

Abstract
We define the target, mechanism, and structural basis of inhibition of bacterial RNA polymerase (RNAP) by the tetramic-acid antibiotic streptolydigin (Stl). Stl binds to a site adjacent to, but not overlapping, the RNAP active center and stabilizes an RNAP-active-center conformational state with a straight bridge helix. The results provide direct support for the proposals that alternative straight-bridge-helix and bent-bridge-helix RNAP-active-center conformations exist, and that cycling between straight-bridge-helix and bent-bridge-helix RNAP-active-center conformations is required for RNAP function. The results set bounds on models for RNAP function and suggest strategies for design of novel antibacterial agents.
The tetramic-acid antibiotic streptolydigin (Stl) inhibits initiation, elongation, and pyrophosphorolysis by bacterial RNA polymerase (RNAP; Siddhikol et al. 1969; Cassani et al. 1971; McClure, 1980). Stl does not inhibit eukaryotic RNAPI, RNAPII, or RNAPIII, which share high three-dimensional structural similarity, but low sequence similarity, with bacterial RNAP (Ebright, 2000; Darst, 2001; Cramer, 2002). Stl exhibits only limited cross-resistance with the inhibitor of bacterial RNAP in current clinical use in antibacterial therapy, rifampicin (Campbell et al. 2001), and exhibits only limited, or no, cross-resistance with other characterized inhibitors of bacterial RNAP, including microcin J25 (MccJ25; Yuzenkova et al. 2002; Adelman et al. 2004; Mukhopadhyay et al. 2004), CBR703 (Artsimovitch et al. 2003), and sorangicin (Campbell et al. 2005). Here, using a combination of genetic, biochemical, and crystallographic approaches, we define the target, mechanism, and structural basis of inhibition of bacterial RNAP by Stl.
Single-amino-acid substitutions resulting in resistance to Stl (Stlr) have been reported in residues 543-546 of the RNAP β subunit (Heisler et al. 1993) and in residues 792-793 of the RNAP β′ subunit (Severinov et al. 1995; Yang and Price, 1995; residues numbered as in Escherichia coli RNAP). In the three-dimensional structure of RNAP (Zhang et al. 1999; Vassylyev et al. 2002), the implicated residues map to two distinct, non-adjacent regions, separated by ∼15 Å. The apparent existence of two distinct, non-adjacent regions important for function of Stl raises the issue of whether Stl interacts with the first region, the second region, or both.
To define systematically the determinants for function of Stl, we performed saturation mutagenesis of the genes encoding the RNAP β and β′ subunits, and isolated and characterized additional Stlr mutants. We performed saturation mutagenesis using a set of twenty “doped” oligodeoxyribonucleotide primers--designed to introduce all possible nucleotide substitutions at all codons for residues that are solvent-exposed and located within 20 Å of the previously reported Stlr substitutions (Supplemental Table S1). We isolated 72 independent Stlr mutants (Supplemental Table S2). Sequencing indicates that 70 of the 72 independent Stlr mutants are single-substitution mutants. The single-substitution mutants comprise 26 distinct substitutions, involving 5 sites within β (543, 544, 545, 570, and 571) and 13 sites within β′ (788, 791, 793, 798, 926, 930, 931, 937, 940, 1136, 1137, 1139, and 1246) (Table 1; Fig. 1A,B). Most Stlr substitutions affect residues that are conserved in bacterial RNAP but are not conserved in eukaryotic RNAPI, RNAPII, and RNAPIII, consistent with the known specificity of Stl for bacterial RNAP (Fig. 1A,B). Each Stlr mutant is able to complement a corresponding temperature-sensitive mutant for growth at the non-permissive temperature, indicating that each Stlr RNAP derivative is functional in transcription--sufficiently functional to support viability (Table 1). Four Stlr RNAP derivatives were purified and verified to be functional in transcription and to exhibit resistance to Stl in vitro (Supplemental Fig. S1).
Table 1. StlR isolates from saturation mutagenesis and selection.
| Amino acid substitution | Number of independent isolates | Ability to complement rpoBts or rpoCts | MIC* (μg/ml) |
|---|---|---|---|
| rpoB | |||
| single-substitution mutants | |||
| 543 Ala→Pro | 1 | + | >20 |
| 543 Ala→Thr | 3 | + | 10 |
| 543 Ala→Val | 6 | + | >20 |
| 544 Gly→Arg | 2 | + | >20 |
| 545 Phe→Cys | 3 | + | >20 |
| 545 Phe→Ile | 2 | + | >20 |
| 545 Phe→Leu | 2 | + | >20 |
| 545 Phe→Ser | 2 | + | >20 |
| 570 Gly→Cys | 2 | + | 10 |
| 571 Leu→Arg | 3 | + | 20 |
| rpoC | |||
| single-substitution mutants | |||
| 788 Leu→Met | 8 | + | >20 |
| 788 Leu→Val | 1 | + | >20 |
| 791 Ala→Gly | 1 | + | >20 |
| 793 Ser→Phe | 6 | + | 20 |
| 798 Arg→Ile | 1 | + | 10 |
| 926 Pro→Leu | 1 | + | 5 |
| 930 Leu→Met | 3 | + | 10 |
| 931 Thr→Ile | 4 | + | 10 |
| 937 Ile→Asn | 3 | + | 20 |
| 940 Ala→Val | 1 | + | 5 |
| 1136 Gly→Arg | 1 | + | 10 |
| 1137 Gly→Asp | 1 | + | 10 |
| 1139 Pro→Arg | 1 | + | >20 |
| 1139 Pro→Leu | 5 | + | 20 |
| 1139 Pro→Ser | 3 | + | 10 |
| 1246 Val→Gly | 4 | + | 10 |
| multiple-substitution mutants | |||
| 1136 Gly→Ser; | 1 | + | 20 |
| 1139 Pro→Ala | |||
| 1138 Leu→Val; | 1 | + | 10 |
| 1139 Pro→Thr |
Minimal inhibitory concentration; 2-3 μg/ml for wild-type rpoB and rpoC.
Fig. 1. Target of Stl.
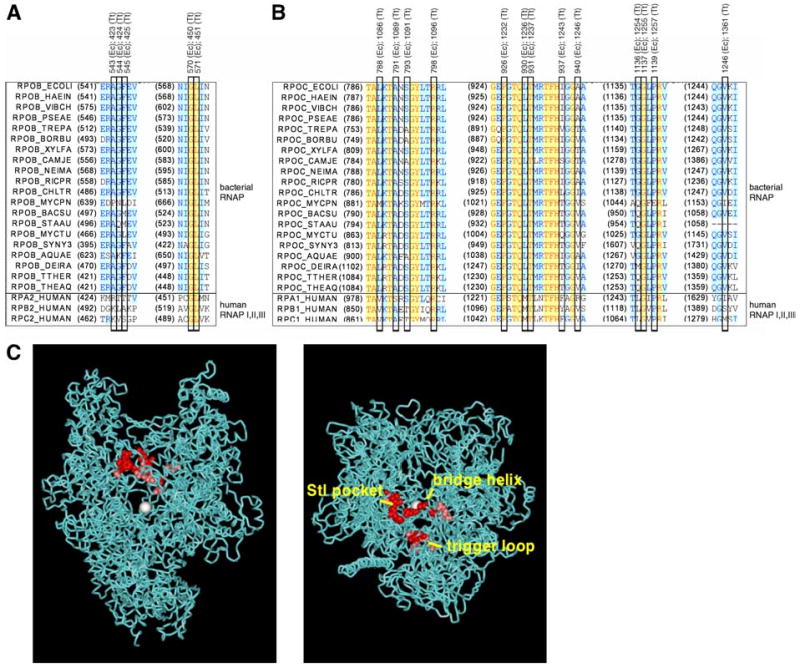
(A,B) Amino-acid sequence alignments for regions of E. coli RNAP β′ subunit (A) and E. coli β subunit (B) in which single-residue substitutions that confer Stl resistance were obtained (Table 1). Sequences for bacterial RNAP are at top; sequences for human RNAPI, RNAPII, and RNAPIII are at bottom; sites of single-residue substitutions that confer resistance to Stl are boxed (with E. coli and T. thermophilus residue numbers). Species names and SwissProt locus identifiers for the sequences are, in order: E. coli (RPOB_ECOLI, RPOC_ECOLI), Haemophilus influenzae (RPOB_HAEIN, RPOC_HAEIN), Vibrio cholerae (RPOB_VIBCH, RPOC_VIBCH), Pseudomonas aeruginosa (RPOB_PSEAE, RPOC_PSEAE), Treponema pallidum (RPOB_TREPA, RPOC_TREPA), Bordetella pertussis (RPOB_BORPE, RPOC_BORPE), Xylella fastidiosa (RPOB_XYLFA, RPOC_XYLFA), Campylobacter jejuni (RPOB_CAMJE, RPOC_CAMJE), Neisseria meningitidis (RPOB_NEIME, RPOC_NEIMA), Rickettsia prowazekii (RPOB_RICPR, RPOC_RICPR), Chlamydia trachomatis (RPOB_CHLTR, RPOC_CHLTR), Mycoplasma pneumoniae (RPOB_MYCPN, RPOC_MYCPN), Bacillus subtilis (RPOB_BACSU, RPOC_BACSU), Staphylococcus aureus (RPOB_STAAU, BACSU, RPOC_STAAU), Mycobacterium tuberculosis (RPOB_MYCTU, RPOC_MYCTU), Synechocystis sp. PCC 6803 (RPOB_SYNY3, RPOC2_SYNY3), Aquifex aeolicus (RPOB_AQUAE, RPOC_AQUAE), Deinococcus radiodurans (RPOB_DEIRA, RPOC_DEIRA), Thermus thermophilus (RPOB_THETH, RPOC_THETH), Thermus aquaticus (RPOB_THEAQ, RPOC_THEAQ), Homo sapiens RNAPI (RPA2_HUMAN, RPA1_HUMAN), Homo sapiens RNAPII (RPB2_HUMAN, RPB1_HUMAN), and Homo sapiens RNAPIII (RPC2_HUMAN, RPC1_HUMAN). (C) Three-dimensional structure of RNAP showing locations of sites of single-residue substitutions that confer resistance to Stl (high-level resistance in red; moderate-level resistance in pink; Table 1). Two orthogonal views are shown: left, view directly into the NTP-uptake channel, toward the active-center Mg2+ (white sphere); right, view directly into the RNAP active-center cleft, toward the active-center Mg2+. Atomic coordinates are for T. thermophilus RNAP holoenzyme (Vassylyev et al. 2002; σ subunit and β′-subunit dispensable region omitted for clarity).
