (Bb). Ca, plot showing inter-spike interval (ISI) during repetitive action potential firing in response to a 200 pA current injection. Cb, summary data showing the ratio of 13th ISI/1st ISI in the control condition and in the presence of SK channel blockers. SFA was evident in the absence and presence of SK channel blockade. #P < 0.05, ##P < 0.01 versus control, NS P > 0.05 (1-way ANOVA). *P < 0.05 UCL1684 versus control, †P < 0.05 apamin versus control (2-way ANOVA). The number of neurones recorded in each group is given inside the corresponding bar. Numbers in parentheses indicate number of animals from which data were obtained.
The role of SK channels in regulating excitability was investigated by comparing the current–spike frequency relationship and the maximum discharge frequency response recorded in the absence and presence of SK channel blockers apamin and UCL1684. Figure 3Ab and c shows examples of action potential trains evoked by a 200 pA current pulse in the presence of apamin or UCL1684. Both blockers significantly increased the maximum firing rate compared to controls. Figure 3Ba plots mean spike frequency elicited by a family of graded depolarizing current steps (25 pA) for groups of neurones in the absence and presence of SK channel blockers. Excitability was also compared by examining differences in the slope of stimulus–response curves. Responses evoked with currents between 0 and 125 pA were linear and the slope increased from 0.14 ± 0.01 Hz pA−1 in the absence of SK channel blockade to 0.25 ± 0.01 Hz pA−1 (P < 0.01) in the presence of apamin and 0.21 ± 0.03 Hz pA−1 (P < 0.05) in the presence of UCL1684 (Fig. 3Bb). It is worth noting that excitability was not significantly different between cells treated with apamin and UCL1684, although the discharge frequency and slope of stimulus–response curves for neurons treated with apamin were slightly greater than for neurones treated with UCL1684.
In the absence of SK channel blockade, neurones had a mean resting Vm of −66 ± 4 mV and a mean hyperpolarizing Rinput of 1.2 ± 0.2 GΩ. The mean current needed to hyperpolarize Vm to near −80 mV was −17 ± 6 pA (n= 7). None of these values were affected by bath application of 100 nm apamin (resting Vm: −66 ± 2 mV; hyperpolarizing Rinput: 1.1 ± 0.2 GΩ; mean current: −16 ± 4 pA; n= 7) or 100 nm UCL1684 (resting Vm: −63 ± 14 mV; hyperpolarizing Rinput: 1.1 ± 0.1 GΩ; mean current: −16 ± 5 pA, n= 5). The hyperpolarizing Rinput was measured by injecting a −20 pA current from a Vm very near to −80 mV.
SK channel blockade does not affect spike-frequency adaptation
Figure 3Aa–c shows that the pattern of inter-spike-interval (ISI) prolongation within action potential trains was not affected by SK channel blockade. In the absence of SK channel blockers (Fig. 3Aa), a 200 pA depolarizing current pulse evoked repetitive action potential firing that exhibited a progressive increase in ISI without causing complete termination of firing. In the presence of either apamin (Fig. 3Ab) or UCL1684 (Fig. 3Ac), mean spike frequency increased (Fig. 3Ba) but ISI prolongation (i.e. SFA) was unchanged, although the average ISI was significantly reduced by apamin (Fig. 3Ca) (P < 0.01) and by UCL1684 (P < 0.05). Summary data for the ratio of 13th ISI and 1st ISI are shown in Fig. 3Cb.
SK channel blockade reduces a medium AHP and reveals an ADP
To explore the mechanism of SK channel mediated suppression of excitability during action potential firing, the effect of SK channel blockade on the mAHP was examined. In current-clamp mode, depolarizing current pulses (150 pA, 500 ms) were injected from a Vm of −60 mV. Traces in Fig. 4Aa–c illustrate the mAHP and ADP recorded in cells with and without SK channel blockade. Under control conditions in the absence of an SK channel blocker (Fig. 4Aa), a train of action potentials was induced that exhibited SFA and a prominent mAHP following termination of action potential firing. In the presence of apamin (Fig. 4Ab) or UCL1684 (Fig. 4Ac) the mAHP was not observed, but a shoulder-shaped ADP was uncovered after the spike train. In the presence of apamin, the ADP was observed in 5 of 7 neurones tested. The peak amplitude was +9 ± 2 mV (range: +3 to +17 mV) (Fig. 4Ba) and the decay time constant was 467 ± 175 ms (range: 158 to 1138 ms) (Fig. 4Bb). The other two cells had smaller amplitude of mAHP (−5 ± 2 mV) compared to those recorded under control condition (−8 ± 2 mV). No ADP was uncovered in these cells. In the presence of UCL1684, an ADP was observed in 5 of 5 neurones tested. Peak ADP amplitude in these cells was +8 ± 1 mV (range: +5 to +13 mV) (Fig. 4Ba) and the decay time constant was 416 ± 123 ms (range: 48–831 ms) (Fig. 4Bb).
