

Abstract
The urokinase-type plasminogen activator receptor (uPAR/CD87) is a glycosylphosphatidylinositol-anchored membrane protein with multiple functions in extracellular proteolysis, cell adhesion, cell migration and proliferation. We now report that cell surface uPAR dimerizes and that dimeric uPAR partitions preferentially to detergent-resistant lipid rafts. Dimerization of uPAR did not require raft partitioning as the lowering of membrane cholesterol failed to reduce dimerization and as a transmembrane uPAR chimera, which does not partition to lipid rafts, also dimerized efficiently. While uPA bound to uPAR independently of its membrane localization and dimerization status, uPA-induced uPAR cleavage was strongly accelerated in lipid rafts. In contrast to uPA, the binding of Vn occurred preferentially to raft- associated dimeric uPAR and was completely blocked by cholesterol depletion.
Keywords: dimerization/GPI/lipid rafts/uPAR/Vn
Introduction
In tissues, extracellular proteolysis is controlled by the production of plasmin which is generated by plasminogen activators, mainly urokinase (uPA, Johnsen et al., 1998). This protease binds with high affinity to a glycosylphosphatidylinositol-anchored membrane receptor, uPAR/CD87 (Ploug et al., 1991). Besides its direct and well described role in extracellular proteolysis, uPAR has important functions in cell adhesion, migration and proliferation (reviewed in Blasi and Carmeliet, 2002). The functions of uPAR in the latter processes are all dependent upon its interaction with other membrane proteins including members of the integrin family (Wei et al., 1996), chemotactic receptors (Resnati et al., 2002), receptor tyrosine kinases like the epidermal growth factor receptor (Liu et al., 2002), as well as with proteins present in the extracellular matrix including vitronectin (Vn, Waltz and Chapman, 1994). Although the many interactions entertained by uPAR are well documented, little is known about the molecular mechanisms involved, how the specificity of these contacts is attained and what are the resulting functional consequences.
Dimerization is the mechanism responsible for activation of most, if not all, transmembrane receptors (for review see Schlessinger, 2000). For these receptors, dimerization follows ligand binding and leads to the activation of protein kinase activity. Receptors attached to the membrane by a GPI-anchor cluster in particular detergent-resistant membrane microdomains known as lipid rafts, where they may make contact with various signalling molecules (reviewed in Simons and Toomre, 2000). The intrinsic dimerization of GPI-anchored membrane receptors, and the possible biological significance, has not been described to date. It has been hypothesized that oligomerization of GPI-anchored proteins may regulate their association with lipid raft membrane domains (Simons and Toomre, 2000). We recently demonstrated that dimerization is an intrinsic property of a soluble uPAR (suPAR) molecule, and that it plays a key role in suPAR interaction with Vn (Sidenius et al., 2002). Dimerization of uPAR may therefore not only determine its association with other proteins, but also direct these interactions to discrete membrane domains.
In this paper we demonstrate that cell surface uPAR partitions to different membrane domains depending on its state of dimerization. In addition, we show that the differential membrane partitioning of uPAR is functionally relevant in directing molecular interactions and biological processes to discrete membrane domains.
Results
uPAR partitions to biochemically distinct membrane domains
To analyze the partitioning of uPAR to lipid rafts we subjected detergent lysates of uPAR-transfected human embryo kidney 293 cells (293/uPAR) to sucrose density gradient centrifugation analysis (flotation) and assayed the fractions for the presence of uPAR by western blotting (Figure 1A). When cell extracts were prepared at 4°C in lysis buffer containing 1% Triton X-100, uPAR was found both in the bottom of the gradient which contains the soluble non-raft membranes, as well as in the top fractions containing the detergent-resistant lipid rafts. Other membrane proteins, including the transferrin receptor (TfR) and β1-integrin (Figure 1A) were recovered exclusively in the non-raft membrane fraction under identical conditions.
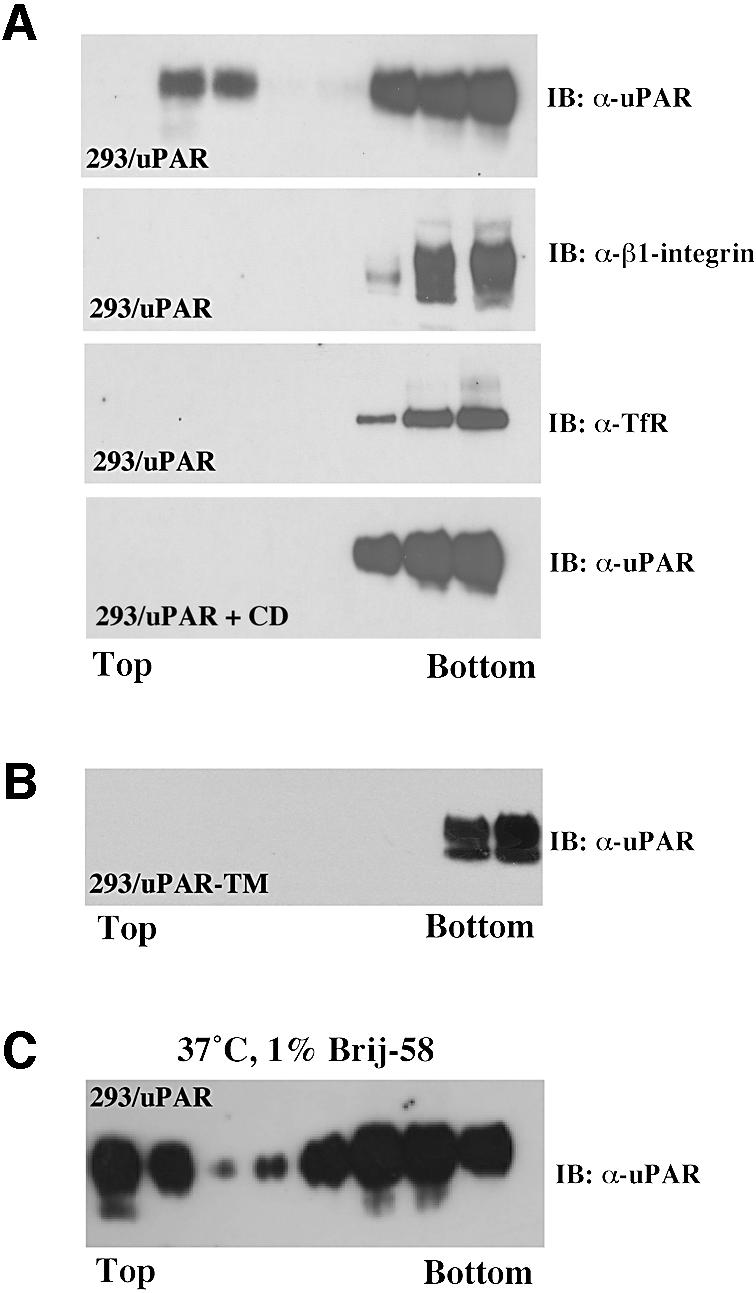
Fig. 1. uPAR partitions to two biochemically distinct membrane domains. (A) Western blot analysis of uPAR membrane localization. 293/uPAR cells were lysed in buffer containing 1% Triton X-100 and subjected to sucrose density gradient ultracentrifugation (see Materials and methods). Equal volumes of the resulting fractions (10 µl) were probed for uPAR using a polyclonal anti-uPAR antibody in immunoblotting. The same fractions were also probed with polyclonal antibodies to β1-integrin and the transferrin receptor, respectively. 293/uPAR cells were first treated with CD (10 mM, 1 h, 37°C) and then subjected to flotation analysis as described above. (B) 293/uPAR-TM-expressing cells were subjected to the same analysis. (C) 293/uPAR cells were detergent lysed in buffer containing 1% Brij-58 at 37°C and subjected to flotation analysis (see Materials and methods). The data presented are representative of several independent experiments.
