

Abstract
Physiological responses to chronic hypoxia include polycythemia, pulmonary arterial remodeling and vasoconstriction. Chronic hypoxia causes pulmonary arterial hypertension leading to right ventricular hypertrophy and heart failure. During pulmonary hypertension, pulmonary arteries exhibit increased expression of smooth muscle-α-actin and -myosin heavy chain. NFATc3 (nuclear factor of activated T cells isoform c3), which is a Ca2+-dependent transcription factor, has been recently linked to smooth muscle phenotypic maintenance through the regulation of the expression of α-actin. The aim of this study was to determine if: a) NFATc3 is expressed in murine pulmonary arteries, b) hypoxia induces NFAT activation, c) NFATc3 mediates the up-regulation of α-actin during chronic hypoxia, and d) NFATc3 is involved in chronic hypoxia-induced pulmonary vascular remodeling. NFATc3 transcript and protein were found in pulmonary arteries. NFAT-luciferase reporter mice were exposed to normoxia (630 torr) or hypoxia (380 torr) for 2, 7 or 21 days. Exposure to hypoxia elicited a significant increase in luciferase activity and pulmonary arterial smooth muscle nuclear NFATc3 localization, demonstrating NFAT activation. Hypoxia induced up-regulation of α-actin and was prevented by the calcineurin/NFAT inhibitor, cyclosporin A (25 mg/Kg/day s.c.). In addition, NFATc3 knockout mice did not showed increased α-actin levels and arterial wall thickness after hypoxia. These results strongly suggest that NFATc3 plays a role in the chronic hypoxia-induced vascular changes that underlie pulmonary hypertension.
As altitude increases, the barometric pressure and atmospheric oxygen partial pressure decrease. This decrease in barometric pressure is the basic cause of all hypoxia-related problems in high-altitude pathophysiology. Similar levels of hypoxia are present in patients with chronic bronchitis, emphysema, cystic fibrosis, asthma and severe restrictive lung diseases (1). Chronic hypoxia (CH1) causes pulmonary hypertension due to pulmonary vasoconstriction, arterial remodeling, and polycythemia which ultimately results in right ventricular (RV) hypertrophy and often heart failure (2).
Pulmonary vasoconstriction is thought to be caused by elevated vascular tone through increased pulmonary arterial smooth muscle cell (PASMC) intracellular Ca2+ ([Ca2+]i) (3–8) and increased sensitivity of the contractile apparatus to Ca2+ (9–12).
Regardless of the cause of pulmonary hypertension, the structural change that is thought to underlie the increased vascular resistance is remodeling of small pulmonary arteries. A prominent feature of this vascular remodeling is medial thickening. In proximal pulmonary vessels, medial enlargement is caused by hypertrophy and hyperplasia of the pre-existing smooth muscle cells [reviewed in (1)]. In addition, differentiation of adventitial fibroblasts into myofibroblasts contributes to medial thickening (13).
Smooth muscle is phenotypically dynamic and maintains its differentiated phenotype through the regulated expression of a repertoire of SM-specific genes [reviewed in (14–17)]. NFAT (nuclear factor of activated T cells), a Ca2+-dependent transcription factor that regulates the expression of genes in both immune and non-immune cells (18;19), has been recently linked to smooth muscle phenotypic maintenance (20–25). NFAT appears to regulate the expression of SM-myosin heavy chain, SM-α-actin, α1 integrin and caldesmon genes (21;22). Recently, we demonstrated that serum response factor (SRF) and NFATc3 cooperatively enhance the expression of SM-α-actin in cultured aortic smooth muscle cells (24). NFATc3 and SRF bind to a region of the first intron of the SM-α-actin gene (24).
SM-α-actin is required for the high force development properties of smooth muscle cells and is the most abundant protein in differentiated SM making up to 40% of total cell protein (26). In adults, expression of SM-α-actin is restricted and is generally activated upon terminal differentiation in cells of myogenic lineage correlating with hypertrophy, whereas increases in non-muscle actins (β- and γ-actin) coincide with cell growth and proliferation (14).
The NFAT family consists of four members (NFATc1, NFATc2, NFATc3, NFATc4) that share the property of Ca2+/calcineurin-dependent nuclear translocation, and a fifth member—NFAT5—which is Ca2+-independent and shares limited homology with the other family members [reviewed in (19;27;28)].
The NFATc3 isoform is specifically implicated in vasculature development (20;25), maintenance of a contractile phenotype (24) and regulation of vascular smooth muscle cell (VSMC) contractility (29).
In smooth muscle, NFAT is activated by Gq/11-coupled receptor agonists, such as uridine triphosphate, endothelin 1 (ET-1) and angiotensin II (Ang II) (23;30–32). This activation is mediated by calcineurin and is dependent on both sarcoplasmic reticulum Ca2+ release through inositol trisphosphate receptors (IP3R) and extracellular Ca2+ influx through voltage-dependent Ca2+ channels (VDCC) (31). Interestingly, during CH, PASMC exhibit increased expression of SM-α-actin (33;34) consistent with PASMC hypertrophy and vascular remodeling (35). In addition, it has been shown that angiotensin II (Ang II), which is elevated during CH (36–38), induces stimulation of SM-α-actin expression at the transcriptional level through a SRF-dependent mechanism in cultured VSMC (36–39). It is thus reasonable to hypothesize that in response to CH, NFAT is activated in PASMC leading to the up-regulation of the SM-α-actin contractile protein and vascular remodeling.
Given the lack of evidence demonstrating hypoxia-induced NFAT regulation of gene transcription, the aims of the present study were to determine if: a) NFATc3 is expressed in murine pulmonary arteries, b) CH induces NFATc3 activation, c) NFATc3 mediates the up-regulation of α-actin during CH, and d) NFATc3 is involved in CH-induced pulmonary vascular remodeling.
In support of this hypothesis, our data demonstrate that CH indeed increases NFAT transcriptional activity and NFATc3 nuclear translocation in mouse pulmonary arteries. Furthermore, pharmacological inhibition of calcineurin activation of NFAT or genetic ablation of NFATc3 prevents CH-induced increases in α-actin expression, arterial wall thickness and right ventricular (RV) hypertrophy. These results suggest that NFATc3 plays a role in the vascular changes observed during CH-induced pulmonary hypertension.
EXPERIMENTAL PROCEDURES
All protocols employed in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico, School of Medicine (Albuquerque, NM).
Animals
Adult male 9x-NFAT-luciferase reporter (NFAT-luc), NFATc3 knockout (NFATc3 KO), BalB/C wild-type (WT) and C57BL/6 (Harland Laboratories) mice (20–25 g) were used. In addition, NFAT-luc were backcrossed with NFATc3 KO for, at least, two generations and NFAT-luc/NFATc3 +/+ (WT), NFAT-luc/NFATc3 +/− (HET) and NFAT-luc/NFATc3 −/− (KO) were used. NFAT-luc mice have been provided by Dr. Jeffery D. Molkentin (Department of Pediatrics, Children’s Hospital Medical Center, Cincinnati, Ohio) (40). NFATc3 KO mice have been kindly provided by Dr. Laurie Glimcher (Harvard University) (41). Heterozygous mice were bred to obtain age-matched WT (Balb/C) and KO. Importantly, it has been shown that the loss of NFATc3 isoform does not induce a compensatory regulation of other NFAT isoforms (42).