The Stlr substitutions define a determinant with approximate dimensions of 30 Å × 15 Å × 10 Å (Fig. 1C). The determinant includes both of the regions previously reported to be important for function of Stl (Heisler et al. 1993; Severinov et al. 1995; Yang and Price, 1993), as well as the intervening region. The determinant encompasses three structural elements within RNAP: the “Stl pocket” (residues 543-545 and 570-571 of β), the “bridge helix” (also referred to as “helix F”; residues 788-798 of β′), and the “trigger loop” (also referred to as the “G-G' loop”; 930-940, 1136-1139, and 1246 of β′) (Fig. 1C). The determinant is located adjacent to, but does not overlap, the RNAP active center (Fig. 1C).
The determinant is located adjacent to, and to a small extent overlaps, the binding site for the inhibitor MccJ25 (Yuzenkova et al. 2002; Adelman et al. 2004; Mukhopadhyay et al. 2004). To determine whether Stl and MccJ25 compete for binding to RNAP, we performed fluorescence-resonance-energy-transfer competition assays (methods as in Mukhopadhyay et al. 2004; Supplemental Fig. S2). The results indicate that interaction of RNAP with Stl and interaction of RNAP with MccJ25 are mutually exclusive. The results further indicate that Kd for interaction of RNAP with Stl is 15±2 μM--a value that equals, within experimental error, Ki for inhibition of RNAP by Stl (18±3 μM; Supplemental Fig. S1; see also Siddhikol et al. 1969; Cassani et al. 1971; McClure, 1980). We infer that the determinant represents, at least in part, the binding site for Stl (as opposed to an allosteric determinant required for function but not for binding).
The fact that Stl inhibits initiation, elongation, and pyrophosphorolysis (the reverse reaction of nucleotide addition) (Siddhikol et al. 1969; Cassani et al. 1971; McClure, 1980) suggests that Stl inhibits a reaction common to these three processes: i.e., phosphodiester-bond formation/breakage or RNAP translocation. To assess effects of Stl on RNAP translocational state, we performed exonuclease-III DNA-footprinting experiments (Guajardo et al. 1998; Bar-Nahum et al. 2005), analyzing a transcription elongation complex containing a 16 nt RNA product with a non-extendable, 3′-deoxy-3′-amino terminus (Fig. 2). The results indicate that Stl stabilizes the post-translocated and pre-translocated states (state 1 and state 0) relative to the backtracked state (state -1) by a factor of ∼10, and stabilizes the post-translocated state relative to the pre-translocated state by a factor of ∼2 (Fig. 2C,D). Effects of Stl on RNAP translocational state are qualitatively similar to, but quantitatively less than, effects of the incoming NTP on RNAP translocational state (Fig. 2C,D; see Bar-Nahum et al. 2005). We conclude that Stl affects RNAP translocational state and suggest that effects on RNAP translocational state are associated with the mechanism of inhibition.
Fig. 2. Effects of Stl on RNAP translocational state.
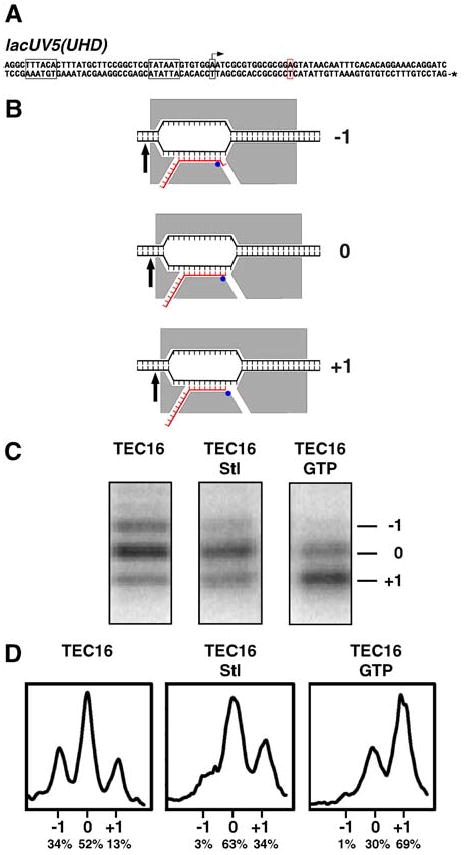
Data are from exonuclease-III DNA footprinting experiments (Guajardo et al. 1998; Bar-Nahum et al. 2005), analyzing a transcription elongation complex containing a 16 nt RNA product with a non-extendable, 3′-deoxy-3′-amino terminus (TEC16).
(A) DNA fragment used in analysis of TEC16. Gray boxes, -35 element, -10 element, and transcription start site (with arrow); red box, halt site (first G in template strand of transcribed region); *, [32P]-phosphate.
(B) Predicted structural organization of the backtracked state (top; state -1), the pre-translocated state (middle; state 0), and the post-translocated state (bottom; state +1) of TEC16, and corresponding predicted 32P-labeled products upon exonuclease-III footprinting [architecture of RNAP (gray), DNA (black), and RNA (red) in transcription elongation complex as in Korzheva et al. 2000; distance between RNAP active center (blue circle) and exonuclease-III stop point at RNAP trailing edge (arrow) as in Metzger et al. 1989; Wang et al. 1995].
(C) Observed 32P-labeled products upon exonuclease-III footprinting of TEC16, TEC16 in the presence of 20 μM Stl, and TEC16 in the presence of 50 μM complementary incoming NTP (GTP) (10 min reactions).
(D) Inferred relative abundances of the backtracked state, the pre-translocated state, and the post-translocated state--shown as scans of bands in (C) (in panels) and as normalized integrated peak areas (beneath panels).
To define the structural basis of inhibition by Stl, we have determined the crystal structure of Thermus thermophilus RNAP holoenzyme in complex with Stl at 3.0 Å resolution (Table 2; Fig. 3B,C; Supplemental Figs. S3-S5). [Stl inhibits T. thermophilus RNAP with Ki = 1.8±0.4 μM (Supplemental Fig. S6).] For direct comparison, we have determined the crystal structure of T. thermophilus RNAP holoenzyme alone, in the identical crystal form, at 3.3 Å resolution (Table 2; Supplemental Figs. S3,S4). The structures define the binding site for Stl and define changes in RNAP-active-center conformation induced by Stl.