Figure 4. SK channel blockade reduces mAHP and uncovers an ADP.

Representative voltage traces show the effect of SK channel blockade on the medium after-hyperpolarizing potential (mAHP). Aa, in the absence of SK channel blockade, a train of action potentials was induced by a depolarizing current pulse (150 pA, 500 ms). Both a mAHP and slow AHP (sAHP) were observed following termination of action potential firing. Ab and Ac, in the presence of apamin (Ab; 100 nm) and UCL1684 (Ac; 100 nm), a shoulder-shaped after-depolarizing potential (ADP) was revealed. Note that the sAHP can still be observed. B, summary data showing that the maximum ADP amplitude (Ba) and the ADP decay time constant (Bb) were similar in the presence of apamin and UCL1684. The number of neurones recorded in each group is given inside the corresponding bar. Numbers in parentheses indicate number of animals from which data were obtained.
SK channel blockade increases depolarizing Rinput without alteration of Vt
To determine whether SK channel blockade affects action potential voltage threshold (Vt) and depolarizing Rinput in PVN–RVLM neurones, ramp current injection was performed. Current ramps induced gradual membrane potential depolarization (0.2 pA ms−1 for 1000 ms duration) and triggered action potentials at Vt (Fig. 5Aa–c, summarized in Fig. 5Bc). Blockade of SK channels increased the rate of depolarization such that Vt was reached earlier during the current ramp (Fig. 5Ab and c). Thus, depolarizing Rinput measured below Vt was significantly increased (P < 0.05) by both apamin and UCL1684 compared to control (Fig. 5Ba). Similar to effects of SK channel blockade on responses to depolarizing current pulses (Fig. 3Aa–c), firing rate of spikes evoked by current ramps was significantly increased in the presence of apamin and UCL1684 (Fig. 5Bb). Note that there was no effect of SK channel blockade on Vt (Fig. 5Bc).
Figure 5. SK channel blockade increases depolarizing input resistance below spike threshold.

Traces show representative responses of PVN–RVLM neurones to ramp current injections in the absence (Aa, control) and presence of the SK channel blockers apamin (Ab, 100 nm) and UCL1684 (Ac, 100 nm). Under each treatment, membrane potential depolarized gradually in response to current injection (0.2 pA ms−1, 1000 ms) and action potential firing began at the potential threshold (Vt). SK channel blockade with apamin or UCL1684 increased subthreshold depolarization rate by increasing the depolarizing input resistance (Rinput). Vt (arrows) for triggering action potentials did not change although the action potential firing rate was increased by bath application of apamin and UCL1684. B, summary data showing that depolarizing Rinput (Ba) and action potential firing rate (Bb) during ramp current injections were significantly increased by SK channel blockade. Blocking SK channels with either apamin or UCL1684 caused a slight depolarizing shift of Vt, but these changes did not reach statistical significance (Bc). *P < 0.05 versus control, **P < 0.01 versus control, NS P > 0.05 (1-way ANOVA). The number of neurones recorded in each group is given inside the corresponding bar. Numbers in parentheses indicate number of animals from which data were obtained.
SK channel blockade does not affect mEPSCs
Traces in Fig. 6Aa and b illustrate mEPSCs recorded in a cell under control conditions (Fig. 6Aa) and after application of 100 nm apamin (Fig. 6Ab). Application of 20 μm CNQX (a glutamate non-NMDA receptor antagonist) effectively abolished mEPSCs (Fig. 6Ac). The insets in Fig. 6Aa and b show the decay time constant (τ) of mEPSCs obtained from control and during bath application of apamin. The decay phase of mEPSCs was best fitted with a one-phase exponential function. The τ was similar during control and bath application of apamin as summarized in Fig. 6Cc. Cumulative probability analysis of mEPSCs in the same cell revealed that SK channel blockade did not affect the distribution pattern of inter-event intervals (Fig. 6Ba) and event amplitudes (Fig. 6Bb). Bath application of apamin did not affect mean frequency and amplitude of mEPSC (Fig. 6Ca and b). Neurones had a mean resting Vm of −53 ± 2 mV and mean holding current of −23 ± 8 pA when the cell was clamped at −80 mV. Neither was significantly affected by apamin (resting Vm: −55 ± 4 mV, holding current: −25 ± 7 pA, n= 4).