The association of uPAR with lipid rafts was membrane cholesterol dependent, as pre-treatment of the cells with methyl-β-cyclodextrin (CD) caused a significant reduction in the fraction of uPAR associated with rafts (Figure 1A). Structurally, the partitioning of uPAR to rafts was dependent upon the presence of the GPI-anchor, as a recombinant uPAR molecule anchored to the membrane by the transmembrane domain from the EGF receptor (uPAR-TM) failed to partition to rafts (Figure 1B). To determine if the raft association of uPAR at 4°C was paralleled by a similar raft association at 37°C (Drevot et al., 2002; Braccia et al., 2003), we repeated the flotation experiments using 1% Brij-58 at 37°C (Figure 1C). Under these conditions a fraction of uPAR, quantitatively comparable to that observed using Triton X-100 (1%) at 4°C, was recovered in the raft fractions. No uPAR-TM, β1-integrin or TfR was detected in the raft fraction when cells were lysed with Brij-58 at 37°C (data not shown).
The detergent solubility of molecules has been shown to change during biosynthesis and we therefore addressed the possibility that the two populations of uPAR might represent molecules at different stages of the biosynthetic pathway. To this end, we repeated the flotation experiments on cells whose surface-exposed proteins were specifically labelled using the membrane impermeable biotinylation reagent sulfo-NHS-biotin prior to cell lysis (Brij-58, 1% at 37°C) and flotation (data not shown). In these experiments, the distribution of biotinylated uPAR between raft and non-raft fractions was very similar, demonstrating that both populations of uPAR represent mature cell surface-exposed uPAR.
Cell surface dimerization of uPAR
We have recently shown that a soluble form of uPAR lacking the GPI-anchor undergoes functional dimerization in vitro (Sidenius et al., 2002) and so we wanted to investigate the possible occurrence of dimeric uPAR in living cells. To this end, intact 293/uPAR cells were treated with the membrane-impermeable chemical cross linker BS3 and the cell lysates analyzed for the presence of uPAR-adducts by immunoblotting (Figure 2A). In these experiments the treatment with BS3 prior to cell lysis gave rise to a single prominent uPAR-adduct with an apparent molecular weight of ∼90 kDa, compatible with a uPAR dimer.

Fig. 2. Dimeric cell surface uPAR revealed by chemical cross linking and co-immunoprecipitation. (A) Western blot analysis of uPAR dimerization by chemical cross linking. 293/uPAR cells were treated with the chemical cross linker BS3 as indicated below the panel, washed and lysed. Equal amounts of total protein were separated by SDS–PAGE and analyzed by immunoblotting using a polyclonal anti-uPAR antibody. Similar data were observed in several experiments. (B) Affinity purification of cross-linked uPAR. Lysates obtained from large-scale cross linking experiments carried out on 293/uPAR cells were loaded onto an anti-uPAR antibody affinity column. After washing, bound protein was eluted, concentrated and separated by SDS–PAGE. The slab gel was silver stained and the band corresponding to the uPAR adduct (marked by an asterisk) excised, subjected to trypsin digestion and analyzed by mass spectrometry (see Supplementary data). (C) Lysates from 293 cells expressing either wild-type uPAR (WT), FLAG-tagged uPAR (FLAG) or both (WT+FLAG) were immunoprecipitated with an anti-uPAR antibody (R4) which recognizes both wild-type and FLAG-tagged uPAR, or with an anti-FLAG antibody (M2) which recognizes only the FLAG-tagged receptor. The immunoprecipitated material was fractionated by SDS–PAGE and analyzed by immunblotting using an anti-uPAR antibody (R2) which recognizes only wild-type uPAR (upper panel). To ensure that appropriate immunoprecipitation had been achieved, the blots were also probed with a polyclonal uPAR antibody which recognizes all forms of the receptor (α-uPAR-PC, lower panel). Similar results were obtained in at least three independent experiments.
To directly confirm that the ∼90 kDa adduct represented a uPAR dimer, a preparative cross-linking experiment was performed and the lysate was purified on an anti-uPAR affinity column (see Supplementary data, available at The EMBO Journal Online). Silver staining of slab gels containing the purified material (Figure 2B) revealed the presence of two major bands corresponding to monomeric uPAR and the uPAR-adduct also observed in immunoblotting. The latter band was excised, subjected to in-gel trypsin digestion and the generated peptides were analyzed by MALDI-TOF mass spectrometry (see Supplementary data). Database searches on the obtained peptide map revealed that the adduct, as expected, contained uPAR (Probability = 1.0, 36% coverage). The peptide map did not show a significant match to any other known protein (Probability < 3.4 × 10–6). Based on the molecular weight of the complex and the absence of any other identifiable protein, we conclude that the adduct represents a uPAR dimer.
To obtain independent evidence for the dimerization of cellular uPAR we generated an epitope-tagged uPAR suitable for use in co-immunoprecipitation experiments. The octameric FLAG-epitope was inserted into uPAR between its third domain and the GPI-anchoring signal (see Supplementary data). Besides exposing an epitope for the monoclonal anti-FLAG antibody M2, this small insertion results in the destruction of the binding epitope for the monoclonal anti-uPAR antibody R2 (Figure 2C, upper panel, lanes 2 and 5; Sidenius et al., 2002; data not shown). We transfected 293 cells with wild-type and FLAG-tagged receptors individually, or with a combination of the two receptors, and selected stable clones expressing comparable levels of the two receptors. To analyze the predicted uPAR/uPAR-FLAG interaction, lysates were immunoprecipitated with the anti-FLAG antibody M2 and analyzed by immunoblotting using the R2 antibody. Indeed, co-immunoprecipitation of uPAR and uPAR-FLAG was observed in lysates prepared from cells transfected with both receptors (Figure 2C, lane 6, upper panel). Control immunoprecipitations with a monoclonal antibody which recognizes both forms of the receptor (R4, Figure 2C, lanes 1–3), and re-probing of the blots with a polyclonal anti-uPAR antibody (Figure 2C, lower panel), demonstrate the specificity of the M2 antibody, the non-reactivity of the R2 antibody with uPAR-FLAG and the comparable expression levels of the two receptors in the different cell clones.