Table 2. Crystallographic data and refinement statistics.
| crystallographic data | ||
|---|---|---|
| RNAP-Stl | RNAP | |
| beamline | BNLS X25 | BNLS X25 |
| space group | P65 | P65 |
| temperature (°C) | -165 | -165 |
| resolution range (Å) | 30.0-3.0 | 30.0-3.3 |
| cell parameters, a (Å) | 237.0 | 236.2 |
| b (Å) | 237.0 | 236.2 |
| c (Å) | 250.0 | 249.9 |
| completeness (%) (highest shell) | 87.8 (74.1) | 98.5 (93.7) |
| reflections (total/unique) | 795,119/139,391 | 532,757/115,975 |
| Rmerge (%) | 12.4 | 13.8 |
| refinement statistics | ||
| RNAP-Stl | RNAP | |
| space group | P32 | P32 |
| resolution range (Å) | 30.0-3.0 | 30.0-3.3 |
| number of reflections | 244,746 | 196,181 |
| R-factor (%) | 26.1 | 28.2 |
| free R-factor (%) | 28.1 | 32.0 |
| number of refined atoms | 54,056 | 53,964 |
| Luzzati error (Å) | 0.60 | 0.57 |
| mean B-factor (Å2) | 72 (RNAP), 49 (Stl) | 74 |
Rfree = Σ|Fo - Fc| / |Fo| for 0.8% of the data with |Fo| ≥ 0 sequestered from the calculations. The crystals have the symmetry of space group P32 with perfect (50%) hemihedral twinning and twinning operator (-h,-k,l), leading to apparent hexagonal (P6/m) intensity symmetry. Processing and scaling were carried out in P65 followed by expansion to P32 for structure solution and refinement.
Fig. 3. Structural basis of inhibition by Stl: interactions with RNAP.
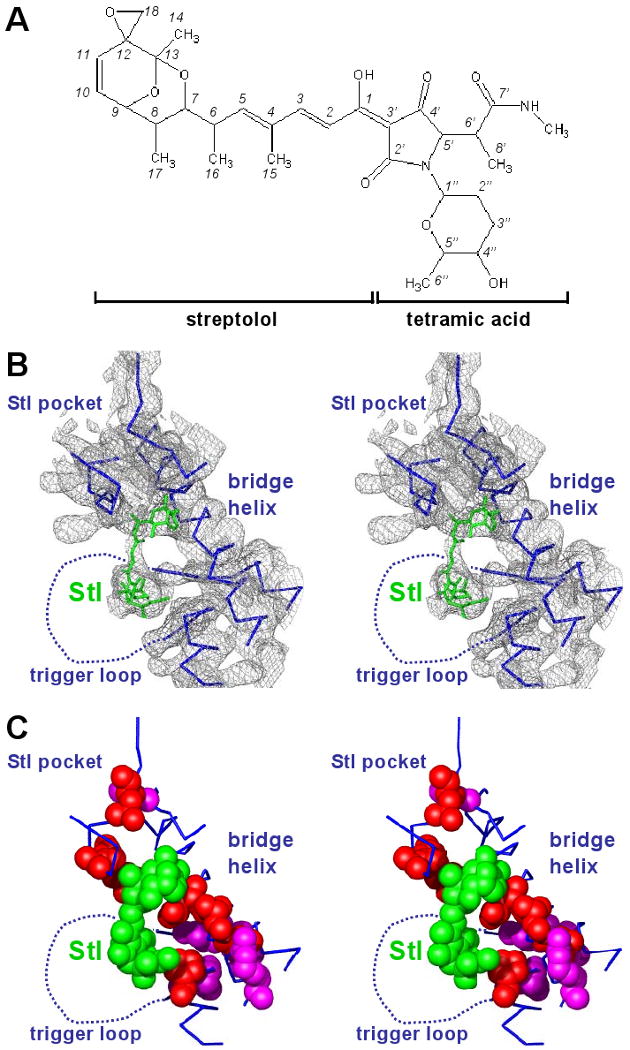
(A) Structure of Stl.
(B) Structure of RNAP-Stl: electron density for Stl binding region. Gray, 3Fo-2Fc electron density map [contoured at 1.0σ; phases from density modification, using noncrystallographic symmetry (NCS) averaging and solvent flipping, prior to inclusion of Stl in the model; Fc from NCS averaging and reconstruction; see Supporting Material]; cyan, RNAP; green, Stl.
(C) Structure of RNAP-Stl: structure of Stl binding region. Cyan, RNAP; cyan dashed line, disordered or poorly ordered RNAP residues; green, Stl; red and pink, substitutions conferring high-level and moderate-level resistance to Stl (Fig. 1A; Table 1).
Stl interacts with three structural elements within RNAP: the Stl pocket, the bridge helix, and the trigger-loop region (Fig. 3B,C). The Stl streptolol moiety interacts with the Stl pocket and bridge helix, and the Stl tetramic-acid moiety interacts with the trigger-loop region (Fig. 3A-C).
Stl is located on the floor of the RNAP active-center cleft, adjacent to, but not overlapping, the RNAP active center. Stl is remote from the NTP-uptake channel (Zhang et al. 1999; Adelman et al. 2004; Mukhopadhyay et al. 2004) and the modeled positions of the NTP entry site (Sosunov et al., 2003; Westover et al. 2004a; also referred to as “E site”), the NTP pre-insertion site (Kettenberger et al. 2004), and the NTP insertion site (Korzheva et al. 2000; Sosunov et al., 2003; Westover et al. 2004a; Kettenberger et al. 2004) (Fig. 4; >9 Å from each); thus Stl is not positioned to interfere directly with NTP uptake, NTP entry, NTP loading, or NTP binding. Stl also is remote from the active-center Mg2+ and active-center-Mg2+ coordinating ligands (Zhang et al. 1999) and from the modeled position of the RNA 3′ end (Korzheva et al. 2000; Sosunov et al. 2003; Westover et al. 2004a; Ketteberger et al. 2004) (Fig. 4; >15 Å from each); thus Stl is not positioned to interfere directly with phosphodiester bond formation/breakage. Stl is close to the modeled position of the downstream fork junction (the downstream separation point of the DNA template and non-template strands; Naryshkin et al. 2000; Korzheva et al. 2000; Kettenberger et al. 2004)--with the Stl tetramic-acid moiety close to, and possibly makes a hydrogen bond with, the sugar-phosphate backbone of the DNA non-template-strand at the downstream fork junction (Fig. 4); thus Stl is positioned potentially to interfere with RNAP function through direct Stl-DNA interactions. Stl also is close to key structural elements that flank the active center and are proposed to undergo conformational changes in RNAP function and regulation (Cramer et al. 2001; Gnatt et al. 2001; Vassylyev et al. 2002; Epshtein et al. 2002; Holmes and Erie, 2003; Bushnell and Kornberg, 2003; Bar-Nahum et al. 2005), and thus is positioned potentially to interfere with RNAP function through effects on RNAP-active-center conformation (see below).
Fig. 4. Structural basis of inhibition by Stl: modeled interactions with transcription elongation complex.
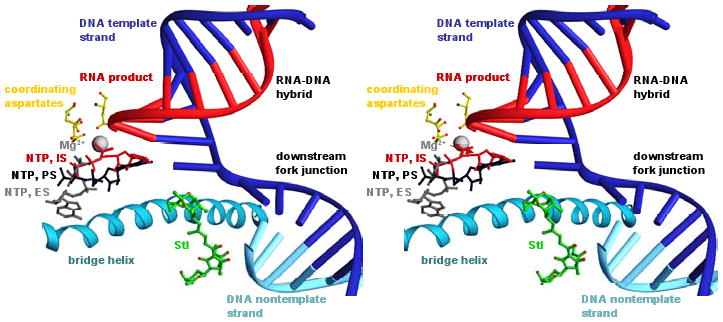
Modeled structure of transcription elongation complex containing Stl. To model positions of the DNA template strand, the DNA nontemplate strand, and the RNA product (blue, cyan, and red nucleic acids), nucleic acids from a structure of the yeast RNAPII transcription elongation complex (Kettenberger et al. 2004; PDB accession 1Y1W) were built into the structure of RNAP-Stl (superimposition based on Cα atoms of residues 482, 822, and 829 of yeast RNAPII RPB1 and Cα atoms of residues 740, 1079, and 1086 of T. thermophilus RNAP β′). To model positions of an NTP at the insertion-site, IS, an NTP at the entry site proposed in Westover et al. 2004a, ES, and an NTP at the pre-insertion site proposed in Kettenberger et al. 2004, PS (red, gray, and black NTPs), NTPs from structures of yeast RNAPII transcription elongation complexes containing bound NTPs (Westover et al., 2004a; Kettenberger et al. 2004; PDB accessions 19RS, 1R9T and 1Y77) were built into the structure (superimposition as above).
Stl contacts, or is close to, the majority of residues at which high-level Stlr substitutions are obtained and for which ordered density is observed in the structure (Figs. 3C, 5C). We infer that these residues represent a binding subdeterminant for Stl (Fig. 5C). Stl is remote from two residues at which high-level Stlr substitutions are obtained and for which ordered density is observed: β′ residues 791 and 793. We infer that these residues represent an allosteric functional subdeterminant for Stl (Fig. 5C).
Fig. 5. Structural basis of inhibition by Stl: stabilization of straight-bridge-helix active-center conformation.