Figure 6. Lack of effect of SK channel blockade on mEPSCs in PVN–RVLM neurones.

Representative traces from a PVN–RVLM neurone show miniature excitatory postsynaptic currents (mEPSCs) during voltage-clamp recording at a holding potential of −80 mV. Recordings were performed in the presence of TTX (1 μm) and picrotoxin (100 μm). Aa and Ab, spontaneous mEPSCs under control condition and in the presence of apamin (100 nm). Inset in Aa and Ab shows the decay time constant (τ) of mEPSCs obtained from control and during bath application of apamin. The decay phase of mEPSCs was best fitted with one-phase exponential function. The decay time constant was similar during control and bath application of apamin (Cc). Ac, bath application of 20 μm CNQX effectively abolished mEPSCs. Cumulative probability plots of mEPSCs from the same neurone show that the distribution of the inter-event intervals (Ba) and amplitudes (Bb) during control condition and in the presence of apamin. Summary data show the lack of effect of apamin on the frequency (Ca), amplitude (Cb), or decay time constant (Cc) of mEPSCs in PVN–RVLM neurones. The number of neurones recorded in each group is given inside the corresponding bar. Numbers in parentheses indicate number of animals from which data were obtained.
Discussion
Although synaptic and intrinsic mechanisms contribute to spontaneous action potential firing in PVN–RVLM neurones (Sonner & Stern, 2007; Li et al. 2008), mechanisms controlling their excitability have not been extensively studied. Here we report that PVN–RVLM neurones express an apamin-sensitive SK current. We found that depolarizing step current injections evoked trains of action potentials that underwent SFA. Upon termination of current injection, a prominent mAHP was observed. Blockade of SK channels did not change the rate of SFA, but did increase the frequency of current-induced spiking. SK channel blockade also abolished the mAHP and revealed a prominent ADP. The increased rate of current-induced spiking during SK channel blockade, therefore, is likely to be due to the loss of mAHP-mediated opposition of the ADP. SK channel blockade did not change resting Vm, but did increase depolarizing Rinput at membrane potentials below spike threshold. Thus SK channel activation in PVN–RVLM neurons appears to occur between resting Vm and spike threshold. Consistent with effects of SK channel blockade being mediated postsynaptically, bath application of apamin had no effect on the frequency, amplitude or decay time constant of mEPSCs. Taken together, these findings provide new evidence that activation of SK channels can modulate intrinsic membrane properties of presympathetic PVN–RVLM neurones and can strongly suppress their in vitro excitability.
Excitability of PVN–RVLM neurones is regulated by SK current
In this study, PVN–RVLM neurones displayed consistent SFA with a near linear rate of ISI prolongation (Fig. 3Ca) that was essentially unchanged by SK channel blockade. This suggests that SFA in PVN–RVLM neurons is largely controlled by channels other than SK channels. Consistent with this conclusion was our observation that PVN–RVLM neurones exhibited an obvious sAHP (Fig. 4). Therefore, an apamin-insensitive IsAHP (Vogalis et al. 2003; Teagarden et al. 2008) might be responsible for the SFA in these neurones. Alternatively, M current could be involved (Yue & Yaari, 2004; Gu et al. 2005; Wu et al. 2008), although we are unaware of evidence that PVN–RVLM neurones express KCNQ channels. Whatever channels mediate SFA in PVN–RVLM neurones, it should be emphasized that although SK channel blockade did not alter the rate of ISI prolongation, it did significantly increase current-evoked discharge frequency overall. Therefore, we would speculate that negative modulation or loss of SK channel function in vivo could lead to increased discharge of PVN–RVLM neurons.
Another observation in the present study was that blocking SK channels increased sub-threshold depolarizing Rinput without altering action potential threshold. Together these effects increased the rate of depolarization and reduced the spike latency. The mechanism responsible for increasing depolarizing Rinput during SK channel blockade is not known. It seems likely, however, that block of SK channels is a direct contributing factor since our data indicate that they activate at membrane potentials between resting Vm and spike threshold. The fact that blockade of SK channels had no effect on resting Vm supports the view that they are activated by the increase in calcium influx through voltage-dependent calcium channels during depolarization.