C57B6 mice were s.c. administered 25 mg/kg/day of cyclosporin A (CsA, Calbiochem) or Cremophor EL (vehicle) for 1 week.
Chronic hypoxia exposure
Animals designated for exposure to CH were housed in a hypobaric chamber with barometric pressure maintained at ~380 mmHg for 2, 7 or 21 days. The chamber was opened one time per week to provide animals with fresh food, water, and clean bedding. Control animals were housed at ambient barometric pressure (Normoxia, N, ~630 mmHg). All animals were maintained on a 12:12-h light-dark cycle.
Assessment of polycythemia and RV hypertrophy
Blood samples were obtained by direct cardiac puncture at the time of lung isolation for measurement of hematocrit. RV hypertrophy was assessed as an index of CH-induced pulmonary hypertension with previously described methods (43;44). Briefly, after isolation of the heart, the atria and major vessels were removed from the ventricles. The RV was dissected from the left ventricle and septum, and each was weighed. The degree of RV hypertrophy was expressed as the ratio of RV to total ventricle weight(T).
Vascular Morphometry
Animals were anesthetized with 5% Isoflurane in O2 and perfused via the left ventricle with ~20 ml of modified physiological saline solution (HEPES-PSS, 134 mM NaCl, 6 mM KCl, 1 mM MgCl, 10 mM HEPES, 2 mM CaCl2, 0.026 mM EDTA and 10 mM glucose) containing heparin, 4% albumin (Sigma) and 10−4 M papaverine (Sigma), at a rate close to normal cardiac output in mice (17 ml/minute) to maximally dilate and flush the circulation of blood. Then, mice were perfused with 4% formaldehyde (Polyscience) and 10−4 M papaverine (Sigma) in PBS at the same rate. Following fixation, the lungs were inflated via the trachea to a pressure of 23 cmH2O with fixative. The tissue was then dehydrated in increasing concentrations of ethanol, with a final dehydration in xylene, and then embedded in paraffin. Lung sections (5 μm) were stained with hematoxylin/iodine/eosin (Van Gieson staining, Sigma) which stains nuclei, elastic laminae, collagen fibers and cellular cytosol to distinguish arteries from veins. A total of 1291 arteries from 13 mice were analyzed. Vessels were examined with a ×40 objective on a Nikon Diaphot 300 microscope, and images were generated with a cooled digital CCD camera (Photometrics SenSys 1400). Vessel images were processed with Metamorph imaging system hardware and software (Universal Imaging). Vessels sectioned at oblique angles were excluded fromanalysis. Media and wall thickness were measured and compared between groups using the following equation: Wall thickness (%) = perimeterext − perimeterint/perimeterext × 100, where perimeterext and perimeterint are the circumferences bounded by external and internal elastic laminae, respectively (45).
Luciferase activity
NFAT-luc mice were euthanized with an overdose of pentobarbital solution (200 mg/kg i.p.). Following euthanasia lungs, brain and small intestines were removed and placed into ice-cold HEPES-PSS. Aorta, intrapulmonary, cerebral and mesenteric arteries were isolated and lysed using tissue lysis buffer from Promega according to manufacturer’s protocol. Lysate was centrifuged for 10 min at 10,000 rcf. Both luciferase activity and protein content were determined in the supernatant. Luciferase activity was measured using a Luciferase Assay System kit (Promega), and light detected with a luminometer (TD20/20, Turner). Protein content was determined by the Bradford method (BioRad) and used to normalize luciferase activity per sample.
Immunofluorescence confocal microscopy
Isolated lungs were formaldehyde fixed (4% in phosphate buffer saline, PBS), cryoprotected with 30 % sucrose in PBS, embedded in OCT media and frozen. Cryostat sections (10 μm) were permeabilized and blocked for nonspecific binding, primary antibodies [rabbit polyclonal anti-NFATc3 (1:100); Santa Cruz and anti-α-actin (1:250); Sigma] were prepared in 0.2% gelatin in PBS and applied overnight at 4°C. Secondary antibodies [anti-rabbit Cy5 and anti-mouse Cy3 (1:500); Jackson Immunoresearch Laboratories] were prepared in 0.2% gelatin in PBS and applied for one hour at room temperature. Nuclei were stained using SYTOX green (1:5,000 in PBS; Molecular Probes). Sections were examined using a 40X objective on a Zeiss 510 laser scanning confocal microscope. Specificity of immune staining was confirmed by the absence of fluorescence in tissues incubated with primary or secondary antibodies alone. For scoring of NFATc3-positive nuclei, multiple fields for each vessel were imaged and counted by two independent observers using Metamorph software (Universal Imaging Corp). The software was programmed so that individual pixels appear white instead of yellow if the green nucleic acid stain and red NFATc3 stain co-localized. Thus, a cell was considered positive if co-localization (white) was uniformly distributed in the nucleus and negative if no co-localization (green only) was observed.
Western blot analysis
Isolated intrapulmonary arteries were homogenized in lysis buffer (10 mM tris-HCl pH 7.4, 1 mM EDTA, 1% IGEPAL, 0.1% sodium deoxycholate, 500 nM sodium orthovanadate, 50 nM NaF, 1 μg/ml pepstatin/leupeptin/aprotinin, 1 mM phenylmethtylsulfonyl fluoride) at 4°C and centrifuged at 16,000 rcf for 2 min. Protein concentration was determined in the supernatant using Bradford’s method (BioRad) as recommended by the manufacturer. To perform Western blot analysis, supernatants (2 μg/lane) were resolved by SDS-PAGE and proteins transferred to PVDF membranes. The membranes were blocked with Tris-buffered (50 mM) saline solution (pH 7.6) with 0.05% Tween containing 5% milk powder at room temperature for 1 h. The membranes were incubated with primary anti SM-α-actin antibody (1:5000, Sigma) or anti β-actin antibody (1:5000) at 4°C overnight, washed and incubated with a peroxidase-conjugated secondary antibody (Pierce) for 1 h at RT. Specifically-bound antibody was detected by enhanced chemiluminescence detection (ECL, Pierce). Relative content of the antigen protein was evaluated using a Gene Gnome imaging system and Genesnap software (Syngene, Cambridge, UK). Band densities were normalized to total protein loaded per lane as determined by Coomasie blue or Sypro Red II staining of the membrane (NIH Image software).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Intrapulmonary arteries were stored in RNAlater (Ambion). Total RNA was isolated using the RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol, with the additional steps of DNase treatment (DNase turbo from Ambion) and RNeasy MinElute Cleanup Kit (Qiagen) to remove possible genomic DNA contamination. Total RNA was reverse transcribed to cDNA using TaqMan Reverse Transcription kit (A&B). For real time detection of SM-α-actin transcripts (Mm01546133_mi) and reference gene (18S, 4319413E-0502018), TaqMan Gene Expression Assays (A&B) were used. PCR was performed using Applied Biosystems 7500 Fast Real-Time PCR System. The normalized gene expression method (2−ΔCT) for relative quantification of gene expression was used (46).