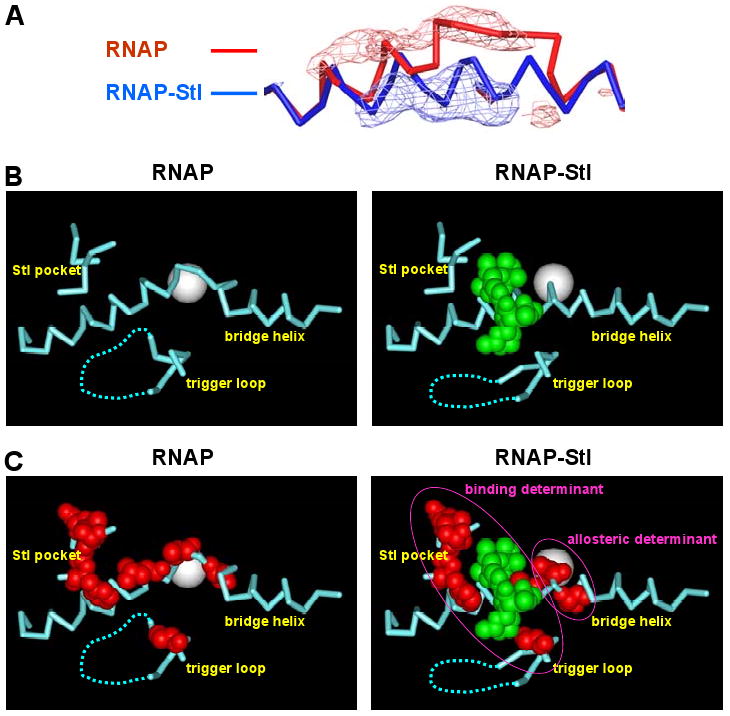
(A) Bridge-helix conformations in RNAP (red) and RNAP-Stl (blue) and superimposed Fo,-Fo difference electron density map (contoured at 3.0σ; calculated using datasets for RNAP-Stl and RNAP and phases from the refined structure of RNAP-Stl; positive difference density in blue; negative difference density in red).
(B) Active-center conformations in RNAP (left) and RNAP-Stl (right). Cyan, RNAP; cyan dashed line, disordered or poorly ordered RNAP residues; white sphere, active-center Mg2+; green, Stl.
(C) Active-center conformations in RNAP (left) and RNAP-Stl (right), showing sites of substitutions conferring high-level Stl resistance (red; Fig. 1A, Table 1), and the inferred binding subdeterminant and allosteric subdeterminant (magenta ovals).
Main-chain and side-chain atoms of β residues 543, 544, and 545 are located directly adjacent to the C4 through C7 atoms and the C15 and C16 methyl groups of the streptolol moiety of Stl (atoms of Stl numbered as in Lee and Rinehart, 1980; Fig. 3A). The side chains of Ala543 and Phe545 make Van der Waals interactions with Stl. The observed Stlr mutations Ala→Pro, Thr, and Val are expected to cause steric conflict with the C15 methyl group of streptolydigin, leading to possible loss of other nearby interactions such as a β Arg548 side-chain guanidine nitrogen hydrogen bond with the non-bridging oxygen of the Stl dioxabicyclonane ring system and Phe545 side-chain Van der Waals interactions with the Stl C16 methyl group. Mutations at β Phe545→Ile, Leu, and Ser would be expected to disfavor Stl binding by loss of hydrophobic association of Stl and the side-chain phenyl group.
Side-chain atoms of β′ residue Leu 788 are located directly adjacent to the C10 and C11 atoms and the epoxide group of the streptolol moiety of Stl. The epoxide ring makes Van der Waals interactions with main-chain atoms of the bridge helix residues Asp784 and Ala787. Apart from a possible weak hydrogen-bonding interaction of the Stl N-methyl amide moiety with the Asn792 (Asp in T. thermophilus) side-chain carbonyl oxygen, these comprise the major stabilizing contacts of Stl with the bridge helix. The Leu788→Val Stlr substitution introduces a β-branched side chain likely to result in steric conflict with Stl binding (in a manner reminiscent of Met184→Val and Met184→Ile substitutions in HIV-1 reverse transcriptase, which result in resistance to the important anti-AIDS drug Lamivudine (3TC) through a steric-hindrance mechanism; see Sarafianos et al. 1999). Side-chain atoms of β′ residue Pro1139 are located directly adjacent to the C2” and C3” atoms of the six-membered ring pendant from the tetramic-acid moiety of Stl. Stlr substitutions obtained at these positions (Table 1; Fig 1A,B) are expected to remove favorable hydrophobic interactions with Stl and/or to introduce unfavorable steric interactions with Stl. Four of the five positions β 543, 544, and 545, and β′ 788 and 1139 show sequence differences with eukaryotic RNAPI, RNAPII, and RNAPIII (Fig. 1A,B), consistent with the known lack of sensitivity of eukaryotic RNAPI, RNAP II, and RNAP III to Stl.
Comparison of the structures of RNAP and of the RNAP-Stl complex reveal that binding of Stl is accompanied by striking changes in the conformations of the bridge helix (Fig. 5A-C) and the trigger loop (Fig 5B,C)--changes that occur in the absence of other, global changes in conformation (Supplemental Fig. S3). In the structure of RNAP in the absence of Stl, the bridge helix is bent and adopts a conformation essentially identical to that reported by Vassylyev et al. 2002 for unliganded RNAP (Fig. 5A; Fig. 5B,C, left panels; Supplemental Fig. S4). In contrast, in the structure of the RNAP-Stl complex, the bridge helix is straight (Fig. 5A; Fig. 5B,C, right panels; Supplemental Fig. S4). In the structure of RNAP in the absence of Stl, the trigger loop interacts with residues of the bridge helix (Fig. 5B,C, left panels). In contrast, in the structure of the RNAP-Stl complex, the trigger loop must move in order to accommodate Stl, and the base of the trigger loop adopts a different conformation (Fig. 5B,C, right panels).
A bent bridge-helix conformation has been observed in all previously reported structures of bacterial RNAP and complexes thereof (Zhang et al. 1999; Campbell et al. 2001, 2005; Vassylyev et al. 2002; Murakami et al. 2002a, 2002b; Artsimovitch et al. 2004; PDB accession numbers 1HQM, 1I6V, 1IW7, 1L9U, 1L9Z, 1SMY, 1YNJ, and 1YNN), whereas a straight bridge-helix conformation has been observed in all previously reported structures of eukaryotic RNAPII and complexes thereof (Cramer et al. 2001; Gnatt et al. 2001; Bushnell and Kornberg, 2003; Bushnell et al. 2002, 2003; Westover et al. 2004a, 2004b; Kettenberger et al. 2004; PDB accession numbers 1I3Q, 1I50, 1I6H, 1K83, 1NIK, 1NT9, 1PQV, 1R5U, 1R9S, 1R9T, 1SFO, 1TWA, 1TWC, 1TWF, 1TWG, 1TWH, 1Y1V, 1Y1W, 1Y1Y, 1Y77, and 1WCM.). Kornberg and co-workers have hypothesized that alternative straight and bent bridge-helix conformations exist in both bacterial RNAP and eukaryotic RNAPII, and have hypothesized that cycling between alternative straight and bent bridge-helix conformations accompanies, and possibly drives, RNAP translocation--hypothesizing that the transition between the pre-translocated state and the post-translocated state is accompanied by, and possibly requires, bending and re-straightening of the bridge helix (Cramer et al. 2001; Gnatt et al. 2001). The “swing-gate” model and “two-pawl-ratchet” model have elaborated on this hypothesis (Epshtein et al. 2002; Bar-Nahum et al. 2005): the “swing-gate” model proposed a specific mechanism by which bridge-helix conformational cycling could mediate both RNAP translocation and NTP entry (Epshtein et al. 2002); the “two-pawl-ratchet” model proposed specific mechanisms by which bridge-helix conformational cycling could serve one of two “pawls” mediating RNAP translocation and maintaining RNAP translocational state (Bar-Nahum et al. 2005). Nevertheless, it previously has not been possible to distinguish between the hypothesis that the different bridge-helix conformations in structures of bacterial RNAP and structures of eukaryotic RNAPII reflect solely a species difference incidental to RNAP function, and the hypothesis that alternative bridge-helix conformational states exist, and may be functionally relevant, for both bacterial RNAP and eukaryotic RNAPII. Crosslinking results consistent with the existence of alternative bridge-helix conformational states in alternative RNAP translocational states have been reported, but these results have permitted no firm conclusions regarding the character of said states (Epshtein et al. 2002; Bar-Nahum et al. 2005). The present structures provide direct, unequivocal evidence for the existence of alternative bridge-helix conformational states in a single RNAP in a single crystal form.