It should be mentioned that the excitability of cells treated with apamin was greater than cells treated with UCL1684 even though the difference did not reach statistical significance. The explanation for this could be that the concentration of UCL1684 we used was lower than that of apamin owing to the former being less soluble in aqueous solution.
Resting Vm and spontaneous discharge are unaffected by SK channel blockade
In GnRH neurones (Liu & Herbison, 2008) and suprachiasmatic nucleus neurones (Teshima et al. 2003), SK channels have been reported to contribute to resting Vm and to restrain in vitro spontaneous discharge. In this study, SK channel blockade had no effect on either resting Vm or spontaneous discharge of PVN–RVLM neurones. Our findings are consistent with those of Li & Pan (2006) who showed that even a 500 nm concentration of apamin failed to alter spontaneous firing of PVN–RVLM neurones. Collectively, these findings indicate that SK channels are not tonically active in PVN–RVLM neurons at resting Vm in brain slices. It is not known if the same holds true for neurons in vivo. In brain slices, extrinsic synaptic inputs are eliminated and therefore loss of excitatory inputs could reduce Ca2+ influx, thereby causing in vitro activity of SK channels to be artificially low. In the case of PVN–RVLM neurones, eliminating glutamatergic input from the forebrain (Grob et al. 2003) and other hypothalamic nuclei (Boudaba et al. 1997; Csáki et al. 2000) would reduce Ca2+ influx through NMDA receptors (Ngo-Anh et al. 2005) and possibly through low- and high-voltage activated Ca2+ channels, thereby possibly preventing tonic activation of SK channels.
In the present study all but two neurones were quiescent and remained so during SK channel blockade. By contrast, previous slice studies reported that most PVN–RVLM neurones fired spontaneously and had values of resting Vm that were depolarized compared to those in the present study (Cato & Toney, 2005; Li & Pan, 2006; Park et al. 2007). These discrepancies are likely to be explained by the fact that the previous studies were carried out during blockade of glutamatergic and GABAergic inputs. Indeed, GABA-A receptor blockade with bicuculline methiodide (BMI) has been reported to depolarize Vm of PVN–RVLM neurones and to increase their in vitro firing rate (Li & Pan, 2006) and their excitability (Park et al. 2007). Given that the GABA-A receptor antagonist BMI can also block SK channels, it is possible that effects could have involved actions at both sites. However, the lack of effect of apamin and UCL1684 to change resting Vm or to induce discharge in the present study suggests that previously reported actions of BMI are most likely to have been due to blockade of GABA-A receptors. Consistent with this, the present study found that resting Vm was relatively depolarized (−53 mV) when recordings were performed in the presence of another GABA-A receptor antagonist, picrotoxin (see Fig. 6).
ImAHP in PVN–RVLM neurones is mediated by SK channels
Large amplitude and long duration AHPs, including the mAHP, develop as a consequence of intracellular Ca2+ accumulation during repetitive action potential firing (Stocker, 2004; Bond et al. 2005). Interestingly, blockade of SK channels in the present study not only reduced the amplitude of the mAHP, but revealed the presence of an ADP. Thus, it appears that opposing actions of the mAHP and the ADP normally participate in setting the rate of firing in response to stimulation. Typically, voltage-dependent Ca2+ influx activates Ca2+-dependent K+ conductance that underlies AHPs. In addition to inhibiting neuronal firing, depolarization and Ca2+ influx can also increase firing rate through activation of a non-selective cation current (Ghamari-Langroudi & Bourque, 2002; Teruyama & Armstrong, 2007). The current underlying the ADP has been proposed to be a Ca2+-activated current rather than a Ca2+ current per se as it is abolished by strongly buffering intracellular Ca2+ with BAPTA (Teruyama & Armstrong, 2007). Consistent with our findings, blockade of SK channels with apamin has been reported to also reveal an ADP and increase the firing of gonadotropin-releasing hormone neurones (Liu & Herbison, 2008) and neocortical layer pyramidal neurones (Higgs & Spain, 2009). These findings indicate that the ability of SK channels to limit excitability of PVN–RVLM neurones is likely to result from their role in evoking/modulating a mAHP that oppose the ADP in these neurones.