Qualitative RT-PCR
Total RNA from intrapulmonary arteries (isolated as mentioned above) was reverse transcribed (IScript, cDNA synthesis Kit, Biorad). PCR was performed using the iTaq DNA polymerase (Biorad) with the following sets of primer pairs: for NFATc3, 5′-CTACTGGTGGCCATCCTGTTGT-3′ and 5′-AGCTCGTGGGCAGAGCGCTGAGAGCACTC-3′; and for cyclophilin, 5′-CAGACAAAGTTCCAAAGACA-3′ and 5′-AGTTCGACCGTCTTCTCAGC-3′. Amplification conditions were 94 °C for 10 min, 45 cycles at 94 °C for 1 min, 55 °C for 1 min, 72 °C for 2 min, and extension for 10 min at 72 °C. Amplified PCR products were separated by agarose gel electrophoresis and detected by ethidium bromide staining.
Statistical analysis
Data were expressed as mean ± SEM. Statistical significance was tested at 95 % (p<0.05) confidence level using unpaired t-test, one-way ANOVA followed by Newman-Keuls multiple comparisons test or two-way ANOVA followed by Bonferroni multiple comparisons test.
RESULTS
NFATc3 isoform is expressed in mouse pulmonary arteries
We have used RT-PCR analysis, immunoblotting and immunofluorescence confocal microscopy to identify NFATc3 expression in native pulmonary arteries isolated from mice. We found transcripts for NFATc3 (Fig. 1A) and protein by western blot (Fig. 1B). NFATc3 immunofluorescence staining was mainly localized in PASMC (3A).
Fig. 1.

NFATc3 is expressed in mouse pulmonary arteries. A. RT-PCR analysis of NFATc3 expression in isolated intrapulmonary arteries from 9xNFAT-luc mice. Lane 1 shows amplification of NFATc3. Amplification of cyclophilin (control) from pulmonary arteries is shown in lane 2. Lane 3 corresponds to a control without the reverse transcription step. The experiment was repeated at least three times and the same bands were detected in pulmonary arteries from all the mouse strains used in this study. B. Western analysis of NFATc3 protein expression in thymus (C57Bl6), pulmonary arteries (9xNFAT-luc), and human Burkitt’s lymphoma-derived Ramos cells (Santa Cruz). Thymus and Ramos cells were used as positive control.
CH increases NFAT activity and NFATc3 nuclear accumulation in pulmonary arteries
Most of the mediators of NFAT activation are up-regulated during CH, including PASMC [Ca2+]i, Ang II (36–38) and endothelin 1 (ET-1) (4;6–8;47;48), although there are no reports of hypoxic activation of NFAT either in vivo or in vitro. Therefore, we examined the effect of different exposure times to CH on NFAT activity in tissues from NFAT-luc reporter mice. We found that CH elicited a significant increase in luciferase activity in pulmonary arteries (Fig. 2A). Interestingly, although luciferase activity decreased in a time dependent manner, it remained significantly elevated over normoxic values even at day 21. No increase in activity was observed in any of the other tissues studied (data not shown). These results for the first time demonstrate that NFAT is activated by CH in pulmonary arteries, suggesting that it is an early response transcription factor under these conditions.
Fig. 2.

NFAT is activated by CH in pulmonary arteries. A. Pulmonary arteries were isolated from 9xNFAT-luc mice exposed to normoxia (N), or to 2, 7 or 21 days of CH. RLU. μg−1= luciferase activity normalized by μg of total protein. *p<0.05 vs. N; #p<0.05 vs. 2 days, n=5 animals. B. Pulmonary arteries were isolated from 9xNFAT-luc/NFATc3 WT, HET and KO mice exposed to 2 days of CH. Luciferase activity was normalized by μg of total protein and expressed as percent change from N WT (average=1.36±0.19). *p<0.01 vs. N WT, #p<0.05 vs. CH WT and HET, n=4–8 animals.
Since NFATc3 is expressed in PASMC, we sought to establish the contribution of NFATc3 to CH-increased NFAT activity. Therefore, we determined the effect of 2 days of hypoxia on reporter expression in pulmonary arteries isolated from NFAT-luc/NFATc3 WT, NFAT-luc/NFATc3 HET and NFAT-luc/NFATc3 KO mice. Consistent with the demonstrated expression of NFATc3 in pulmonary arteries (fig. 1A, 1B and 3A) and the significant contribution of this isoform to SMC function (23;24;29), we found that the reporter response to hypoxia was significantly impaired in pulmonary arteries isolated from NFAT-luc/NFATc3 KO mice (Fig. 2B). As expected, HET arteries displayed an intermediate phenotype. The reporter activity in NFAT-luc/NFATc3 WT arteries after 2 days of hypoxia was similar to the values obtained in CH NFAT-luc arteries (3.03±0.82 vs. 4.62±0.84). Furthermore, the values obtained in the NFAT-luc/NFATc3 KO arteries were similar to the luciferase activity in both NFAT-luc and NFAT-luc/NFATc3 WT arteries under normoxia. These results demonstrate that NFATc3 significantly contributes to CH-induced NFAT transcriptional activity.
Fig. 3.

NFATc3 nuclear accumulation increases in pulmonary arterial smooth muscle cells after CH. A. Representative images showing cytosolic localization of NFATc3 under normoxic (N) conditions and nuclear localization following exposure to CH for 2, 7, or 21 days. Lung sections from 9xNFAT-luc mice were co-stained with the DNA-binding dye SYTOX (green), anti-NFATc3 (red) and anti-SM-α-actin (blue). Pulmonary arterial smooth muscle nuclear co-localization of NFATc3 is shown in white. Arrows indicate examples of NFATc3 positive nuclei. B. Summary of effects of CH on NFATc3 nuclear accumulation over time. *p<0.05 vs. N, #p<0.05 vs. 7 and 21 days, n=14 images from at least 3 animals/group (4 arteries/animal). C. Summary of effects of CsA on CH-induced NFATc3 nuclear accumulation. C57B6 mice were treated with vehicle (V) or CsA (25mg/Kg/day) and exposed to N or 7 days of CH. Lung sections were stained as in A. *p<0.05 vs. N V, #p<0.05 vs. CH V, n= 6 images from at least 3 animals/group (2 arteries/animal).
Consistent with the luciferase activity data, PASMC NFATc3 nuclear accumulation increased significantly at every time point of hypoxia exposure (Fig. 3A and B). However, the highest value of NFAT activity was at day 2 whereas NFATc3 nuclear accumulation was at day 7. Administration of CsA significantly attenuated the CH-induced increase in NFATc3 nuclear accumulation in PASMC (Fig. 3C). In addition, it reduced basal NFATc3 nuclear localization. CsA is an immunosuppressant that inhibits calcineurin, the Ca2+/calmodulin-dependent phosphatase that dephosphorylates NFAT allowing its nuclear import (19).