Dimeric uPAR partitions preferentially to lipid rafts
A dimeric GPI-anchored protein may be considered a single protein unit with two GPI-anchors and we therefore next addressed the possibility that uPAR dimerization is responsible for its differential lipid raft partitioning. To this end we performed flotation analysis on cells which had been subjected to chemical cross linking prior to cell lysis at 4°C in Triton X-100 buffer and analyzed the fractions for the presence of different dimeric forms of uPAR by western blotting (Figure 3). In contrast to non cross-linked uPAR, which was found to be predominantly detergent soluble as shown in Figure 1A, the cross-linked dimeric uPAR displayed an inverse distribution, with the majority being associated with the lipid rafts (Figure 3A, upper panel). When the same experiment was performed on CD-treated cells, the shift towards detergent solubility of dimeric uPAR was reduced as compared with monomeric uPAR, indicative of a stronger interaction of the dimeric molecule with rafts. Similar results were obtained using an independent 293 clone expressing >10-fold less uPAR receptor, excluding the possibility that uPAR dimerization and the differential raft partitioning is an artefact of high level expression (results not shown). The reciprocal detergent solubility of monomeric and dimeric uPAR was even more dramatic when the lysis was performed using Brij-58 at 37°C (Figure 3B). In this case, the most abundant form of uPAR in the raft fractions was the dimer, possibly suggesting that the association of monomeric uPAR with rafts may, at least partially, be an artefact caused by detergent solubilization at 4°C. Control experiments in which the cross linking was performed on the individual fractions obtained after the flotation demonstrated that both monomeric and dimeric uPAR displayed a distribution in the gradients identical to that observed when the cross linking was performed on intact cells (data not shown). Additional evidence for the preferential raft partitioning of dimeric uPAR was obtained by co-immunoprecipitation analysis of fractions obtained from flotation experiments on 293 cells transfected with both wild-type and FLAG-tagged uPAR (Figure 3C). In these experiments, the majority of co-immunoprecipitated uPAR was found in the raft fractions even though the majority of receptor was located in the non-raft fraction.

Fig. 3. Dimerization regulates uPAR partitioning to lipid rafts. (A) Differential raft partitioning of monomeric and dimeric uPAR. 293/uPAR cells were incubated in the presence (lower panel) or absence (upper panel) of CD (10 mM, 1 h, 37°C), treated with BS3 (0.5 mM, 30 min, 4°C), and analyzed by flotation and immunoblotting. (B) Flotation analysis of BS3-treated 293/uPAR cells lysed at 37°C in buffer containing 1% Brij-58. (C) Differential raft-partitioning of dimeric uPAR analyzed by co-immunoprecipitation. Flotation analysis was carried out on lysates of 293 cells expressing both wild-type and FLAG-tagged uPAR. Raft fractions (2, 3 and 4) and non-raft fractions (6, 7 and 8) were pooled and subjected to co-immunoprecipitation analysis as described in the legend to Figure 2C. The total uPAR present in each membrane compartment was visualized by immunoblotting with a polyclonal anti-uPAR antibody. Prior to immunoprecipitation, all fractions were equalized for detergent and sucrose concentration. The results presented are representative of two (C) or more (A and B) independent experiments.
Dimerization of uPAR is independent of its association with lipid rafts
The preferential partitioning of dimeric uPAR to lipid rafts prompts the question of whether uPAR dimerization causes raft association, or if the association of monomeric uPAR with rafts causes dimerization. It has been shown that clustering of GPI-anchored proteins in lipid rafts is cholesterol dependent (Friedrichson and Kurzchalia, 1998), and we therefore assessed the effect of CD on chemical uPAR/uPAR cross linking (Figure 4A) and co-immunoprecipitation (Figure 4B). In this analysis, CD failed to inhibit subsequent chemical cross linking or co-immunoprecipitation of uPAR, demonstrating that uPAR dimers remain stable even after raft disruption. To address the possibility that an initial lipid raft association may have caused the uPAR dimer formation, we repeated the chemical cross linking experiments on cells expressing the transmembrane anchored uPAR (Figure 4C). However, even though this receptor fails to partition to rafts (Figure 1B), the chemical cross linking consistently resulted in the appearance of an adduct with the size expected for a uPAR-TM dimer.
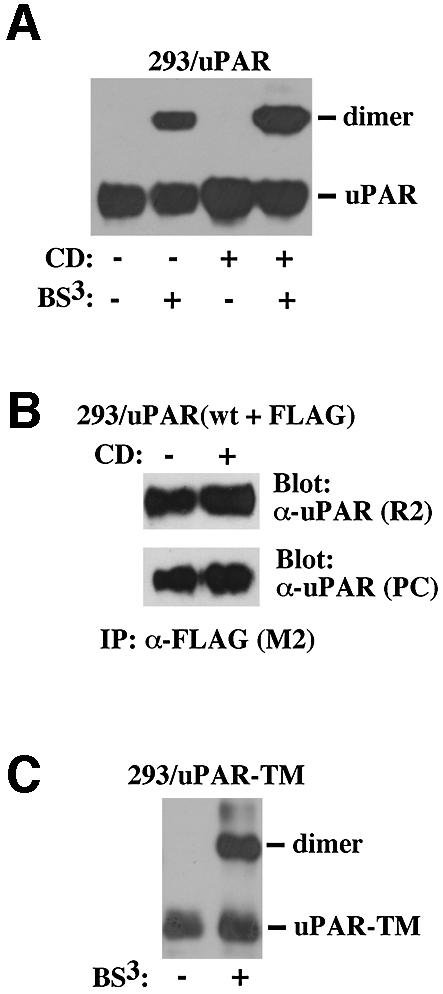
Fig. 4. Dimerization of uPAR does not require its association with lipid rafts. (A) The effect of CD treatment on chemical cross linking. 293/uPAR cells were cholesterol depleted using CD and treated with BS3 as indicated below the panel, and the cell lysates analyzed by western blotting. (B) The effect of CD treatment on uPAR co-immunoprecipitation. 293 cells transfected with a combination of uPAR-WT and uPAR-FLAG were treated with CD as indicated and analyzed by co-immunoprecipitation as described in the legend to Figure 2C. (C) Chemical cross linking of uPAR-TM. 293/uPAR-TM cells were treated with chemical cross linker as indicated and cell lysates analyzed by western blotting as above.