The structures of RNAP and RNAP-Stl herein were obtained in identical crystal forms with nearly identical unit-cell dimensions (Table 2). Therefore, the difference in bridge-helix conformation in the structure of RNAP-Stl results can be directly attributed to the presence of Stl. The difference in bridge-helix conformation in the structure of RNAP-Stl can be understood in terms of two specific RNAP-Stl interactions: (1) Stl directly interacts with residues of the bridge helix (Figs. 4,5), and (2) Stl sterically displaces the trigger loop from its position in the absence of Stl and, concomitantly, disrupts potentially stabilizing interactions with the bent bridge helix that occur in the absence of Stl (Fig. 5B,C). We conclude that Stl stabilizes an RNAP-active-center conformational state having a straight bridge helix.
Strikingly, the residues inferred to represent an allosteric subdeterminant for Stl, β′ residues 791 and 793, are located at the site at which the bridge helix bends and make distinct interactions in straight-bridge-helix and bent-bridge-helix conformational states (“hinge residues”; Fig. 5C). Also, strikingly, both Stlr substitutions obtained within this subdeterminant are substitutions expected to disfavor the straight state relative to the bent state: the β′ Ala791→Gly substitution is predicted to decrease helical propensity (Chou and Fasman, 1974), and thus is expected to disfavor the straight state; the β′ Ser793→Phe substitution is predicted to introduce steric clash with trigger-loop residues in the straight state, and thus is expected to disfavor the straight state. We conclude that Stl binding and/or Stl function requires an RNAP-active-center conformational state having a straight bridge helix.
Our results are consistent with two alternative hypotheses. According to the first hypothesis, Stl binds to an RNAP-active-center conformational state having a straight bridge helix and corresponding specific trigger-loop conformation, and Stl inhibits RNAP function by trapping the RNAP active center in this conformational state, thereby interfering with conformational cycling important for RNAP function. According to the second hypothesis, Stl binds to and favors an RNAP-active-center conformational state having a straight bridge helix and corresponding specific trigger-loop conformation, but Stl inhibits RNAP function by another mechanism (e.g., direct, steric interference with nucleic acid preventing translocation, or non-bridge-helix-related allosteric effects on NTP binding, catalysis, or translocation). We favor the simpler hypothesis: i.e., Stl inhibits RNAP function by trapping a straight-bridge-helix RNAP-active-center conformational state, thereby interfering with conformational cycling important for RNAP function. A similar model has been proposed for α-amanitin, an inhibitor of eukaryotic RNAPII that interacts with the bridge helix from a site within the NTP-uptake channel (interacting with the “underside” of the bridge helix in the view orientation in Fig. 4B,C; Bushnell and Kornberg, 2003). However, in that case, no direct evidence has been presented for conformational cycling of the bridge helix, and the possibility that the inhibitor instead functions by at least partly obstructing the NTP-uptake channel and interfering with NTP uptake (like MccJ25; Adelman et al. 2004; Mukhopadhyay et al. 2004) has not been excluded. The possibility also remains that a structure of RNAP complexed with Stl in the presence of bound nucleic acid, which is not yet available, would yield unexpected insights in terms of conformation and/or access of substrates that would modify our current perspective.
The structures have important implications for understanding the mechanism of RNAP function. Specifically:
The structures provide direct support for proposals that alternative straight-bridge-helix and bent-bridge-helix RNAP-active-center conformational states exist (Cramer et al. 2001; Gnatt et al. 2001; Vassylyev et al. 2002; Epshtein et al. 2002; Bushnell and Kornberg, 2003; Bar-Nahum et al. 2005).
In conjunction with the known inhibition of RNAP function by Stl (Siddhikol et al. 1969; Cassani et al. 1971; McClure, 1980) and our results indicating that bridge-helix hinge residues constitute an allosteric determinant for Stl (Fig. 5C), the structures provide direct support for proposals that cycling between straight-bridge-helix and bent-bridge-helix RNAP-active-center conformational states is important for RNAP function (Cramer et al. 2001; Gnatt et al. 2001; Vassylyev et al. 2002; Epshtein et al. 2002; Bushnell and Kornberg, 2003; Bar-Nahum et al. 2005).
In conjunction with our results regarding effects of Stl on RNAP translocational states (Fig. 2), the structures suggest that transition to a straight-bridge-helix RNAP-active-center conformation disfavors, and possibly excludes, backtracked states.
In conjunction with our results regarding effects of Stl on RNAP translocational states (Fig. 2), the structures suggest that transition to a straight-bridge-helix RNAP-active-center conformation disfavors the pre-translocated state relative to the post-translocated state.
Taken together, the results appear to support the basic proposals of Kornberg and co-workers regarding bridge-helix conformational cycling in RNAP function (Cramer et al. 2001; Gnatt et al. 2001), argue against two more specific models for bridge-helix conformational cycling in RNAP function (Epshtein et al. 2002; Bar-Nahum et al. 2005), and set bounds on any future more specific model for bridge-helix conformational cycling in RNAP function. The “swing-gate” model (Epshtein et al. 2002) proposed that transition to a straight-bridge-helix active-center conformation would favor backtracked states and pre-translocated states relative to the post-translocated state (and expressly predicted that Stl would stabilize a bent-bridge-helix active-center conformational state), predictions that are inconsistent with our results. The version of the “two-pawl ratchet” model (Bar-Nahum et al. 2005) that was solved analytically and used for quantitative simulations in published work [a version in which parameter Kb+/Kb-, was much less than parameter KFb+/KFb- and much less than 1 (parameters defined as in Supplemental Fig. S1 of Bar-Nahum et al. 2005)] proposed that transition to a straight-bridge-helix active-center conformational state would favor the pre-translocated state relative to the post-translocated state--and thus appears to be inconsistent with our results. However, other versions of the two-pawl-ratchet model, with different parameters, are not excluded; such other versions of the two-pawl-ratchet model are consistent with all results in Bar-Nahum et al. 2005 and all results herein.
These results also have implications for development of novel antibiotics. The proximity of the Stl tetramic-acid moiety to the modeled position of the downstream fork junction (Fig. 4) suggests that attachment of a DNA binding moiety to the tetramic-acid moiety should result in increased affinity for transcription complexes, and that attachment of a sequence-specific DNA binding moiety should result in sequence-specific, potentially gene-specific, increased affinity for transcription complexes. The recent total synthesis of a tetramic-acid antibiotic with an aromatic moiety pendant on the tetramic-acid moiety provides a potential route to such compounds (Iwata et al. 2005).
Experimental Procedures
Stl
Stl was provided by E. Steinbrecher of Pharmacia & Upjohn, Kalamazoo MI.
Plasmids
Plasmid pREII-NHα encodes N-terminally hexahistidine-tagged E. coli RNAP α subunit under control of tandem lpp and ′lacUV5 promoters (Niu et al. 1996). Plasmid pRL706 encodes C-terminally hexahistidine-tagged E. coli RNAP β subunit under control of the trc promoter (Severinov et al. 1997). Plasmid pRL663 encodes C-terminally hexahistidine-tagged E. coli RNAP β′ subunit under control of the tac promoter (Wang et al. 1995).
RNAP: E coli
E. coli RNAP core and holoenzyme were prepared as in Niu et al. 1996. Derivatives of RNAP core and holoenzyme containing substitutions in β were prepared from strain XE54 (thi; Tang et al. 1994) transformed with pRL706 derivatives, using analogous procedures. Derivatives of RNAP core and holoenzyme containing substitutions in β′ were prepared from strain 397c [rpoCts397 argG thi lac (λcI857h80St68dlac+); Christie et al. 1996] transformed with pRL663 derivatives, using analogous procedures. Purities typically were >95%.
RNAP: T. thermophilus
RNAP holoenzyme was isolated from T. thermophilus strain HB-8 as in Vassylyeva et al. 2002, with an overall yield of ∼8 mg of electrophoretically homogeneous protein (>95% purity) per 100 g of wet cell paste. Quantitative in vitro transcription assays (methods as in Vassylyeva et al. 2002; Laptenko and Borukhov, 2003) revealed that the preparation contains ∼80% catalytically active molecules.