Calcineurin/NFAT inhibition with CsA and genetic deletion of the NFATc3 isoform prevent CH-induced RV hypertrophy but not polycythemia
RV hypertrophy is a consequence of increased pulmonary arterial pressure and is used as an index of pulmonary hypertension (2). To assess CH induced RV hypertrophy, we determined the change of RV/T weight as a % of normoxic control values. RV hypertrophy was determined in vehicle and CsA-treated C57B6 male mice exposed to 1 week hypobaric CH or normoxia. Greater RV/T ratios were observed for CH vehicle-treated mice compared with normoxic controls (fig. 4A), thus demonstrating RV hypertrophy indicative of pulmonary hypertension. Interestingly, CsA treatment attenuated CH-induced RV hypertrophy (fig. 4A).
Fig. 4.

Calcineurin/NFATc3 mediates CH-induced RV hypertrophy but not polycythemia. A. Percent change in RV/T in vehicle (V)- and CsA-treated and WT and KO mice exposed to 7 and 21 days of CH, respectively, normalized to normoxia (N) values. *p<0.01 vs. V or WT. n= see columns. B. Hematocrit (%) in vehicle (V)- and CsA-treated and WT and KO mice exposed to 7 and 21 days of CH, respectively, normalized to normoxia (N) values. *p<0.01 vs. N, n= see columns.
Since calcineurin can dephosphorylate other cellular proteins (19) and NFATc3 is the isoform that is activated by CH (fig. 2B), we determined changes in RV/T weight in WT and NFATc3 KO exposed to 3 weeks CH or normoxia. As expected, CH induced RV hypertrophy in WT mice, whereas in the KO the hypertrophic response was significantly attenuated (Fig. 4A). These findings suggest that NFATc3 contributes to the development of pulmonary arterial hypertension.
In addition, CH mice exhibited polycythemia as evidenced by a significantly greater hematocrit compared with normoxic animals (fig. 4B). This polycythemic response was similar in vehicle- and CsA-treated and in WT and KO CH mice, demonstrating that NFATc3 is not involved in the polycythemic response to CH.
Calcineurin/NFAT inhibition with CsA and genetic deletion of the NFATc3 isoform prevent CH-induced increases in expression of SM-α-actin in pulmonary arteries
It has been previously demonstrated that CH increases the expression of contractile proteins, such as SM-α-actin, and it mediates PASMC hypertrophy (33). NFATc3 regulates SM-α-actin expression in aortic smooth muscle cells in culture by binding to the intronic region of its promoter (24). To determine if NFAT mediates SM-α-actin up-regulation during CH in vivo, we measured SM-α-actin transcript and protein levels in intrapulmonary arteries isolated from vehicle and CsA-treated C57B6 male mice exposed to 1 week hypobaric CH or normoxia. SM-α-actin transcript (real-time PCR) and protein levels (western blot) significantly increased after 1 week of CH. NFAT inhibition with CsA completely prevented this response (Fig. 5A and B), suggesting that CH-induced activation of NFAT in pulmonary arteries mediates SM-α-actin up-regulation.
Fig. 5.
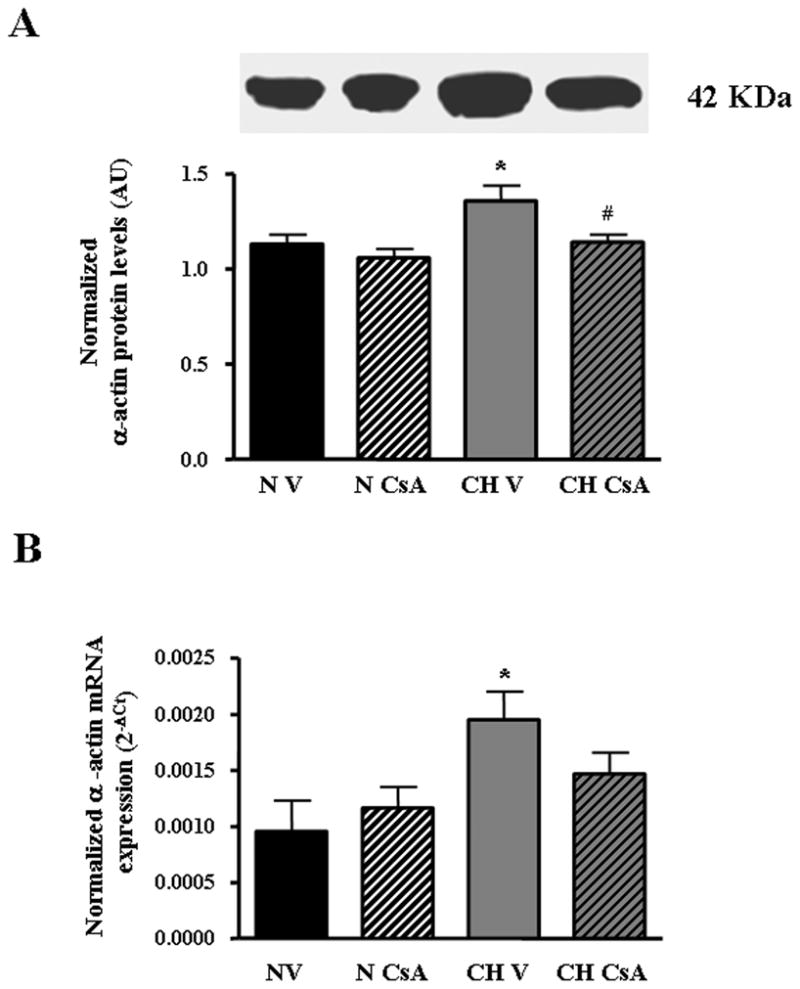
Calcineurin/NFAT mediates CH-induced up-regulation of SM-α-actin in pulmonary arteries. A. Western blot analysis of SM-α-actin protein expression in isolated pulmonary arteries from C57B6 mice treated with vehicle (V) or CsA and exposed to normoxia (N) or CH for 7 days. SM-α-actin band densities were normalized to total protein loaded per lane as determined by Coomasie blue or Sypro Red II staining of the membrane. AU= arbitrary units. *p<0.05 vs. N V, #p<0.05 vs. CH V, n=6 animals, B. Summary of qRT-PCR analysis of SM-α-actin transcript expression in isolated pulmonary arteries from the same animals described in A. Threshold crossing value (CT) was calculated using well factor background subtraction, and gene expression normalized to 18S for each sample. *p<0.05 vs. N V, n=5 animals.
Since calcineurin NFATc3 is the isoform demonstrated to regulate SM-α-actin (24), we evaluated S M-α-actin protein levels (immunoblot) in NFATc3 KO and WT littermates exposed to hypoxia. In contrast to the effect of 1 week CH exposure on α-actin expression in C57BC mice (Fig. 5), a similar period of hypoxic acclimation did not alter α-actin protein levels in NFATc3 WT BalB/C mice (data not shown). However, after 3 weeks of CH, SM-α-actin protein levels significantly increased in pulmonary arteries from CH WT mice, whereas no changes were observed in arteries from CH NFATc3 KO (Fig. 6A). Although the response time appears to be different in BalB/C and C57B6 mice, these findings clearly demonstrate that NFATc3 mediates up-regulation of SM-α-actin during CH. Together with the CsA experiments, these results further demonstrate that CH-induced up-regulation of SM-α-actin is mediated by calcineurin activation.