Blockade of SK channels does not alter glutamatergic activity in PVN–RVLM neurones
It should be emphasized that although we observed a prominent SK current in PVN–RVLM neurones, increased excitability resulting from bath application of SK channel blockers may not be entirely due to postsynaptic actions. We therefore investigated whether bath application of apamin was accompanied by increased excitatory synaptic input that might also contribute to increased excitability. Such seems unlikely given our observation of increased depolarizing Rinput in the presence of apamin. Nevertheless, SK3 channels have been localized to nerve terminals in cultured hippocampal neurones (Obermair et al. 2003) and at the neuromuscular junction (Roncarati et al. 2001), and this raises at least the possibility that they modulate terminal release of transmitter. However, apamin treatment did not affect the frequency, amplitude or decay time constant of mEPSCs recorded from PVN–RVLM neurones. Therefore it seems most likely that SK channels expressed postsynptically in PVN–RVLM neurones are the main site at which blockers acted in this study to increase excitability.
Conclusions
In summary, findings from this study strongly suggest that SK channels play an important role in regulating excitability of PVN–RVLM neurones. Blockade of SK channels increases in vitro spike discharge of PVN–RVLM neurones in response to current injections. The mechanism appears to involve both an increase in the depolarizing Rinput below spike threshold and reduced mAHP during repetitive action potential firing. The latter may oppose a prominent ADP expressed in these cells.
When SK channels of PVN–RVLM neurones are blocked, how does this affect the activity of downstream neurones in the RVLM? This will be an important question to answer in order to understand how regulation of this pathway affects sympathetic nervous system outflow under physiological and disease conditions. It has been reported that activation of the PVN increases the discharge of bulbospinal vasomotor neurones in the RVLM (Yang & Coote, 1998; Yang et al. 2001) and a recent study reported that water deprivation induced activation of a prominent glutamatergic pathway from the PVN to the RVLM (Stocker et al. 2006). Considering the fact that blockade of SK channels in PVN–RVLM neurones increased their excitability, we speculate that negative modulation or loss of SK channel function in these neurones could increase glutamatergic activation of RVLM vasomotor neurones. Such an effect could play an important role in PVN-mediated activation of sympathetic vasomotor tone that occurs under physiological conditions (Stocker et al. 2005; Cruz et al. 2008; Reddy et al. 2007), in metabolic disorders (Perin et al. 2001; Zheng et al. 2006; Stocker et al. 2007) and in cardiovascular diseases (Zucker et al. 2001; Felder et al. 2001; Sonner et al. 2008).
Acknowledgments
This study was funded by NIH grants HL-076312 (GMT) and HL071645 (GMT) and American Heart Association grant AHA0865107F (QHC). We thank Dr Robert Brenner for critically reviewing the manuscript. The authors gratefully acknowledge Mary Ann Andrade and Alfredo S. Calderon for technical assistance.
Glossary
Abbreviations
- ADP
after-depolarization potential
- AHP
after-hyperpolarization potential
- 8CPT-cAMP
cAMP analogue 8-(4-chlorophenylthio) 3′,5′-cyclic adenosine monophosphate
- ISI
inter-spike interval
- mEPSC
miniature excitatory postsynaptic current
- PVN
paraventricular nucleus
- RVLM
rostral ventrolateral medulla
- SFA
spike-frequency adaptation
- SK
small-conductance Ca2+-activated K+
- SNA
sympathetic nerve activity
Author contributions
Q.-H.C. designed and performed the research, analysed and interpreted data and wrote the manuscript. G.M.T. interpreted data and revised the manuscript. These experiments were performed in the Department of Physiology, University of Texas Health Science Center at San Antonio.