StlR mutants: isolation
Saturation mutagenesis and selection of antibiotic-resistant mutants was performed by a variation of the method in Mukhopadhyay et al. 2004. A set of “doped” oligodeoxyribonucleotide primers corresponding to codons 158-165, 166-173, 443-447, 512-522, 523-532, 535-541, 542-549, and 563-573 of the rpoB gene of plasmid pRL706, and codons 325-335, 336-346, 425-429, 726-740, 741-754, 775-790, 791-799, 922-933, 1133-1137, 1138-1146, 1147-1152, and 1239-1248 of the rpoC gene of plasmid pRL663, was synthesized on an AB392 automated synthesizer (Applied Biosystems) using solid-phase β-cyanoethylphosphoramidite chemistry (sequences in Supplemental Table S1). The level of “doping” (nucleotide misincorporation) was selected to yield an average of 0.4-1 substitution per molecule of oligodeoxyribonucleotide primer (equations in Hermes et al. 1989, 1990). Thus, the nucleotides corresponding to codons 443-447 and 542-649 of rpoB, and 424-429, 1133-1137, and 1147-1152 of rpoC, were synthesized using phosphoramidite reservoirs containing 92% of the correct phosphoramidite and 8% of a 1:1:1:1 mix of dA, dC, dG, and dT phosphoramidites (i.e., 94% total correct phosphoramidite and 6% total incorrect phosphoramidite); the nucleotides corresponding to codons 158-165, 166-173, 512-522, 523-532, 535-541, and 563-573 of rpoB, and codons 325-335, 336-346, 726-740, 741-754, 775-790, 791-799, 922-933, 1138-1146, and 1239-1248 of the rpoC, were synthesized using phosphoramidite reservoirs containing 98% of the correct phosphoramidite and 2% of a 1:1:1:1 mix of dA, dC, dG, and dT phosphoramidites (i.e., 98.5% total correct phosphoramidite and 1.5% total incorrect phosphoramidite); and all other nucleotides were synthesized using phosphoramidite reservoirs containing 100% of the correct phosphoramidite. Primer-extension mutagenesis reactions were performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene), with a “doped” oligodeoxyribonucleotide primer, a complementary wild-type oligodeoxyribonucleotide primer, and pRL706 or pRL663 as template (primers at 75 nM; all other components at concentrations as specified by the manufacturer). Mutagenized plasmid DNA was introduced by transformation into strain XL1-Blue [hsdR17 recA1 endA1 relA1 gyrA96 lac thi supE44 (F' proAB lacIqZΔM15 Tn10); Stratagene], transformants (∼104 cells) were applied to LB-agar plates (Sambrook and Russel, 2001) containing 200 μg/ml ampicillin, plates were incubated 16 h at 37°C, and plasmid DNA was prepared from the pooled resulting colonies. The resulting passaged mutagenized plasmid DNA was introduced by transformation into strain D21f2/tolC∷Tn10 (tolC:Tn10 rfa lac28 proA23 trp30 his51 rpsL173 ampC tsx81; a strain with cell-envelope defects resulting in increased susceptibility to hydrophobic agents, including Stl; Fralick and Burns-Keliher, 1994; unpublished data), transformants (∼104 cells) were applied to LB-agar plates containing 2 μg/ml Stl, 200 μg/ml ampicillin, and 1mM IPTG, and plates were incubated 16-24 h at 37°C. For each StlR clone (identified as a clone yielding a colony on the original selective plate and also yielding colonies when re-streaked to the same medium and incubated 16 h at 37°C), plasmid DNA was prepared, plasmid DNA was introduced by transformation into strain D21f2/tolC∷Tn10, transformants (∼104 cells) were applied to LB-agar plates containing 2 μg/ml Stl, 200 μg/ml ampicillin, and 1mM IPTG and, in parallel, to LB-agar plates containing 200 μg/ml ampicillin and 1mM IPTG, and plates were incubated 16 h at 37°C. For each plasmid-linked StlR clone (identified as a clone yielding comparable numbers of colonies on the plates with and without Stl), the nucleotide sequence of the mutagenized rpoB or rpoC segment was determined by dideoxy nucleotide sequencing.
StlR mutants: complementation assays
For StlR mutants in rpoB, temperature-sensitive strain RL585 [rpoBamcI supDts43,74 Δ(recA-srl)306 lacZam 2110 galEKam leuam trpam sueA rpsL tsx srl301∷Tn10-84; Landick et al. 1990] was transformed with a pRL706 derivative, transformants (∼104 cells) were applied to LB-agar plates containing 200 μg/ml ampicillin, and 10 μg/ml tetracycline, plates were incubated 16 h at 37°C, and bacterial growth was scored. For StlR mutants in rpoC, temperature-sensitive strain 397c (Christie et al. 1996) was transformed with a pRL663 derivative, transformants (∼104 cells) were applied to LB-agar plates containing 200 μg/ml ampicillin, plates were incubated 16 h at 37°C, and bacterial growth was scored.
StlR mutants: minimal inhibitory concentration (MIC) assays
Single colonies of strain D21f2/tolC∷Tn10 (Fralick and Burns-Keliher, 1994) transformed with a pRL706 or pRL663 derivative were inoculated into 3 ml LB containing 200 μg/ml ampicillin, and incubated 3-4 h at 37°C with shaking. Diluted aliquots (105 cells in 10 μl LB; concentration determined using OD600 = 1 for 109 cells) were subcultured in 1 ml LB containing 0, 1.25, 2.5, 5, 10, or 20 μg/ml Stl and incubated 16 h at 37°C with shaking. The MIC was defined as the lowest concentration of Stl that reduced OD600 by ≥95%.
RNAP translocational state: TEC16
Reaction mixtures for preparation of transcription elongation complexes containing a 16 nt RNA product with a non-extendable, 3′-deoxy-3′-amino terminus (TEC16) contained (200 μl): 10 nM hexahistidine-tagged RNAP holoenzyme, 40 nm DNA fragment lacUV5(UHD) 32P-labeled at the 5′ end of the DNA template strand (sequence in Figure 2A; prepared by PCR amplification, using duplex of annealed 86 nt synthetic oligodeoxyribonucleotides, nontemplate-strand primer, and 32P-5′-end-labeled template-strand primer), 1 mM ApA (Sigma-Aldrich), 50 μM 3′-amino-3′-deoxy ATP (IBA) 25 μM CTP, 25 μM GTP, and 25 μM UTP in buffer A (30 mM HEPES, pH 8.0, 100 mM NaCl, 10 mM MgCl2, 1 mM β-mercaptoethanol, and 0.05% Tween-20). Reactions were initiated by addition of ApA and NTPs (in 10 μl buffer A) to other components (in 190 μl buffer A; pre-equilibrated 10 min at 37°C) and were allowed to proceed for 10 min at 37°C. TEC16 was adsorbed onto Ni2+-NTA-agarose by addition of 200 μl Ni2+-NTA-agarose slurry (Qiagen; washed twice with 2 ml buffer A and re-suspended in 200 μl buffer A), incubated 10 min at 37°C, and centrifuged at 14,000 × g for 1 min at 4°C; was washed three times with 1 ml buffer A containing 1.1 M KCl (each time with incubation 10 min at 4°C, followed by centrifugation at 14,000 × g for 1 min at 4°C); and was washed four times with 1 ml buffer A (each time with incubation 30 s at 4°C, followed by centrifugation at 14,000 × g for 1 min at 4°C). The resulting Ni2+-NTA-agarose-immobilized, NTP-free TEC16 was re-suspended in 180 μl buffer A at 25°C, and subdivided into 16 μl aliquots.
RNAP translocational state: exonuclease-III DNA footprinting
To a 16 μl aliquot of Ni2+-NTA-agarose-immobilized, washed, NTP-free TEC16 (preceding section; used immediately after preparation) at 25°C, was added 2 μl buffer A, 2 μl 200 μM Stl in buffer A, or 2 μl 500 μM GTP in buffer A, and, after 15 s at 25°C, 2 μl 5 U/ml exonuclease III (New England Biolabs). Reactions were allowed to proceed for 2.5, 5, or 10 min at 37°C, and were terminated by addition of 20 μl 80% formamide, 10 mM EDTA, 0.04% bromophenol blue, and 0.04% xylene cyanol, and incubation 10 min at 65°C. Products of the reaction, and, in parallel, products of a G+A nucleotide-sequencing reaction (Maxam and Gilbert, 1980) of the same DNA fragment, were resolved by urea-PAGE (Sambrook and Russel, 2001), and quantified using an x/y storage-phosphor scanner (Storm 860; Molecular Dynamics).
Structure determination: crystallization and cryocooling
Crystallization and crystal-handling procedures were based on protocols described in Vassylyeva et al. 2002. A stock crystallization solution was prepared by adding T. thermophilus RNAP holoenzyme (10 mg/ml) in 20 mM Tris-HCl, pH 7.7, 100 mM NaCl, and 1% glycerol to an equal volume of 33 mM magnesium formate containing 40 μM ZnCl2. A 2 μl aliquot of this solution was placed in a microbridge sitting-drop or vapor-diffusion hanging-drop crystallization apparatus. Small (∼0.05-0.1 mm) hexagonal crystals formed within one to two days and grew to a final size of ∼.35 × .35 × .2 mm within four to five days. The largest RNAP holoenzyme crystals were transferred to a solution of 20 mM MES, pH 5.85, 13 mM magnesium formate, 2 mM spermine, 2 mM DTT, 5% PEG 8000, and 10-15% MPD, with or without 1 mM streptolydigin, for 10-15 min, then removed and flash-cooled in liquid nitrogen.