Fig. 6.

NFATc3 mediates CH-induced up-regulation of SM-α-actin in pulmonary arteries. A. Western blot analysis of SM-α-actin protein expression in isolated pulmonary arteries from WT and NFATc3 KO mice exposed to normoxia (N) or CH for 21 days. SM-α-actin band densities were normalized to total protein loaded per lane as determined by Coomasie blue or Sypro Red II staining of the membrane. AU= arbitrary units. *p<0.05 vs. N WT, #p<0.05 vs. CH WT, n=4 animals. B. Western blot analysis of β-actin protein expression in isolated pulmonary arteries from the same animals described in A. Band densities were normalized to total protein loaded per lane as determined by Sypro Red II staining of the membrane. n=4 animals.
Neither CH nor genetic deletion of NFATc3 isoform affects expression of β-actin in pulmonary arteries
To confirm that NFATc3 is specifically regulating the expression of SM-α-actin during CH and that the effects seen are not simply due to an overall increase in protein synthesis associated with pulmonary arterial remodeling, we determined β-actin protein levels in pulmonary arteries from WT and NFATc3 KO exposed to 3 weeks of CH. β-actin protein levels did not change in any of the groups (Fig. 6B). These results demonstrate that the effect of CH is specific to the contractile protein α-actin and not to the related cytoskeletal protein β-actin.
Genetic deletion of NFATc3 isoform prevents CH-induced increases in arterial wall thickness
The structural changes that are thought to underlie the increased vascular resistance during CH include remodeling of small pulmonary arteries. A prominent feature of vascular remodeling is medial thickening (1). In proximal pulmonary vessels, medial enlargement is caused by hypertrophy and hyperplasia of the pre-existing smooth muscle cells whereas distal arteries that were previously non-muscularized become muscularized (1). Our data suggest that NFATc3 is involved in CH-induced pulmonary arterial remodeling since CH-increased α-actin expression in pulmonary arteries, a marker of smooth muscle differentiation and hypertrophy (17), was prevented by inhibiting calcineurin/NFATc3. To further address this possibility, changes in pulmonary arterial wall thickness were evaluated in WT and KO mice exposed to 3 weeks CH or normoxia. The analysis was performed in arteries from 3 different diameter ranges, 70–150, 151–230, 231–310 and 311–450 μm. As previously demonstrated (49–51), arterial wall thickness increased in CH WT animals. Figures 5A and B show the changes in the % of arterial wall thickness for arterial size range 231–310 μm, similar results were obtained in all size ranges (data not shown). Consistent with our hypothesis, NFATc3 KO did not show increased arterial wall thickness after been exposed to CH. Together with the prevented increase in α-actin expression, these results suggest that NFATc3 is involved in the PASMC hypertrophic response that is, in part, responsible of the arterial remodeling that underlies the development of pulmonary hypertension.
DISCUSSION
The present study examined effects of CH on NFAT activity in pulmonary arteries and determined the role of NFAT in the vascular remodeling that underlies CH-induced pulmonary hypertension.
The major findings of this study are that: 1) NFATc3 transcript and protein are present in murine pulmonary arteries; 2) CH induces an increase in NFAT activity and this activation is impaired in pulmonary arteries from NFAT-luc/NFATc3 KO mice; 3) NFAT activation is followed by an increase in NFATc3 nuclear accumulation in PASMC; 4) pharmacological inhibition of calcineurin/NFAT pathway or genetic deletion of NFATc3 significantly attenuated CH-induced RV hypertrophy, up-regulation of SM-α-actin and increased pulmonary arterial wall thickness. These findings suggest that NFATc3 is involved in the mechanisms that lead to pulmonary hypertension during CH.
NFATc3 is expressed in PASMC
NFATc3 has been previously suggested to play a major role in SM contractile phenotype maintenance through the regulation of the expression of contractile proteins and potassium channels (24;29;52). Therefore, we first investigated if the NFATc3 isoform is expressed in murine pulmonary arteries. Indeed, NFATc3 transcript and protein were detected in isolated intrapulmonary arteries from all the mouse strains used in this study. Interestingly, Yaghi et al recently found that only the NFATc4 isoform is expressed in rat pulmonary arteries (53). However, extensive studies of the cerebral vasculature also revealed the presence of NFATc3 and c4, but not NFATc1 or c2 (23;31;32;54). In contrast, NFATc1, c2 and/or c3 have been described in thoracic aortic smooth muscle cells from both rats and humans (21;55). Recently, NFATc1, c3 and c4 were described in human myometrial arteries, no c2 was detected (56). Thus, NFAT isoform expression in VSMC appears to be somewhat species- and vascular bed-specific but this study clearly shows expression of the NFATc3 isoform in mouse PASMC.
CH induces NFATc3 activation in pulmonary arteries
This study provides evidence for a novel mechanism of NFATc3 activation. Our results demonstrate that CH increases NFAT activity and this activation is prevented in arteries from NFAT-luc/NFATc3 KO. In addition, CH increases NFATc3 nuclear accumulation in PASMC. However, the time course of NFAT activation and NFATc3 accumulation are slightly different. NFAT activity was maximal at day 2 whereas NFATc3 was maximally accumulated in the nucleus of PASMC at day 7. It is possible that other NFAT co-factors, such as AP-1 or GATA that are up-regulated by CH (57;58), are increasing more at day 2, thus enhancing NFATc3 binding to the DNA. Also, it is possible that other NFAT isoforms expressed in PASMC contribute to the highest NFAT activity observed at day 2 of hypoxia exposure since the activity assay is not isoform specific. However, this possibility is unlikely based on the results demonstrating that NFAT activation is dramatically impaired in arteries from NFAT-luc/NFATc3 KO mice.
Our data further suggest that hypoxic activation of NFATc3 is mediated by Ca2+/calcineurin because CsA prevented CH-increased NFATc3 nuclear accumulation. These results are consistent with the reported increase in PASMC [Ca2+]i during CH (3–8).
Exposure to hypoxia causes hypoxic vasoconstriction due to acute inhibition of O2-sensing voltage-dependent K channels (KV channels) (3). Hypoxic inhibition of KV channels causes PASMC membrane depolarization leading to extracellular Ca2+ influx through voltage-dependent Ca2+ channels (VDCC) (3). This increase in PASMC [Ca2+]i may contribute to the early calcineurin/NFAT activation at 2 days.