Binding of uPA to uPAR is independent of receptor dimerization and raft association
A major function of uPAR is to promote pericellular plasminogen activation through the high-affinity binding of the plasminogen activator uPA to the cell surface, and we therefore determined the capacity of uPAR to direct the binding of uPA to the two different membrane compartments. To this end, we subjected 293/uPAR cells incubated with 125I-uPA to flotation analysis and assayed the resulting fractions for the presence of radiolabel by γ-counting (Figure 5A). In these experiments, the distribution of 125I-uPA between the raft and non-raft fractions was similar to that of the receptor itself, with approximately one quarter of the 125I-uPA being found associated with the lipid rafts. This suggests that uPA binds to uPAR with a similar affinity, independently of where the receptor is located in the membrane. In control experiments in which the cells had been incubated with 125I-labeled receptor associated protein (RAP), very few counts were found associated with the raft fractions. This is consistent with the fact that RAP binds with high affinity to members of the LDL-related receptor family, which partition exclusively to the detergent-soluble membrane fraction (Marynen et al., 1984). To verify that the 125I-uPA recovered in the different fractions was indeed associated with uPAR, we subjected the pooled raft and non-raft fractions to immunoprecipitation using an α-uPAR or a control antibody and assayed the precipitates by γ-counting (Figure 5B).

Fig. 5. Lipid raft- and dimerization-independent binding of uPA to uPAR. (A) 293/uPAR cells were incubated with 125I-labeled uPA (1 nM, filled bars) or RAP (3 nM, open bars). After removal of unbound reagents the cells were lysed in Triton X-100 buffer and subjected to flotation analysis. Fractions were collected and the radioactivity was determined by γ-counting. The level of radioactivity in each fraction is shown as percentage of the sum of radioactivity in all fractions. The data represents the mean ±SD of two independent binding/flotation experiments. The percentage of total radioactivity associated with the raft and non-raft fractions is indicated below each graph. (B) To determine the amount of 125I-labeled uPA specifically associated with uPAR, raft and non-raft fractions were pooled and subjected to immunoprecipitation using a polyclonal antibody against uPAR (α-uPAR) or an irrelevant antibody (control). After washing, precipitated radioactivity was determined by γ-counting. (C) 293/uPAR cells were treated with BS3 (upper panel) or CD and BS3 (lower panel), as described previously, and subsequently to flotation analysis. Fractions were immunoprecipitated with biotinylated uPA and analysed by SDS–PAGE and immunoblotting using an anti-uPAR antibody.
The symmetrical distribution of uPAR and uPAR-associated uPA suggests that uPA interacts with both monomeric and dimeric uPAR. To test this possibility directly, the fractions obtained after flotation of cross-linked 293/uPAR cells (see Figure 3A) were subjected to pull-down analysis using biotinylated uPA, and assayed for uPAR by immunoblotting (Figure 5C). In this analysis, uPA was found to interact with both monomeric and dimeric uPAR, independently of whether these forms of the receptor were located in the raft or non-raft membrane fractions. Cholesterol depletion resulted in a shift of the material pulled down towards the non-raft fractions but did not significantly alter the forms, or quantity, of uPAR that was immunoprecipitated.
uPA-catalyzed uPAR cleavage is accelerated in lipid rafts
An important consequence of uPA binding to its receptor is the subsequent cleavage of uPAR between domains 1 and 2 to produce the GPI-linked anchored uPAR fragment D2D3 (Høyer-Hansen et al., 1992). To investigate if uPAR cleavage was affected by receptor localization and/or dimerization, we carried out time-course experiments of uPA-induced cleavage of cell surface uPAR (Figure 6). After incubation with uPA, cells were lysed and subjected to flotation analysis. As shown in Figure 6A, at t = 0 most uPAR is intact with the amount of D2D3 present subsequently increasing with the time of incubation with uPA. Intact uPAR and D2D3 were observed in both detergent-resistant as well as soluble fractions. To measure the rate of uPAR cleavage more accurately, the fractions corresponding to detergent-soluble and detergent-resistant membranes were pooled and analyzed by western blotting after deglycosylation (Figure 6B), and the blots quantified by densitometry (Figure 6C). The rate of uPAR cleavage in the lipid raft compartment was clearly much more rapid (half-life ∼10 min) when compared with non-raft membranes (half-life >1 h). In addition, uPAR cleavage was more complete in the rafts with no, or little, full-length uPAR remaining after 1 h in comparison with the almost 50% remaining in the detergent-soluble membrane compartments. The uPAR cleavage was strictly dependent upon the uPA/uPAR interaction, as the inclusion of a 100-fold excess of non-catalytic growth factor-like domain of uPA completely inhibited cleavage (data not shown). To address the possibility that uPAR cleavage affects uPAR dimerization, we performed co-immunoprecipitation experiment on cells exposed to uPA for different lengths of time (Figure 6D). In these experiments, the degree of uPAR cleavage was paralleled by a reduction in uPAR–uPAR interactions, since the amounts of R2-detected uPAR in M2 immunoprecipitates decreased with time (Figure 6D).

Fig. 6. uPA-mediated uPAR cleavage is accelerated in lipid rafts. (A) 293/uPAR cells were treated for various times (0, 1, 5, 15, 60 and 120 min) at 37°C with 3 nM uPA in DMEM supplemented with 0.1% BSA. After the incubation, cells were moved to ice, washed in ice-cold PBS, lysed in Triton X-100 containing buffer, and subjected to flotation analysis as described above. uPAR and cleaved uPAR (D2D3) were identified by immunoblotting analysis using a polyclonal anti-uPAR antibody. (B) Flotation fractions corresponding to the raft and non-raft material were pooled, aliquots were reduced and deglycosylated, fractionated by SDS–PAGE and immunoblotted with a polyclonal anti-uPAR antibody. (C) The intensity of the bands corresponding to full-length uPAR in (B) was quantified by densitometry and plotted as a percentage of the intensity observed in cells not exposed to uPA (0 min). The data represents the mean value of two independent experiments. (D) Lysates of 293/uPAR cells exposed to uPA (3 nM) for different lengths of time were immunoprecipitated with M2 and blotted with R2 to reveal the co-immunoprecipitated uPAR, or with a polyclonal anti-uPAR antibody to reveal total uPAR.