Structure determination: data collection and reduction
Diffraction data for RNAP-Stl and RNAP crystals were collected at Brookhaven National Laboratory National Synchrotron Light Source (BNL-NSLS) beamline X-25 and Cornell High Energy Synchrotron Source (CHESS) beamline F1. Data were processed and scaled using HKL 2000 (Otwinowski and Minor, 1997; crystallographic statistics in Table 2).
Structure determination: structure solution and refinement
Initial solution of the RNAP-Stl and RNAP structures was carried out using molecular-replacement methods in CNS 1.1 (Brünger et al. 1998) using PDB code 1IW7 (Vassylyev et al. 2002) as the search model. The data were found to have perfect hemihedral twinning [consistent with the prior description of this crystal form (Vassylyeva et al. 2002; Vassylyev et al. 2002); twin operator = -h,-k,l; twin fraction = 0.5] and were de-twinned (Yeates, 1997) prior to structure solution and crystallographic refinement, resulting in a change of effective space group symmetry from P65 to P32 (with two RNAP molecules in the asymmetric unit). Early stages of refinement of both the RNAP-Stl and RNAP structures included rigid body refinement of each RNAP molecule in the asymmetric unit, followed by rigid body refinement of each subunit of each RNAP molecule, followed by rigid body refinement of 40 subdivisions (consisting of ∼15-200 amino acid residue segments) of each molecule. Initial electron density maps were improved by applying density modification and solvent flattening using program DM in the CCP4 program suite [Collaborative Computational Project, Number 4; locally modified to handle the large number of noncrystallographic symmetry (NCS) transformations]. Two-fold NCS averaging was performed for 37 subdivisions in β′, β, ω, and σ subunits, and four-fold NCS averaging was performed for 3 subdivisions in αI and αII subunits. Phases used for computing the density map shown in Fig. 3 (which showed minimal bias from the input model coordinates) were derived from phase extension from 8 Å to 3 Å in 1000 cycles, with 0.25 weight applied to the input model-derived phases (Stl coordinates were not included in any model prior to and at this stage) when combining with NCS-averaged and solvent-flattened phases. Electron density maps generated by other approaches, including phase extension from 4 to 3 Å in 100-1000 cycles, were less clear. Rounds of refinement in CNS 1.1 used torsion angle annealing, followed by 20 cycles of positional refinement. The density-modified, NCS-averaged electron density maps produced from the refined coordinates were used to model Stl and to carry out manual model building of RNAP using the program O (Jones et al. 1991). The most plausible and energetically stable conformers of Stl were constructed and analyzed by conducting a systematic of Stl conformational space using molecular dynamics simulations in modeling program MOLOC (Gerber and Muller, 1995). The ten lowest energy conformers were analyzed for the best fit to the electron density map. Among these one could be fitted with only minor bond rotation adjustments to the density in both RNAP molecules. Subsequent refinement and model building cycles were performed, leading to the current crystallographic model, with a standard crystallographic residual of R = 0.261 and Rfree = 0.281 computed using all data with |Fo|>0 between 30 and 3.0 Å resolution (Table 2). Successive rounds of de-twinning, as prescribed in Yeates, 1997, and implemented in CNS, resulted in considerable density-map improvement and permitted unambiguous fitting of Stl into maps calculated using density modification using DM, model refinement using CNS, and solvent flipping using CNS following model refinement. Density consistent with ordered C-terminal extensions in αII subunits permitted extension of the model by an additional six residues (B230-B235 and L230-L235) relative to the input model (Vassylyev et al. 2002). Analogous procedures were used to refine the RNAP dataset, but, in view of the lower resolution of the RNAP dataset than the RNAP-Stl dataset (3.3 Å vs. 3.0 Å), only torsion angle refinement was applied. Atomic coordinates and structure factors for RNAP-Stl and RNAP have been deposited in the PDB (PDB accession numbers XXX and YYY).
Supplementary Material
Acknowledgments
We thank E. Steinbrecher and Pharmacia & Upjohn, Inc. for streptolydigin, J. Fralick and R. Landick for bacterial strains, staff members at BNLS and CHESS and S. Ginell at APS for assistance with synchrotron data collection, and H. Berman, K. Das, J. Ding, C. Lawson, J. Patel, and members of our laboratories for assistance and discussions. This work was supported by NIH grants GM41376 to R.H.E., AI27690 and GM66671 to E.A., GM54098 to S.B., and GM21589 to H. Berman (supported A.N.), and by a Howard Hughes Medical Investigatorship to R.H.E.
References
- Adelman K, Yuzenkova J, La Porta A, Zenkin N, Lee J, Lis J, Borukhov S, Wang M, Severinov K. Molecular mechanism of transcription inhibition by peptide antibiotic Microcin J25. Mol Cell. 2004;14:753–762. doi: 10.1016/j.molcel.2004.05.017. [DOI] [PubMed] [Google Scholar]
- Armache K, Kettenberger H, Cramer P. Architecture of initiation-competent 12-subunit RNA polymerase II. Proc Natl Acad Sci USA. 2003;100:6964–6968. doi: 10.1073/pnas.1030608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artsimovitch I, Chu C, Lynch A, Landick R. A new class of bacterial RNA polymerase inhibitor affects nucleotide addition. Science. 2003;302:650–654. doi: 10.1126/science.1087526. [DOI] [PubMed] [Google Scholar]
- Artsimovitch I, Patlan V, Sekine S, Vassylyeva M, Hosaka T, Ochi K, Yokoyama S, Vassylyev D. Structural basis for transcription regulation by alarmone ppGpp. Cell. 2004;117:299–310. doi: 10.1016/s0092-8674(04)00401-5. [DOI] [PubMed] [Google Scholar]
- Bar-Nahum G, Epshtein V, Ruckenstein A, Rafikov R, Mustaev A, Nudler E. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- Brünger A, Adams P, Clore G, DeLano W, Gros P, Grosse-Kunstleve R, Jiang JS, Kuszewski J, Nilges M, Pannu N, et al. Crystallographic and NMR system: a new software suite for macromolecular structure determination. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Bushnell D, Cramer P, Kornberg R. Structural basis of transcription: alpha-amanitin-RNA polymerase II cocrystal at 2.8 A resolution. Proc Natl Acad Sci USA. 2002;99:1218–1222. doi: 10.1073/pnas.251664698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell D, Kornberg R. Complete, 12-subunit RNA polymerase II at 4.1-A resolution: implications for the initiation of transcription. Proc Natl Acad Sci USA. 2003;100:6969–6973. doi: 10.1073/pnas.1130601100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell D, Westover K, Davis R, Kornberg R. Structural basis of transcription: an RNA polymerase II-TFIIB cocrystal at 4.5 angstroms. Science. 2004;303:983–988. doi: 10.1126/science.1090838. [DOI] [PubMed] [Google Scholar]
- Campbell E, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst S. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104:901–912. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Campbell E, Pavlova O, Zenkin N, Leon F, Irschik H, Jansen R, Severinov K, Darst S. Structural, functional, and genetic analysis of sorangicin inhibition of bacterial RNA polymerase. EMBO J. 2005;24:674–682. doi: 10.1038/sj.emboj.7600499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassani G, Burgess R, Goodman H, Gold L. Inhibition of RNA polymerase by streptolydigin. Nature New Biol. 1971;230:197–200. doi: 10.1038/newbio230197a0. [DOI] [PubMed] [Google Scholar]
- Chou P, Fasman G. Prediction of protein conformation. Biochem. 1974;13:222–245. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- Christie G, Cale S, Iraksson L, Jin D, Xu M, Sauer B, Calendar R. Escherichia coli rpoC397 encodes a temperature-sensitive C-terminal frameshift in the b' subunit of RNA polymerase that blocks growth of bacteriophage P2. J Bacteriol. 1996;178:6991–6993. doi: 10.1128/jb.178.23.6991-6993.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;D54:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cramer P. Multisubunit RNA polymerases. Curr Opin Struct Biol. 2002;12:89–97. doi: 10.1016/s0959-440x(02)00294-4. [DOI] [PubMed] [Google Scholar]
- Cramer P, Bushnell D, Kornberg R. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863–1876. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- Darst S. Bacterial RNA polymerase. Curr Opin Structl Biol. 2001;11:155–162. doi: 10.1016/s0959-440x(00)00185-8. [DOI] [PubMed] [Google Scholar]
- Ebright R. RNA polymerase: structural similarities between bacterial RNA polymerase and eukaryotic RNA polymerase II. J Mol Biol. 2000;304:687–698. doi: 10.1006/jmbi.2000.4309. [DOI] [PubMed] [Google Scholar]