Prolonged exposure to hypoxia reduces KV channel expression (5;8), re-sets PASMC Em to a more depolarized state, and enhances agonist-mediated vasoconstriction (4). It has been shown that PASMC from primary pulmonary hypertensive patients exhibit decreased KV mRNA and currents, with a resultant increase in [Ca2+]i compared with controls (5). In addition, it has been recently demonstrated that up-regulation of transient receptor potential channels (TRPC) contribute to the elevated basal Ca2+ and vascular tone observed in pulmonary hypertension (6;7;59;60). Interestingly, Ca2+ influx through TRPC is an important determinant of NFAT activity in skeletal muscle (61). However, it is controversial whether CH increases PASMC [Ca2+]i. Naik et al (62) have demonstrated no differences in pulmonary arterial wall Ca2+ between rats exposed to 4 weeks of CH or normoxia. However, PASMC membrane potential was more depolarized in CH arteries. Therefore, we cannot discard the possibility that other mechanisms besides Ca2+ mediate the CH-induced activation of calcineurin/NFAT. In addition, other humoral factors that are up-regulated by hypoxia could contribute to the increased NFAT activity demonstrated in this study, such as ET-1 and Ang II (4;36;37;63–70).
Calcineurin/NFATc3 is involved in the mechanism of CH-induced RV hypertrophy
Sustained high pulmonary arterial resistance to blood flow induced by CH causes an increase in the right ventricular (RV) afterload, which leads to RV hypertrophy (71). Our results show that two different strains of mice (C57B6 and BalbC) have RV hypertrophy after been exposed to CH, suggesting they develop pulmonary hypertension. In addition, they are also polycythemic in response to CH. Interestingly, calcineurin inhibition with CsA and the genetic deletion of NFATc3 significantly attenuated the development of RV hypertrophy without affecting the polycythemic response. These results are consistent with a recent report showing that CsA prevents CH-induced RV hypertrophy in rats (72). In this study, polycythemia was also prevented and the upregulation of hypoxia inducible factor 1 (HIF-1)-mediated genes. Surprisingly the authors did not address if NFAT inhibition was involved in the effects of CsA.
In addition, a few clinical studies reported that CsA improves pulmonary function of patients with primary pulmonary hypertension (73;74) further supporting our findings.
Calcineurin/NFATc3 mediates CH-induced up-regulation of SM-α-actin in pulmonary arteries
CH-induced pulmonary hypertension is caused by increased pulmonary arterial vasoconstriction and remodeling. It has been previously shown that pulmonary arterial α-actin and myosin heavy chain expression increase after exposure to CH (2;33;34). In agreement with these previous findings, we have found that SM-α-actin transcript and protein levels significantly increased after CH exposure. Interestingly, NFAT inhibition with CsA completely prevented this response. Furthermore, SM-α-actin protein levels increased in pulmonary arteries from CH WT but not from CH NFATc3 KO mice. These results are consistent with the expression of this isoform in pulmonary arteries and the reported regulation of α-actin expression by NFATc3 (21;24). In addition, since calcineurin can dephosphorylate other cellular proteins (19), the results obtained with the NFATc3 KO confirm that the effect of CsA was specific for NFAT.
The present study demonstrates a novel effect of CH to upregulate SM-α-actin through a calcineurin/NFATc3-dependent pathway. In support of the selective regulation of SM-α-actin expression by NFATc3, no changes were observed in β-actin protein levels.
These results suggest that NFATc3 is involved in PASMC hypertrophy which underlies pulmonary arterial remodeling.
It is interesting that NFATc3-mediated CH-induced increase in SM-α-actin expression does not coincide with the maximum NFAT activity at day 2 but rather overlaps with the intermediate level of activation at days 7 and 21. Therefore, it is possible that the expression of other genes that increase or decrease during CH may be transcriptional regulated by NFATc3. For example, KV 2.1 has been shown to be downregulated by activated NFATc3 in mouse cerebral arteries smooth muscle (29) and interestingly, it is also downregulated after 2 days of CH (8;75).
NFATc3 mediates CH-induced pulmonary arterial remodeling
Medial thickening caused in part by PASMC hypertrophy is one of the prominent features of vascular remodeling induced by CH [reviewed in (1)]. Our studies demonstrate pulmonary arterial remodeling (increased wall thickness) in mice after CH. NFATc3 contributes to that process evidenced by the lack of remodeling in NFATc3 KO after been exposed to CH. These results are also in agreement with the prevented upregulation of SM-α-actin, further supporting a role for NFATc3 in the development of pulmonary hypertension.
Differentiation of adventitial fibroblasts into myofibroblasts also contributes to medial thickening (13), a process dependent on increased expression of contractile proteins (differentiation markers) such as α-actin. Further studies are needed to determine whether the NFATc3-mediated CH-induced increase in α-actin is due to a) PASMC hypertrophy which requires increased contractile protein expression and/or b) increased myofibroblasts in the medial layer which express α-actin.
In summary, our results for the first time demonstrate that NFATc3 is expressed in PASMC and is activated by CH in a calcineurin-dependent manner. Most importantly, CH-induced RV hypertrophy, up-regulation of SM-α-actin and vascular remodeling are mediated by calcineurin/NFATc3. These findings strongly suggest that NFATc3 may be involved in the vascular changes that underlie the development of pulmonary hypertension.
Fig. 7.

NFATc3 mediates CH-induced increases in pulmonary arterial wall thickness. A. Representative cross sections from normoxic (N) and CH (21 days) WT and KO mice. Arrows point internal and external elastic laminae. B. Percent arterial wall thickness of arteries with diameter of 231–310 μm. *p<0.001. Arteries n= see columns, number of mice 3–4.
Acknowledgments
We gratefully acknowledge Dr. Laurie Glimcher for giving us the NFATc3 KO mice. We would also like to thank Dr. Molkentin for providing the 9xNFAT-luciferase mice. We thank Tamara Howard for her contribution in the vascular morphometric studies. In addition, we thank Drs. Nancy Kanagy, Thomas Resta and Jessica Filosa for helpful discussions. This work was supported by Dedicated Health Research Funds of the UNM SOM and a Postdoctoral Fellowship from “Ministerio de Educación y Ciencia”, Spain to Sergio de Frutos.
Footnotes
ABBREVIATIONS USED: CH: chronic hypoxia; PASMC: pulmonary arterial smooth muscle cells; [Ca2+]i: intracellular Ca2+ concentration; SM: smooth muscle; NFAT: nuclear factor of activated T cells; SRF: serum response factor; VSMC: vascular smooth muscle cells; Ang II: angiotensin II; NFAT-luc: 9xNFAT-luciferase mice; NFATc3 KO: NFATc3 knockout mice; WT: wild type mice; CsA: cyclosporin A; ECL: enhanced chemiluminescence detection; qRT-PCR: quantitative real-time polymerase chain reaction; ET-1: endothelin 1; KV channels: voltage-dependent K channels; VDCC: voltage-dependent Ca2+ channels; Em: membrane potential; T: total ventricle weight; TRCP: transient receptor potential channels; HIF-1: hypoxia inducible factor 1.