Dimerization directs the selective binding of the extracellular matrix protein Vn to uPAR in lipid rafts
We recently showed that dimerization of soluble uPAR is a prerequisite for its high-affinity interaction with the extracellular matrix protein Vn (Sidenius et al., 2002). As dimeric uPAR partitions preferentially to lipid rafts, we predicted that uPAR-dependent cell-surface Vn binding would occur selectively to the raft fraction. To address this possibility, we incubated 293/uPAR cells with 125I-labeled Vn, subjected detergent lysates to flotation analysis and analyzed the fractions for the presence of 125I-Vn as described above (Figure 7A). In these experiments, approximately one third of the 125I-Vn was recovered associated with the rafts, and the remainder in the fractions corresponding to the detergent-soluble material. Transfected uPAR is not the only binding site for Vn on 293 cells, as this cell line has been shown to express Vn-binding members of the integrin family (Bodary and McLean, 1990). To determine the amount of Vn specifically associated with uPAR in the different membrane fractions, we performed anti-uPAR immunoprecipitations and analyzed the precipitates by γ-counting (Figure 7B). In these experiments, 125I-Vn was found to associate much more efficiently with the uPAR present in rafts as compared with uPAR located in the non-raft membrane fractions (Figure 7B). To determine if the selective high-affinity binding of Vn to raft-associated uPAR was caused by the particular lipid environment and/or uPAR dimerization, fractions obtained after flotation of cross-linked 293/uPAR cells were subjected to pull-down analysis using a biotinylated recombinant Vn protein. This construct contained the 97 N-terminal residues of mature Vn, which encompasses the entire somatomedin B domain which has been shown to be responsible for the interaction with uPAR (Okumura et al., 2002). This protein was used instead of intact Vn because it is more easily biotinylated, and less prone to aggregation and loss due to absorption. Control experiments demonstrated that the Vn(1–97) protein interacts with soluble uPAR in a dimerization-dependent manner exactly as previously described for intact Vn (data not shown; Sidenius et al., 2002). In this analysis (Figure 7C), Vn(1–97) was found to preferentially precipitate dimeric uPAR, and predominantly from the raft fractions, supporting the notion that Vn interacts preferentially with dimeric uPAR. When the cells had been cholesterol depleted prior to cross linking and flotation, the Vn(1–97) did not precipitate any uPAR, suggesting that not only dimerization but also the lipid environment is crucial for the uPAR/Vn interaction. To determine if the reduced Vn(1–97) binding observed biochemically was paralleled by a reduction in cell adhesion (see Supplementary data for methods) we performed adhesion assays on 293/uPAR cells which had been treated with CD to disrupt cholesterol-dependent microdomains (Figure 7D). In these experiments, a significant reduction (P = 0.027, Student’s paired t-test) in cellular adhesion to Vn was observed after CD treatment. The reduction appeared to be specific for Vn, as integrin-mediated adhesion to fibronectin was unaltered by CD treatment. Control experiments using anti-uPAR antibodies demonstrated that the adhesion to Vn was uPAR dependent (data not shown).

Fig. 7. Dimeric uPAR directs the selective binding of Vn to lipid rafts. (A) 293/uPAR cells incubated with 125I-labeled Vn were lysed and subjected to flotation analysis. The resulting fractions were assayed for radioactivity. The level of radioactivity in each fraction is shown as a percentage of the sum of radioactivity in all fractions. The data represents the mean ±SD of two independent binding/flotation experiments. The percentage of total radioactivity associated with the raft and non-raft fractions is indicated. (B) To determine the amount of 125I-labeled Vn specifically associated with uPAR, the raft and non-raft fractions were pooled and subjected to immunoprecipitation using a polyclonal antibody against uPAR (α-uPAR) or an irrelevant antibody (ct. Ab). After washing, the radioactivity bound to the beads was determined by γ-counting. (C) Cells treated with BS3 (upper panel) or CD followed by BS3 (lower panel) were lysed, subjected to flotation analysis and aliquots of the resulting fractions were immunoprecipitated with biotinylated Vn(1–97). uPAR was visualized using an anti-uPAR polyclonal antibody. (D) Adhesion of 293/uPAR cells to Vn- or Fn-coated wells was allowed to proceed for 30 min at 37°C, with or without pre-treatment of cells with 10 mM CD for 1 h at 37°C (see Materials and methods). Adherent cells were quantified after fixing by staining with crystal violet. The data represent the mean ±SD of three independent experiments, each carried out in quadruplicate.
Discussion
Using chemical cross linking and co-immunoprecipitation we show that uPAR exists both in monomeric and dimeric form on the cell surface. Do these techniques allow us to estimate the fraction of uPAR that exists as a dimer? Cross linking may overestimate dimerization as the amount of cross-linked uPAR formed reflects not only the presence of stable uPAR dimers but also stochastic encounters between these molecules during treatment. In fact, cholesterol depletion fractionally increases the amount of uPAR that is cross linked. This possibly reflects an increase in stochastic encounters caused by the increased mobility of the receptors in the membrane. However, cross linking may also underestimate the extent of dimerization, as a fraction of uPAR may be dimeric, yet not react constructively with the cross linker to form stable dimers detectable by our methods. Co-immunoprecipitation is not prone to the artefacts observed due to stochastic encounters and is neither affected by cholesterol depletion. However, the degree of dimerization as estimated by co-immunoprecipitation is affected by differences in the levels of expression of the two receptors, the relative efficiency of the antibodies used in immunoprecipitation, as well as association/dissociation events that routinely occur during the precipitation procedure. Nevertheless, both techniques yield congruent results, namely that a minor fraction of uPAR (10–30%) is present at the cell surface in dimeric form.
Detergent solubility is a widely accepted technique that is routinely used in the analysis of membrane raft proteins. However, recent studies have clearly underscored the important limitations and pitfalls of this technique (Schuck et al., 2003). The current study is based on the assumption that the two pools of uPAR obtained by detergent fractionation correspond to two biochemically and biologically different membrane domains also present in the intact living cell. Much evidence supports this assumption. The cholesterol dependence of the association of uPAR with the detergent-resistant membrane fraction directly demonstrates that this fraction of uPAR is indeed associated with a cholesterol-dependent type of membrane domain, i.e. lipid rafts. A recent proteomic study, based on cholesterol depletion, has clearly identified uPAR as a ‘true’ raft protein (Foster et al., 2003). In our experiments most of uPAR is recovered in the fractions containing the detergent-soluble membranes. Although we generically term this material as ′non-raft′ it is important to note that it cannot be ruled out that these receptors are also associated with a type of lipid raft less resistant to detergent extraction. Indeed, the co-existence of different types of lipid rafts in the same cell membrane has been documented both in mammalian cells (Gomez-Mouton et al., 2001) and in yeast (Bagnat and Simons, 2002). Most importantly, the data presented demonstrate that the two populations of uPAR separated by detergent solubility actually represent functionally distinct membrane domains before the detergent extraction. The chemical cross linking is performed before cell lysis, yet the uPAR dimerization pattern is very different between the raft and non-raft fractions. The radioactively labelled Vn binding is performed before cell lysis, yet binding is largely confined to the raft fraction. The cleavage of uPAR by uPA is performed on intact cells, yet the cleavage kinetics are much faster in the raft fraction.
Our data clearly demonstrate that monomeric and dimeric uPAR are differentially distributed between the two types of membrane domain. Nevertheless, the distribution of the two forms of uPAR between the two membrane domains appears to be incomplete, as we observe both monomeric uPAR in the lipid raft fractions and dimeric uPAR in the non-raft fractions. However, several observations suggest that the differential partitioning in vivo may be more pronounced. First, a fraction of the dimeric uPAR associated with the detergent soluble membranes may derive from solubilized lipid rafts. Secondly, some of the monomeric uPAR found in lipid rafts may be dimeric uPAR which has escaped constructive cross linking. Thirdly, the differential partitioning is more pronounced when the cell lysis is performed at physiological temperature. It has previously been suggested that dimerization and oligomerization of membrane proteins may have a fundamental regulatory function in their association with lipid rafts (Simons and Toomre, 2000). Indeed, the induced clustering of different membrane proteins has been shown to enhance their partitioning to lipid rafts (Harder et al., 1998; Janes et al., 1999; Abrami et al., 2003). To our knowledge, however, our data represent the first demonstration that dimerization, the elementary form of oligomerization, is sufficient to determine receptor association with lipid rafts.