References
- Akine A, Montanaro M, Allen AM. Hypothalamic paraventricular nucleus inhibition decreases renal sympathetic nerve activity in hypertensive and normotensive rats. Auton Neurosci. 2003;108:17–21. doi: 10.1016/j.autneu.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Allen AM. Inhibition of the hypothalamic paraventricular nucleus in spontaneously hypertensive rats dramatically reduces sympathetic vasomotor tone. Hypertension. 2002;39:275–280. doi: 10.1161/hy0202.104272. [DOI] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP. SK channels in excitability, pacemaking and synaptic integration. Curr Opin Neurobiol. 2005;15:305–311. doi: 10.1016/j.conb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Schrader LA, Tasker JG. Physiological evidence for local excitatory synaptic circuits in the rat hypothalamus. J Neurophysiol. 1997;77:3396–3400. doi: 10.1152/jn.1997.77.6.3396. [DOI] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- Cato MJ, Toney GM. Angiotensin II excites paraventricular nucleus neurons that innervate the rostral ventrolateral medulla: an in vitro patch-clamp study in brain slices. J Neurophysiol. 2005;93:403–413. doi: 10.1152/jn.01055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QH, Toney GM. Discharge properties and osmotic responsiveness of hyperthalamic PVN neurons projecting to the rostral ventrolateral medulla (abstract) FASEB J. 2000;14:A626. [Google Scholar]
- Chen QH, Toney GM. Identification and characterization of two functionally distinct groups of spinal cord-projecting paraventricular nucleus neurons with sympathetic-related activity. Neuroscience. 2003;118:797–807. doi: 10.1016/s0306-4522(03)00033-2. [DOI] [PubMed] [Google Scholar]
- Chen QH, Haywood JR, Toney GM. Sympathoexcitation by PVN-injected bicuculline requires activation of excitatory amino acid receptors. Hypertension. 2003;42:725–731. doi: 10.1161/01.HYP.0000085197.20043.44. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Bonagamba LG, Machado BH, Biancardi VC, Stern JE. Intermittent activation of peripheral chemoreceptors in awake rats induces Fos expression in rostral ventrolateral medulla-projecting neurons in the paraventricular nucleus of the hypothalamus. Neuroscience. 2008;157:463–472. doi: 10.1016/j.neuroscience.2008.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csáki A, Kocsis K, Halász B, Kiss J. Localization of glutamatergic/aspartatergic neurons projecting to the hypothalamic paraventricular nucleus studied by retrograde transport of [3H]D-aspartate autoradiography. Neuroscience. 2000;101:637–655. doi: 10.1016/s0306-4522(00)00411-5. [DOI] [PubMed] [Google Scholar]
- Jenkinson DH. Potassium channels – multiplicity and challenges. Br J Pharmacol. 2006;147:S63–S71. doi: 10.1038/sj.bjp.0706447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder RB, Francis J, Weiss RM, Zhang ZH, Wei SG, Johnson AK. Neurohumoral regulation in ischemia-induced heart failure. Role of the forebrain. Ann N Y Acad Sci. 2001;940:444–453. doi: 10.1111/j.1749-6632.2001.tb03697.x. [DOI] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Bourque CW. Flufenamic acid blocks depolarizing afterpotentials and phasic firing in rat supraoptic neurones. J Physiol. 2002;545:537–542. doi: 10.1113/jphysiol.2002.033589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob M, Trottier JF, Drolet G, Mouginot D. Characterization of the neurochemical content of neuronal populations of the lamina terminalis activated by acute hydromineral challenge. Neuroscience. 2003;122:247–257. doi: 10.1016/j.neuroscience.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Haywood JR, Mifflin SW, Craig T, Calderon A, Hensler JG, Hinojosa-Laborde C. γ-Aminobutyric acid (GABA)-A function and binding in the paraventricular nucleus of the hypothalamus in chronic renal-wrap hypertension. Hypertension. 2001;37:614–618. doi: 10.1161/01.hyp.37.2.614. [DOI] [PubMed] [Google Scholar]
- Higgs MH, Spain WJ. Conditional bursting enhances resonant firing in neocortical layer 2–3 pyramidal neurons. J Neurosci. 2009;29:1285–1299. doi: 10.1523/JNEUROSCI.3728-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney MJ, Weiss ML, Patel KP, Wang Y, Fels RJ. Paraventricular nucleus bicuculline alters frequency components of sympathetic nerve discharge bursts. Am J Physiol Heart Circ Physiol. 2001;281:H1233–1241. doi: 10.1152/ajpheart.2001.281.3.H1233. [DOI] [PubMed] [Google Scholar]