REFERENCE LIST
- 1.Hopkins N, McLoughlin P. Journal of Anatomy. 2002;201:335–348. doi: 10.1046/j.1469-7580.2002.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Am J Physiol Heart Circ Physiol. 1979;236:H818–H827. doi: 10.1152/ajpheart.1979.236.6.H818. [DOI] [PubMed] [Google Scholar]
- 3.Weir EK, Archer SL. FASEB J. 1995;9:183–189. doi: 10.1096/fasebj.9.2.7781921. [DOI] [PubMed] [Google Scholar]
- 4.Li KX, Fouty B, McMurtry IF, Rodman DM. Am J Physiol Heart Circ Physiol. 1999;277:H363–H370. doi: 10.1152/ajpheart.1999.277.1.H363. [DOI] [PubMed] [Google Scholar]
- 5.Yuan JXJ, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr, Gaine SP, Orens JB, Rubin LJ. Circulation. 1998;98:1400–1406. doi: 10.1161/01.cir.98.14.1400. [DOI] [PubMed] [Google Scholar]
- 6.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JXJ. PNAS. 2004;101:13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin MJ, Leung GPH, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JSK. Circ Res. 2004;95:496–505. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Juhaszova M, Rubin LJ, Yuan XJ. J Clin Invest. 1997;100:2347–53. doi: 10.1172/JCI119774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jernigan NL, Walker BR, Resta TC. Am J Physiol Lung Cell Mol Physiol. 2004. [DOI] [PubMed] [Google Scholar]
- 10.Nagaoka T, Gebb SA, Karoor V, Homma N, Morris KG, McMurtry IF, Oka M. J Appl Physiol. 2006;100:996–1002. doi: 10.1152/japplphysiol.01028.2005. [DOI] [PubMed] [Google Scholar]
- 11.Abe K, Shimokawa H, Morikawa K, Uwatoku T, Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K, Takeshit A. Circ Res. 2004;94:385–393. doi: 10.1161/01.RES.0000111804.34509.94. [DOI] [PubMed] [Google Scholar]
- 12.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Am J Physiol Lung Cell Mol Physiol. 2004;287:L656–L664. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- 13.Short M, Nemenoff RA, Zawada WM, Stenmark KR, Das M. Am J Physiol Cell Physiol. 2004;286:C416–C425. doi: 10.1152/ajpcell.00169.2003. [DOI] [PubMed] [Google Scholar]
- 14.Owens GK. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 15.Owens GK, Vernon SM, Madsen CS. J Hypertens Suppl. 1996;14:S55–S64. [PubMed] [Google Scholar]
- 16.Owens GK. Acta Physiol Scand. 1998;164:623–635. doi: 10.1111/j.1365-201x.1998.tb10706.x. [DOI] [PubMed] [Google Scholar]
- 17.Owens GK, Kumar MS, Wamhoff BR. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 18.Emmel EA, Verweij CL, Durand DB, Higgins KM, Lacy E, Crabtree GR. Science. 1989;246:1617–1620. doi: 10.1126/science.2595372. [DOI] [PubMed] [Google Scholar]
- 19.Rao A, Luo C, Hogan PG. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 20.Graef IA, Chen F, Crabtree GR. Curr Opin Genet Dev. 2001;11:505–512. doi: 10.1016/s0959-437x(00)00225-2. [DOI] [PubMed] [Google Scholar]
- 21.Wada H, Hasegawa K, Morimoto T, Kakita T, Yanazume T, Abe M, Sasayama S. J Cell Biol. 2002;156:983–991. doi: 10.1083/jcb.200106057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohkawa Y, Hayashi K, Sobue K. Biochem Biophys Res Commun. 2003;301:78–83. doi: 10.1016/s0006-291x(02)02965-0. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez Bosc LV, Wilkerson MK, Bradley KN, Eckman DM, Hill-Eubanks DC, Nelson MT. J Biol Chem. 2004;279:10702–10709. doi: 10.1074/jbc.M312920200. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez Bosc LV, Layne J, Nelson MT, Hill-Eubanks DC. J Biol Chem. 2004;280:26113–26120. doi: 10.1074/jbc.M411972200. [DOI] [PubMed] [Google Scholar]
- 25.Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Cell. 2001;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- 26.Fatigati V, Murphy RA. J Biol Chem. 1984;259:14383–14388. [PubMed] [Google Scholar]
- 27.Hill-Eubanks DC, Gomez MF, Stevenson AS, Nelson MT. Trends Cardiovasc Med. 2003;13:56–62. doi: 10.1016/s1050-1738(02)00212-8. [DOI] [PubMed] [Google Scholar]
- 28.Hogan PG, Chen L, Nardone J, Rao A. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 29.Amberg GC, Rossow CF, Navedo MF, Santana LF. J Biol Chem. 2004:M408789200. doi: 10.1074/jbc.M408789200. [DOI] [PubMed] [Google Scholar]
- 30.Abbott KL, Loss JR, Robida AM, Murphy TJ. Mol Pharmacol. 2000;58:946–953. doi: 10.1124/mol.58.5.946. [DOI] [PubMed] [Google Scholar]
- 31.Gomez MF, Stevenson AS, Bonev AD, Hill-Eubanks DC, Nelson MT. J Biol Chem. 2002;277:37756–37764. doi: 10.1074/jbc.M203596200. [DOI] [PubMed] [Google Scholar]
- 32.Stevenson AS, Gomez MF, Hill-Eubanks DC, Nelson MT. J Biol Chem. 2001;276:15018–15024. doi: 10.1074/jbc.M011684200. [DOI] [PubMed] [Google Scholar]
- 33.Durmowicz AG, Stenmark KR. Pediatr Rev. 1999;20:e91–e102. [PubMed] [Google Scholar]
- 34.Jones R, Jacobson M, Steudel W. American Journal of Respiratory Cell and Molecular Biology. 1999;20:582–594. doi: 10.1165/ajrcmb.20.4.3357. [DOI] [PubMed] [Google Scholar]
- 35.Owens GK, Rabinovitch PS, Schwartz SM. Proc Natl Acad Sci USA. 1981;78:7759–7763. doi: 10.1073/pnas.78.12.7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chassagne C, Eddahibi S, Adamy C, Rideau D, Marotte F, Dubois-Rande JL, Adnot S, Samuel JL, Teiger E. American Journal of Respiratory Cell and Molecular Biology. 2000;22:323–332. doi: 10.1165/ajrcmb.22.3.3701. [DOI] [PubMed] [Google Scholar]
- 37.Morrell NW, Atochina EN, Morris KG, Danilov SM, Stenmark KR. J Clin Invest. 1995;96:1823–1833. doi: 10.1172/JCI118228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orte C, Polak JM, Haworth SG, Yacoub MH, Morrell NW. J Pathol. 2000;192:379–384. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH715>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 39.Hautmann MB, Thompson MM, Swartz EA, Olson EN, Owens GK. Circ Res. 1997;81:600–10. doi: 10.1161/01.res.81.4.600. [DOI] [PubMed] [Google Scholar]