Binding experiments show that the differential membrane distribution of uPAR is paralleled by a similar distribution of binding sites for uPA, implying a dimerization- and lipid-environment-independent interaction between these two molecules. Pull-down experiments confirm this idea, as uPA is found to interact with both monomeric and dimeric uPAR, independently of membrane localization. The weak reduction in the binding of uPA to dimeric uPAR, as compared with the monomeric receptor, may possibly reflect minor differences in the affinity between uPA and the two forms of uPAR, but may also be caused by steric hindrance introduced in the presence of the cross linker. Cholesterol depletion causes a shift of the uPA binding sites towards the non-raft membrane fraction but does not significantly alter the amount, or type, of uPAR precipitated. Although the data demonstrates that uPA interacts equally well with uPAR independently of where in the membrane the receptor is located, they do not allow us to extrapolate that plasminogen activation, a major function of the uPA/uPAR interaction, will also display the same distribution. In fact, plasminogen activation requires the concomitant binding of both uPA and plasminogen to the cell surface (Ellis et al., 1991) and probably to the same membrane domain. Studies specifically addressing the role of raft association in uPAR’s function as a plasminogen activator are currently under way.
Binding of uPA to cell surface uPAR causes cleavage of the receptor in the linker region connecting domains 1 and 2 (Høyer-Hansen et al., 1992) and we now show that this cleavage is strongly accelerated in lipid rafts. At least two possible explanations may account for this observation. First, both the substrate (uPAR) and the enzyme (uPA) are presumed to be present at a higher density in lipid rafts as compared with the rest of the cell membrane, thereby favouring the cleavage reaction. Secondly, it is possible that the conformation of dimeric uPAR is more prone to cleavage by uPA. Our data do not allow us to distinguish between these two possibilities with certainty. Raft disruption by cholesterol treatment only marginally affects the overall cleavage rate of uPAR (data not shown), suggesting that it is dimerization rather than the lipid environment which is responsible for the acceleration of uPAR cleavage. However, even if the acceleration of raft uPAR cleavage was completely lipid environment dependent, the overall change in cleavage rate would be marginal as only a minor fraction of uPAR is located in lipid rafts. Specific inhibitors of uPAR dimerization will be required to directly investigate the possible role of dimerization in the acceleration of uPAR cleavage.
The functional importance of the rapid uPAR cleavage in lipid rafts has yet to be determined. Our data clearly demonstrate that uPAR cleavage is likely to regulate uPAR dimerization, and by extension also uPAR’s lipid raft association. However, the generated D2D3 fragments remain raft associated, at least throughout the timescale of our experiments. This residual affinity of cleaved uPAR for lipid rafts may possibly be explained by a residual weak dimerization and/or a slow diffusion rate out of these membrane domains. Cleavage of uPAR may cause a rapid location-specific loss of uPA, as the D2D3 fragments have a 1000-fold lower affinity for uPA (Ploug et al., 1994) and Vn binding activity. This may not only limit local plasminogen activation and hence extracellular proteolysis but may also destroy uPAR-Vn interactions which require intact uPAR (Høyer-Hansen et al., 1997; Sidenius and Blasi, 2000). Beside its effects on uPA and Vn binding, uPAR cleavage generates the potent chemotactic uPAR fragment, D2D3 (Resnati et al., 2002). The cleavage of uPAR in lipid rafts therefore rapidly generates a high local concentration of D2D3 in a membrane domain known to be enriched in many signalling molecules necessary to initiate cell migration, including G-protein coupled receptors and their downstream signalling molecules (Foster et al., 2003). That the raft association of uPAR is important for its function in signal transduction is indeed supported by recent studies showing that in neutrophils uPAR clustering is required for uPA-dependent increase in Ca2+ flux, and that cholesterol depletion blocks subsequent uPA-induced Ca2+ flux (Sitrin et al., 2003).
In contrast to uPA, which binds uPAR independently of its membrane localization and dimerization state, we clearly demonstrate that Vn binds preferentially to dimeric, raft-associated, uPAR. Although cholesterol depletion has little or no effect on the extent of uPAR dimerization, the treatment completely blocks the uPAR/Vn interaction in pull-down experiments. Even if the inhibition of the uPAR/Vn interaction by cholesterol depletion correlates with the disruption of lipid rafts, detailed analysis of the data suggests that it is not the disruption of the lipid raft per se that blocks the uPAR/Vn interaction. First, cholesterol depletion only causes a partial disruption of lipid rafts, as indicated by the shift of uPAR towards the non-raft fractions, yet it causes a complete inhibition of the uPAR/Vn interaction as evaluated by pull-down assays. Secondly, even in untreated cells a minor fraction of dimeric receptor recovered in the non-raft membrane fractions is pulled down by Vn(1–97). After cholesterol treatment, the same fractions contain more dimeric uPAR, yet none of it can be pulled down by Vn. Thirdly, some dimeric uPAR is recovered in the raft membrane fractions even after cholesterol depletion, yet this uPAR cannot be pulled down by Vn. Besides demonstrating a direct role of the lipid environment in the uPAR/Vn interaction, these experiments strongly suggest that it is not the integrity of the lipid rafts (as evaluated by buoyancy) that determines if the uPAR dimer is in a Vn-binding conformation or not, but rather the lipid microenvironment more closely associated with the receptor. Consistently with the Vn(1–97) pull-down assays, cholesterol depletion is associated with a significant reduction in cell adhesion to immobilized Vn, although the inhibition in this case is incomplete. The partial inhibition of adhesion may possibly be explained by cholesterol repopulation of the cell membrane during the adhesion assay, but this possibility was not addressed experimentally.
It is evident from our data that dimerization controls uPAR interaction with different proteins and membrane domains, but how is uPAR dimerization itself regulated? We have previously shown that soluble uPAR dimerizes in vitro, but only in the presence of uPA (Sidenius et al., 2002). However, unlike suPAR, dimerization of cell surface uPAR apparently does not require uPA as it is observed in 293/uPAR cells which produce no uPA. One possible explanation is that the high level of uPAR expressed by 293/uPAR cells uncouples the ligand dependence. However, we also observe efficient uPAR dimerization in 293/uPAR clones expressing significantly reduced levels of receptor (data not shown). A second possibility is that the interaction between uPAR and another protein(s), apart from uPA, controls dimerization. One obvious candidate is Vn, as this protein interacts preferentially with dimeric uPAR and is present in high quantities in the serum-containing media used to culture the 293 cells. A third possibility is that membrane-anchored uPAR has a conformation favouring dimerization and similar to that otherwise provided by the presence of the ligand. Indeed, it has recently been shown that soluble and GPI-anchored uPAR have different conformations (Høyer-Hansen et al., 2001; Andolfo et al., 2002).