- Kleiber AC, Zheng H, Schultz HD, Peuler JD, Patel KP. Exercise training normalizes enhanced glutamate-mediated sympathetic activation from the PVN in heart failure. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1863–1872. doi: 10.1152/ajpregu.00757.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Pan HL. Plasticity of GABAergic control of hypothalamic presympathetic neurons in hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H1110–1119. doi: 10.1152/ajpheart.00788.2005. [DOI] [PubMed] [Google Scholar]
- Li DP, Pan HL. Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension. 2007;49:916–925. doi: 10.1161/01.HYP.0000259666.99449.74. [DOI] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM, Pan HL. Pre- and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J Physiol. 2008;586:1637–1647. doi: 10.1113/jphysiol.2007.149732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Herbison AE. Small-conductance calcium-activated potassium channels control excitability and firing dynamics in gonadotropin-releasing hormone (GnRH) neurons. Endocrinology. 2008;149:3598–3604. doi: 10.1210/en.2007-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovick TA, Coote JH. Electrophysiological properties of paraventriculo-spinal neurones in the rat. Brain Res. 1988;454:123–130. doi: 10.1016/0006-8993(88)90810-4. [DOI] [PubMed] [Google Scholar]
- Martin DS, Segura T, Haywood JR. Cardiovascular responses to bicuculline in the paraventricular nucleus of the rat. Hypertension. 1991;18:48–55. doi: 10.1161/01.hyp.18.1.48. [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Obermair GJ, Kaufmann WA, Knaus HG, Flucher BE. The small conductance Ca2+-activated K+ channel SK3 is localized in nerve terminals of excitatory synapses of cultured mouse hippocampal neurons. Eur J Neurosci. 2003;17:721–731. doi: 10.1046/j.1460-9568.2003.02488.x. [DOI] [PubMed] [Google Scholar]
- Pakhomov AG, Semenov I, Brenner R, Toney GM. Hydraulically coupled microejection technique for precise local solution delivery in tissues. J Neurosci Methods. 2006;155:231–240. doi: 10.1016/j.jneumeth.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Park JB, Skalska S, Son S, Stern JE. Dual GABAA receptor-mediated inhibition in rat presympathetic paraventricular nucleus neurons. J Physiol. 2007;582:539–551. doi: 10.1113/jphysiol.2007.133223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Kulik A, Muller M, Ballanyi K, Stocker M. Molecular determinants of Ca2+-dependent K+ channel function in rat dorsal vagal neurones. J Physiol. 2000;527:283–290. doi: 10.1111/j.1469-7793.2000.t01-1-00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin PC, Maule S, Quadri R. Sympathetic nervous system, diabetes, and hypertension. Clin Exp Hypertens. 2001;23:45–55. doi: 10.1081/ceh-100001196. [DOI] [PubMed] [Google Scholar]
- Porter JP, Brody MJ. Neural projections from paraventricular nucleus that subserve vasomotor functions. Am J Physiol Regul Integr Comp Physiol. 1985;248:R271–281. doi: 10.1152/ajpregu.1985.248.3.R271. [DOI] [PubMed] [Google Scholar]
- Reddy MK, Schultz HD, Zheng H, Patel KP. Altered nitric oxide mechanism within the paraventricular nucleus contributes to the augmented carotid body chemoreflex in heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H149–157. doi: 10.1152/ajpheart.00117.2006. [DOI] [PubMed] [Google Scholar]
- Roncarati R, Di Chio M, Sava A, Terstappen GC, Fumagalli G. Presynaptic localization of the small conductance calcium-activated potassium channel SK3 at the neuromuscular junction. Neuroscience. 2001;104:253–262. doi: 10.1016/s0306-4522(01)00066-5. [DOI] [PubMed] [Google Scholar]
- Sonner PM, Stern JE. Functional role of A-type potassium currents in rat presympathetic PVN neurones. J Physiol. 2007;582:219–238. doi: 10.1113/jphysiol.2007.134379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonner PM, Filosa JA, Stern JE. Diminished A-type potassium current and altered firing properties in presympathetic PVN neurones in renovascular hypertensive rats. J Physiol. 2008;586:1605–1622. doi: 10.1113/jphysiol.2007.147413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JE. Electrophysiological and morphological properties of pre-autonomic neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2001;537:161–177. doi: 10.1111/j.1469-7793.2001.0161k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M. Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci. 2004;5:758–770. doi: 10.1038/nrn1516. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker SD, Cunningham JT, Toney GM. Water deprivation increases Fos immunoreactivity in PVN autonomic neurons with projections to the spinal cord and rostral ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol. 2004a;287:R1172–1183. doi: 10.1152/ajpregu.00394.2004. [DOI] [PubMed] [Google Scholar]