- 40.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oukka M, Ho IC, de la Brousse FC, Hoey T, Grusby MJ, Glimcher LH. Immunity. 1998;9:295–304. doi: 10.1016/s1074-7613(00)80612-3. [DOI] [PubMed] [Google Scholar]
- 42.Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Mol Cell Biol. 2002;22:7603–13. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Resta TC, Gonzales RJ, Dail WG, Sanders TC, Walker BR. Am J Physiol Heart Circ Physiol. 1997;272:H806–H813. doi: 10.1152/ajpheart.1997.272.2.H806. [DOI] [PubMed] [Google Scholar]
- 44.Resta TC, Chicoine LG, Omdahl JL, Walker BR. Am J Physiol Heart Circ Physiol. 1999;276:H699–H708. doi: 10.1152/ajpheart.1999.276.2.H699. [DOI] [PubMed] [Google Scholar]
- 45.Meyrick B, Reid L. Anat Rec. 1979;193:71–97. doi: 10.1002/ar.1091930106. [DOI] [PubMed] [Google Scholar]
- 46.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 47.Ferri C, Bellini C, De Angelis C, De Siati L, Perrone A, Properzi G, Santucci A. J Clin Pathol. 1995;48:519–524. doi: 10.1136/jcp.48.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang X, Chen W, Chen J. Chin Med J(Engl) 1997;110:104–108. [PubMed] [Google Scholar]
- 49.Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, Schermuly RT. Circulation. 2006;113:286–295. doi: 10.1161/CIRCULATIONAHA.105.581405. [DOI] [PubMed] [Google Scholar]
- 50.Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns HH, Kummer W, Braun-Dullaeus RC. Respir Res. 2007;8:15. doi: 10.1186/1465-9921-8-15. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Resta T, Sanders T, Eichinger M, Crowley M, Walker B. Respiration Physiology. 1998;114:161–173. doi: 10.1016/s0034-5687(98)00086-3. [DOI] [PubMed] [Google Scholar]
- 52.Nieves-Cintron M, Amberg GC, Nichols CB, Molkentin JD, Santana LF. J Biol Chem. 2006:M608822200. doi: 10.1074/jbc.M608822200. [DOI] [PubMed] [Google Scholar]
- 53.Yaghi A, Sims SM. Am J Physiol Lung Cell Mol Physiol. 2005;289:L1061–L1074. doi: 10.1152/ajplung.00096.2005. [DOI] [PubMed] [Google Scholar]
- 54.Gomez MF, Gonzalez Bosc LV, Stevenson AS, Wilkerson MK, Hill-Eubanks DC, Nelson MT. J Biol Chem. 2003;278:46847–46853. doi: 10.1074/jbc.M304765200. [DOI] [PubMed] [Google Scholar]
- 55.Boss V, Abbott KL, Wang XF, Pavlath GK, Murphy TJ. J Biol Chem. 1998;273:19664–19671. doi: 10.1074/jbc.273.31.19664. [DOI] [PubMed] [Google Scholar]
- 56.Nilsson LM, Sun ZW, Nilsson J, Nordstrom I, Chen YW, Molkentin JD, Wide-Swensson D, Hellstrand P, Lydrup ML, Gomez MF. Am J Physiol Cell Physiol. 2007;292:C1167–C1178. doi: 10.1152/ajpcell.00590.2005. [DOI] [PubMed] [Google Scholar]
- 57.Semenza GL. Cell. 1999;98:281–284. doi: 10.1016/s0092-8674(00)81957-1. [DOI] [PubMed] [Google Scholar]
- 58.Kallio PJ, Okamoto K, O’Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. The EMBO Journal. 1998;17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang J, Weigand L, Lu W, Sylvester JT, Semenza L, Shimoda LA. Circ Res. 2006;98:1528–1537. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 60.Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. The FASEB Journal: Official Publication Of The Federation Of American Societies For Experimental Biology. 2006;20:1660–1670. doi: 10.1096/fj.05-5560com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenberg P, Hawkins A, Stiber J, Shelton JM, Hutcheson K, Bassel-Duby R, Shin DM, Yan Z, Williams RS. PNAS. 2004;101:9387–9392. doi: 10.1073/pnas.0308179101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naik JS, Earley S, Resta TC, Walker BR. J Appl Physiol. 2005;98:1119–1124. doi: 10.1152/japplphysiol.00819.2004. [DOI] [PubMed] [Google Scholar]
- 63.Ip SP, Chan YW, Leung PS. Pancreas. 2002;25:296–300. doi: 10.1097/00006676-200210000-00013. [DOI] [PubMed] [Google Scholar]
- 64.Shimoda LA, Sham JS, Sylvester JT. Physiol Res. 2000;49:549–560. [PubMed] [Google Scholar]
- 65.Blumberg FC, Wolf K, Arzt M, Lorenz C, Riegger GAJ, Pfeifer M. J Appl Physiol. 2003;94:446–452. doi: 10.1152/japplphysiol.00239.2002. [DOI] [PubMed] [Google Scholar]
- 66.El Gamal Y, Hossny E, Awwad K, Mabrouk R, Boseila N. Ann Allergy Asthma Immunol. 2002;88:370–373. doi: 10.1016/S1081-1206(10)62366-6. [DOI] [PubMed] [Google Scholar]
- 67.Ivy DD, Le Cras TD, Horan MP, Abman SH. Am J Physiol. 1998;274:L535–L541. doi: 10.1152/ajplung.1998.274.4.L535. [DOI] [PubMed] [Google Scholar]
- 68.Nishida M, Eshiro K, Okada Y, Takaoka M, Matsumura Y. J Cardiovasc Pharmacol. 2004;44:187–191. doi: 10.1097/00005344-200408000-00007. [DOI] [PubMed] [Google Scholar]
- 69.Soma S, Takahashi H, Muramatsu M, Oka M, Fukuchi Y. American Journal of Respiratory Cell and Molecular Biology. 1999;20:620–630. doi: 10.1165/ajrcmb.20.4.3356. [DOI] [PubMed] [Google Scholar]
- 70.Yuyama H, Fujimori A, Sanagi M, Koakutsu A, Noguchi Y, Sudoh K, Sasamata M, Miyata K. Vascular Pharmacology. 2005;43:40–46. doi: 10.1016/j.vph.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 71.Cheever KH. J Cardiovasc Nurs. 2005;20:108–116. doi: 10.1097/00005082-200503000-00005. [DOI] [PubMed] [Google Scholar]
- 72.Koulmann N, Novel-Chate V, Peinnequin A, Chapot R, Serrurier B, Simler N, Richard H, Ventura-Clapier R, Bigard X. Am J Respir Crit Care Med. 2006;174:699–705. doi: 10.1164/rccm.200512-1976OC. [DOI] [PubMed] [Google Scholar]
- 73.Morelli S, Giordano M, De Marzio P, Priori R, Sgreccia A, Valesini G. Lupus. 1993;2:367–369. doi: 10.1177/096120339300200606. [DOI] [PubMed] [Google Scholar]
- 74.Padeh S, Laxer RM, Silver MM, Silverman ED. Arthritis Rheum. 1991;34:1575–1579. doi: 10.1002/art.1780341216. [DOI] [PubMed] [Google Scholar]
- 75.Shimoda LA, Manalo DJ, Sham JSK, Semenza GL, Sylvester JT. Am J Physiol Lung Cell Mol Physiol. 2001;281:L202–L208. doi: 10.1152/ajplung.2001.281.1.L202. [DOI] [PubMed] [Google Scholar]