Materials and methods
Materials
Cell culture media and supplements were purchased from Gibco-BRL and plastic ware was from Costar. Methyl-β-cyclodextrin, M2 antibody and general laboratory chemicals were from Sigma-Aldrich. Secondary antibodies and radiolabel were from Amersham Pharmacia Biotech. PVDF membranes used in western blotting were from Millipore. Triton X-100, disuccinimidyl suberate (DSS), bis-sulfodisuccinimidyl suberate (BS3), Iodogen, chemoluminescent substrate and Streptavidin beads were from Pierce. Urea-purified Vn was obtained from Promega and clinical grade uPA from Crinos (Italy). Vn, uPA and RAP were iodinated according to the Iodogen procedure as previously described (Behrendt et al., 1991). The R2, R4 and polyclonal anti-uPAR antibodies were kindly provided by Dr Gunilla Høyer-Hansen (Finsen Laboratory, Copenhagen). The Trx-VN1-97wt vector was kindly provided by David Loskutoff (Scripps Research Institute, CA).
Cell culture, cholesterol depletion, chemical cross linking and binding assays
293 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with penicillin (100 U/ml), streptomycin (100 U/ml), glutamine (5 mM) and 10% fetal bovine serum (FBS) at 37°C in 5% CO2. Transfections were performed using Fugene 6 according to the manufacturer’s instructions (Roche) and stable transfectants were selected in 0.8 mg/ml G418. Individual cell clones were characterized by western blotting using antibodies R2 and/or M2 and maintained in 0.2 mg/ml G418. Cholesterol depletion was performed on semi-confluent cell layers (10 cm dishes) washed twice with serum-free DMEM (0.1% BSA) and incubated in DMEM (0.1% BSA), containing 10 mM CD for 1 h at 37°C. Cells were washed twice with ice-cold PBS and lysed directly or subjected to chemical cross linking. Cross linking was performed by incubating cells with 0.5 mM BS3 in PBS for 30 min at 4°C. Un-reacted BS3 was removed by washing the cells with PBS, and the cells lysed as described below. For binding assays, semi-confluent cells (5 cm dishes) were washed twice in binding buffer (DMEM containing 0.1% BSA and 25 mM HEPES pH 7.5) and incubated with 125I-labeled uPA, Vn or RAP for 90 min at 4°C. Unbound reagents were removed by washing four times with binding buffer and the cells lysed as described below.
Cell lysis, flotation analysis, immunoprecipitation and immunoblotting
For immunoblotting and co-immunoprecipitation experiments cells were lysed in RIPA buffer [0.1% sodium dodecyl sulfate (SDS), 1% Triton X-100, 1% deoxycholate, 0.15 M NaCl and 0.1 M Tris–HCl pH 7.6) containing a cocktail of protease inhibitors (Complete, Roche). After 10 min on ice, lysates were collected by scraping, sonicated for 15 s (0.3 s bursts), centrifuged at 13 000 r.p.m. for 15 min at 4°C and the supernatant recovered. Total protein concentrations were determined using the DC Protein Assay (Bio-Rad) with BSA as standard. For immunoprecipitation experiments using R2 and M2 antibodies, lysates were pre-cleared using Protein A–Sepharose beads (Amersham Pharmacia Biotech) and the supernatant transferred to another aliquot of beads to which the relevant antibody had been pre-bound for 2 h at 4°C. Following immunoprecipitation overnight, the beads were washed three times in RIPA buffer and the adsorbed material eluted by boiling in non-reducing sample buffer. Proteins were separated by SDS–PAGE, transferred to PVDF membranes (Millipore) and probed with antibodies as detailed in the figure legends. Immune complexes were visualized by incubation with peroxidase-conjugated secondary antibodies and chemiluminescent detection. For sucrose density gradient analysis of detergent extracts (flotation), semi-confluent cell layers (10 cm dishes) were washed twice with ice-cold PBS and lysed for 30 min on ice in 1 ml of buffer A (25 mM Tris–HCl pH 7.5, 0.15 M NaCl) containing 1% Triton X-100 and a cocktail of protease inhibitors. Lysates were collected by scraping, transferred to 12 ml ultra centrifugation tubes (Beckman Instruments Inc.) and mixed carefully with 1 ml of 80% sucrose in buffer A. The lysates were overlaid with a step gradient of sucrose (4 ml 30%, 2 ml 5% and 4 ml 0%) prepared in buffer A and centrifuged at 39 000 r.p.m. for 16 h using a SW41Ti rotor in a Beckman L7-55 ultracentrifuge at 4°C. After centrifugation, the top 4 ml of buffer A were removed and the remaining 8 ml were harvested as 1 ml fractions (1–8). Equal volumes (10 µl) of each fraction were analyzed directly by western blotting or pooled as the ‘rafts’ fraction (2, 3 and 4) and the ‘non-raft’ fraction (6, 7 and 8) and analyzed by immunoprecipitation. For immunoprecipitation with R2 and M2, 0.5 ml of the pooled raft and non-raft fractions were added to 0.5 ml of RIPA buffer and immunoprecipitated as described above. For immunoprecipitations using uPA and Vn(1–97), biotinylated proteins (3 µg/sample) were pre-coupled to streptavidin beads for 2 h at 4°C, beads were washed twice with RIPA and aliquots of fractions from flotation assays were added [using 0.2 ml of lysate for uPA immunoprecipitation and 0.5 ml for that with Vn(1–97)] and made up to a final volume of 1 ml with RIPA. Immunoprecipitation and analysis was then carried out as described above. To avoid possible artefacts caused by the different concentration of sucrose and Triton X-100 present in the different fractions, sucrose and Triton X-100 was added to the individual fractions to normalize for these compounds. Complete cell lysis and the separative power of the sucrose gradients was ensured by parallel analysis of cells which had been cholesterol depleted and/or cells expressing the detergent soluble receptor uPAR-TM plated at the same density.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We acknowledge the kind gift of the TrxVN1-97wt vector from David Loskutoff, Scripps Research Institute, La Jolla, CA. This work was performed within the Center of Excellence on Cell Differentiation of the Italian Ministry of University and Research (MIUR) and supported by grants from the Italian Association for Cancer Research (AIRC) and the European Union (Contract No. QLG1-CT-2000-01131).