- Stocker SD, Keith KJ, Toney GM. Acute inhibition of the hypothalamic paraventricular nucleus decreases renal sympathetic nerve activity and arterial blood pressure in water-deprived rats. Am J Physiol Regul Integr Comp Physiol. 2004b;286:R719–725. doi: 10.1152/ajpregu.00494.2003. [DOI] [PubMed] [Google Scholar]
- Stocker SD, Hunwick KJ, Toney GM. Hypothalamic paraventricular nucleus differentially supports lumbar and renal sympathetic outflow in water-deprived rats. J Physiol. 2005;563:249–263. doi: 10.1113/jphysiol.2004.076661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker SD, Simmons JR, Stornetta RL, Toney GM, Guyenet PG. Water deprivation activates a glutamatergic projection from the hypothalamic paraventricular nucleus to the rostral ventrolateral medulla. J Comp Neurol. 2006;494:673–685. doi: 10.1002/cne.20835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker SD, Meador R, Adams JM. Neurons of the rostral ventrolateral medulla contribute to obesity-induced hypertension in rats. Hypertension. 2007;49:640–646. doi: 10.1161/01.HYP.0000254828.71253.dc. [DOI] [PubMed] [Google Scholar]
- Tasker JG, Dudek FE. Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J Physiol. 1991;434:271–293. doi: 10.1113/jphysiol.1991.sp018469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teagarden M, Atherton JF, Bevan MD, Wilson CJ. Accumulation of cytoplasmic calcium, but not apamin-sensitive afterhyperpolarization current, during high frequency firing in rat subthalamic nucleus cells. J Physiol. 2008;586:817–833. doi: 10.1113/jphysiol.2007.141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruyama R, Armstrong WE. Calcium-dependent fast depolarizing afterpotentials in vasopressin neurons in the rat supraoptic nucleus. J Neurophysiol. 2007;98:2612–2621. doi: 10.1152/jn.00599.2007. [DOI] [PubMed] [Google Scholar]
- Teshima K, Kim SH, Allen CN. Characterization of an apamin-sensitive potassium current in suprachiasmatic nucleus neurons. Neuroscience. 2003;120:65–73. doi: 10.1016/s0306-4522(03)00270-7. [DOI] [PubMed] [Google Scholar]
- Toney GM, Chen QH, Cato MJ, Stocker SD. Central osmotic regulation of sympathetic nerve activity. Acta Physiol Scand. 2003;177:43–55. doi: 10.1046/j.1365-201X.2003.01046.x. [DOI] [PubMed] [Google Scholar]
- Villalobos C, Shakkottai VG, Chandy KG, Michelhaugh SK, Andrade R. SKCa channels mediate the medium but not the slow calcium-activated afterhyperpolarization in cortical neurons. J Neurosci. 2004;24:3537–3542. doi: 10.1523/JNEUROSCI.0380-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogalis F, Storm JF, Lancaster B. SK channels and the varieties of slow after-hyperpolarizations in neurons. Eur J Neurosci. 2003;18:3155–3166. doi: 10.1111/j.1460-9568.2003.03040.x. [DOI] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Surmeier DJ, Disterhoft JF. Coupling of L-type Ca2+ channels to KV7/KCNQ channels creates a novel, activity-dependent, homeostatic intrinsic plasticity. J Neurophysiol. 2008;100:1897–1908. doi: 10.1152/jn.90346.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Coote JH. Influence of the hypothalamic paraventricular nucleus on cardiovascular neurons in the rostral ventrolateral medulla of the rat. J Physiol. 1998;513:521–530. doi: 10.1111/j.1469-7793.1998.521bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Bertram D, Coote JH. The role of glutamate and vasopressin in the excitation of RVL neurones by paraventricular neurones. Brain Res. 2001;908:99–103. doi: 10.1016/s0006-8993(01)02593-8. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Mayhan WG, Bidasee KR, Patel KP. Blunted nitric oxide-mediated inhibition of sympathetic nerve activity within the paraventricular nucleus in diabetic rats. Am J Physiol Regul Integr Comp Physiol. 2006;290:R992–1002. doi: 10.1152/ajpregu.00363.2005. [DOI] [PubMed] [Google Scholar]
- Zucker IH, Wang W, Pliquett RU, Liu JL, Patel KP. The regulation of sympathetic outflow in heart failure. The roles of angiotensin II, nitric oxide, and exercise training. Ann N Y Acad Sci. 2001;940:431–443. [PubMed] [Google Scholar]