- Epshtein V, Mustaev A, Markovtsov V, Bereshchenko O, Nikiforov V, Goldfarb A. Swing-gate model of nucleotide entry into the RNA polymerase active center. Mol Cell. 2002;10:623–634. doi: 10.1016/s1097-2765(02)00640-8. [DOI] [PubMed] [Google Scholar]
- Fralick J, Burns-Keliher L. Additive effect of tolC and rfa mutations on the hydrophobic barrier of the outer membrane of Escherichia coli K-12. J Bacteriol. 1994;176:6404–6406. doi: 10.1128/jb.176.20.6404-6406.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber PR, Muller K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J Comput Aided Mol Des. 1995;9:251–68. doi: 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
- Gnatt A, Cramer P, Fu J, Bushnell D, Kornberg R. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- Guajardo R, Lopez P, Dreyfus M, Sousa R. NTP concentration effects on initial transcription by T7 RNAP indicate that translocation occurs through passive sliding and reveal that divergent promoters have distinct NTP concentration requirements for productive initiation. J Mol Biol. 1998;281:777–792. doi: 10.1006/jmbi.1998.1988. [DOI] [PubMed] [Google Scholar]
- Heisler L, Suzuki H, Landick R, Gross C. Four contiguous amino acids define the target for streptolydigin resistance in the beta subunit of Escherichia coli RNA polymerase. J Biol Chem. 1993;268:25369–25375. [PubMed] [Google Scholar]
- Hermes J, Blacklow S, Knowles J. Searching sequence space by definably random mutagenesis: improving the catalytic potency of an enzyme. Proc Natl Acad Sci USA. 1990;87:696–700. doi: 10.1073/pnas.87.2.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermes J, Parekh S, Blacklow S, Koster H, Knowles J. A reliable method for random mutagenesis: the generation of mutant libraries using spiked oligodeoxyribonucleotide primers. Gene. 1989;84:143–151. doi: 10.1016/0378-1119(89)90148-0. [DOI] [PubMed] [Google Scholar]
- Holmes S, Erie D. Downstream DNA sequence effects on transcription elongation: allosteric binding of nucleoside triphosphates facilitates translocation via a ratchet motion. J Biol Chem. 2003;278:35597–35608. doi: 10.1074/jbc.M304496200. [DOI] [PubMed] [Google Scholar]
- Iwata Y, Maekawara N, Tanino K, Miyashita M. Tetramic acid antibiotics: stereoselective synthesis of streptolic acid and tirandalydigin. Angew Chem Int Ed. 2005;44:1532–1536. doi: 10.1002/anie.200462300. [DOI] [PubMed] [Google Scholar]
- Jones T, Zou J, Cowan S, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kettenberger H, Armache K, Cramer P. Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell. 2003;114:347–357. doi: 10.1016/s0092-8674(03)00598-1. [DOI] [PubMed] [Google Scholar]
- Kettenberger H, Armache K, Cramer P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell. 2004;16:955–965. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- Korzheva N, Mustaev A, Kozlov M, Malhotra A, Nikiforov V, Goldfarb A, Darst S. A structural model of transcription elongation. Science. 2000;289:619–625. doi: 10.1126/science.289.5479.619. [DOI] [PubMed] [Google Scholar]
- Landick R, Stewart J, Lee DN. Amino acid changes in conserved regions of the beta-subunit of Escherichia coli RNA polymerase alter transcription pausing and termination. Genes Dev. 1990;4:1623–1636. doi: 10.1101/gad.4.9.1623. [DOI] [PubMed] [Google Scholar]
- Laptenko O, Borukhov S. Biochemical assays of Gre factors of Thermus thermophilus. Meths Enzymol. 2003;371:219–232. doi: 10.1016/S0076-6879(03)71016-7. [DOI] [PubMed] [Google Scholar]
- Lee V, Rinehart K. 13C NMR spectra of streptolydigin, tirandmycin, and related degradation products. J Antibiot. 1980;33:408–415. doi: 10.7164/antibiotics.33.408. [DOI] [PubMed] [Google Scholar]
- Maxam A, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Meths Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- McClure W. On the mechanism of streptolydigin inhibition of Escherichia coli RNA polymerase. J Biol Chem. 1980;255:1610–1616. [PubMed] [Google Scholar]
- Metzger W, Schickor P, Heumann H. A cinematographic view of Escherichia coli RNA polymerase translocation. EMBO J. 1989;8:2745–2754. doi: 10.1002/j.1460-2075.1989.tb08416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay J, Sineva E, Knight J, Levy R, Ebright R. Antibacterial peptide microcin J25 inhibits transcription by binding within, and obstructing, the RNA polymerase secondary channel. Mol Cell. 2004;14:739–751. doi: 10.1016/j.molcel.2004.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Masuda S, Darst S. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 Å resolution. Science. 2002a;296:1280–1284. doi: 10.1126/science.1069594. [DOI] [PubMed] [Google Scholar]
- Murakami K, Masuda S, Campbell E, Muzzin O, Darst S. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002b;296:1285–1290. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- Naryshkin N, Revyakin A, Kim Y, Mekler V, Ebright R. Structural organization of the RNA polymerase-promoter open complex. Cell. 2000;101:601–611. doi: 10.1016/s0092-8674(00)80872-7. [DOI] [PubMed] [Google Scholar]
- Niu W, Kim Y, Tau G, Heyduk T, Ebright R. Transcription activation at Class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell. 1996;87:1123–1134. doi: 10.1016/s0092-8674(00)81806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meths Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- Sarafianos SG, Das K, Clark AD, Jr, Ding J, Boyer PL, Hughes SH, Arnold E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with β-branched amino acids. Proc Natl Acad Sci USA. 1999;96:10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinov K, Markov D, Severinova E, Nikiforov V, Landick R, Darst S, Goldfarb A. Streptolydigin-resistant mutants in an evolutionarily conserved region of the beta' subunit of Escherichia coli RNA polymerase. J Biol Chem. 1995;270:23926–23929. doi: 10.1074/jbc.270.41.23926. [DOI] [PubMed] [Google Scholar]
- Severinov K, Mooney R, Darst SA, Landick R. Tethering of the large subunits of Escherichia coli RNA polymerase. J Biol Chem. 1997;272:24137–24140. doi: 10.1074/jbc.272.39.24137. [DOI] [PubMed] [Google Scholar]
- Siddhikol C, Erbstoeszer J, Weisblum B. Mode of action of streptolydigin. J Bacteriol. 1969;99:151–155. doi: 10.1128/jb.99.1.151-155.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov V, Sosunova E, Mustaev A, Bass I, Nikiforov V, Goldfarb A. Unified two-metal mechanism of RNA synthesis and degradation by RNA polymerase. EMBO J. 2003;22 doi: 10.1093/emboj/cdg193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Severinov K, Goldfarb A, Fenyo D, Chait B, Ebright R. Location, structure, and function of the target of a transcription activator protein. Genes Dev. 1994;8:3058–3067. doi: 10.1101/gad.8.24.3058. [DOI] [PubMed] [Google Scholar]
- Vassylyev D, Sekine S, Laptenko O, Lee J, Vassylyeva M, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- Vassylyeva M, Lee J, Sekine S, Laptenko O, Kuramitsu S, Shibata T, Inoue Y, Borukhov S, Vassylyev D, Yokoyama S. Purification, crystallization and initial crystallographic analysis of RNA polymerase holoenzyme from Thermus thermophilus. Acta Cryst. 2002;D50:760–763. doi: 10.1107/S0907444902011770. [DOI] [PubMed] [Google Scholar]
- Wang D, Meier T, Chan C, Feng G, Lee D, Landick R. Discontinuous movements of DNA and RNA in RNA polymerase accompany formation of a paused transcription complex. Cell. 1995;81:341–350. doi: 10.1016/0092-8674(95)90387-9. [DOI] [PubMed] [Google Scholar]
- Westover K, Bushnell D, Kornberg R. Structural basis of transcription: nucleotide selection by rotation in the RNA polymerase II active center. Cell. 2004a;119:481–489. doi: 10.1016/j.cell.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Westover K, Bushnell D, Kornberg R. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 2004b;303:1014–1016. doi: 10.1126/science.1090839. [DOI] [PubMed] [Google Scholar]
- Yang X, Price C. Streptolydigin resistance can be conferred by alterations to either the beta or beta' subunits of Bacillus subtilis RNA polymerase. J Biol Chem. 1995;270:23930–23933. doi: 10.1074/jbc.270.41.23930. [DOI] [PubMed] [Google Scholar]
- Yeates T. Detecting and overcoming crystal twinning. Meths Enzymol. 1997;276:344–358. [PubMed] [Google Scholar]
- Yuzenkova J, Delgado M, Nechaev S, Savalia D, Epshtein V, Artsimovitch I, Mooney R, Landick R, Farias R, Salomon R, Severinov K. Mutations of bacterial RNA polymerase leading to resistance to microcin J25. J Biol Chem. 2002;277:50867–50875. doi: 10.1074/jbc.M209425200. [DOI] [PubMed] [Google Scholar]
- Zhang G, Campbell E, Minakhin L, Richter C, Severinov K, Darst S. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.