References
- Abrami L., Liu,S., Cosson,P., Leppla,S.H. and van der Goot,F.G. (2003) Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol., 160, 321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfo A., English,W.R., Resnati,M., Murphy,G., Blasi,F. and Sidenius,N. (2002) Metalloproteases cleave the urokinase-type plasminogen activator receptor in the D1–D2 linker region and expose epitopes not present in the intact soluble receptor. Thromb. Haemost., 88, 298–306. [PubMed] [Google Scholar]
- Bagnat M. and Simons,K. (2002) Cell surface polarization during yeast mating. Proc. Natl Acad. Sci. USA, 99, 14183–14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrendt N., Ploug,M., Patthy,L., Houen,G., Blasi,F. and Danø,K. (1991) The ligand-binding domain of the cell surface receptor for urokinase-type plasminogen activator. J. Biol. Chem., 266, 7842–7847. [PubMed] [Google Scholar]
- Blasi F. and Carmeliet,P. (2002) uPAR: a versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol., 3, 932–943. [DOI] [PubMed] [Google Scholar]
- Bodary S.C. and McLean,J.W. (1990) The integrin beta 1 subunit associates with the vitronectin receptor alpha v subunit to form a novel vitronectin receptor in a human embryonic kidney cell line. J. Biol. Chem., 265, 5938–5941. [PubMed] [Google Scholar]
- Braccia A., Villani,M., Immerdal,L., Niels-Christiansen,L.L., Nystrom,B.T., Hansen,G.H. and Danielsen,E.M. (2003) Microvillar membrane microdomains exist at physiological temperature: Galectin-4’s role as lipid raft stabilizer revealed by ‘superrafts’. J. Biol. Chem., 278, 15679–15684. [DOI] [PubMed] [Google Scholar]
- Drevot P., Langlet,C., Guo,X.J., Bernard,A.M., Colard,O., Chauvin,J.P., Lasserre,R. and He,H.T. (2002) TCR signal initiation machinery is pre-assembled and activated in a subset of membrane rafts. EMBO J., 21, 1899–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis V., Behrendt,N. and Danø,K. (1991) Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J. Biol. Chem., 266, 12752–12758. [PubMed] [Google Scholar]
- Foster L.J., De Hoog,C.L. and Mann,M. (2003) Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl Acad. Sci. USA, 100, 5813–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrichson T. and Kurzchalia,T.V. (1998) Microdomains of GPI-anchored proteins in living cells revealed by crosslinking. Nature, 394, 802–805. [DOI] [PubMed] [Google Scholar]
- Gomez-Mouton C. et al. (2001) Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc. Natl Acad. Sci. USA, 98, 9642–9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T., Scheiffele,P., Verkade,P. and Simons,K. (1998) Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol., 141, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høyer-Hansen G., Rønne,E., Solberg,H., Behrendt,N., Ploug,M., Lund,L.R., Ellis,V. and Danø,K. (1992) Urokinase plasminogen activator cleaves its cell surface receptor releasing the ligand-binding domain. J. Biol. Chem., 267, 18224–18229. [PubMed] [Google Scholar]
- Høyer-Hansen G., Behrendt,N., Ploug,M., Danø,K. and Preissner,K.T. (1997) The intact urokinase receptor is required for efficient vitronectin binding: receptor cleavage prevents ligand interaction. FEBS Lett., 420, 79–85. [DOI] [PubMed] [Google Scholar]
- Høyer-Hansen G., Pessara,U., Holm,A., Pass,J., Weidle,U., Danø,K. and Behrendt,N. (2001) Urokinase-catalysed cleavage of the urokinase receptor requires an intact glycolipid anchor. Biochem. J., 358, 673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes P.W., Ley,S.C. and Magee,A.I. (1999) Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol., 147, 447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen M., Lund,L.R., Rømer,J., Almholt,K. and Danø,K. (1998) Cancer invasion and tissue remodeling: common themes in proteolytic matrix degradation. Curr. Opin. Cell Biol., 10, 667–671. [DOI] [PubMed] [Google Scholar]
- Liu D., Ghiso,J.A., Estrada,Y. and Ossowski,L. (2002) EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell, 1, 445–457. [DOI] [PubMed] [Google Scholar]
- Marynen P., Van Leuven,F., Cassiman,J.J. and Van den Berghe,H. (1984) Solubilization and affinity purification of the alpha 2-macroglobulin receptor from human fibroblasts. J. Biol. Chem., 259, 7075–7079. [PubMed] [Google Scholar]
- Okumura Y., Kamikubo,Y., Curriden,S.A., Wang,J., Kiwada,T., Futaki,S., Kitagawa,K. and Loskutoff,D.J. (2002) Kinetic analysis of the interaction between vitronectin and the urokinase receptor. J. Biol. Chem., 277, 9395–9404. [DOI] [PubMed] [Google Scholar]
- Ploug M., Rønne,E., Behrendt,N., Jensen,A.L., Blasi,F. and Danø,K. (1991) Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J. Biol. Chem., 266, 1926–1933. [PubMed] [Google Scholar]
- Ploug M., Ellis,V. and Danø,K. (1994) Ligand interaction between urokinase-type plasminogen activator and its receptor probed with 8-anilino-1-naphthalenesulfonate. Evidence for a hydrophobic binding site exposed only on the intact receptor. Biochemistry, 33, 8991–8997. [DOI] [PubMed] [Google Scholar]
- Resnati M., Pallavicini,I., Wang,J.M., Oppenheim,J., Serhan,C.N., Romano,M. and Blasi,F. (2002) The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl Acad. Sci. USA, 99, 1359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell, 103, 293. [DOI] [PubMed] [Google Scholar]
- Schuck S., Honsho,M., Ekroos,K., Shevchenko,A. and Simons,K. (2003) Resistance of cell membranes to different detergents. Proc. Natl Acad. Sci. USA, 100, 5795–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidenius N. and Blasi,F. (2000) Domain 1 of the urokinase receptor (uPAR) is required for uPAR-mediated cell binding to vitronectin. FEBS Lett., 470, 40–46. [DOI] [PubMed] [Google Scholar]
- Sidenius N., Andolfo,A., Fesce,R. and Blasi,F. (2002) Urokinase regulates vitronectin binding by controlling urokinase receptor oligomerization. J. Biol. Chem., 277, 27982–27990. [DOI] [PubMed] [Google Scholar]
- Simons K. and Toomre,D. (2000) Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol., 1, 31–39. [DOI] [PubMed] [Google Scholar]
- Sitrin R.G., Johnson,D.R., Pan,P.M., Harsh,D.M., Huang,J., Petty,H.R. and Blackwood,R.A. (2003) Lipid raft compartmentalization of urokinase receptor signaling in human neutrophils. Am. J. Respir. Cell Mol. Biol., doi: 10.1165/rcmb.2003-0079OC. [DOI] [PubMed] [Google Scholar]
- Waltz D.A. and Chapman,H.A. (1994) Reversible cellular adhesion to vitronectin linked to urokinase receptor occupancy. J. Biol. Chem., 269, 14746–14750. [PubMed] [Google Scholar]
- Wei Y., Lukashev,M., Simon,D.I., Bodary,S.C., Rosenberg,S., Doyle,M.V. and Chapman,H.A. (1996) Regulation of integrin function by the urokinase receptor. Science, 273, 1551–1555. [DOI] [PubMed] [Google Scholar]